

Abstract
Cocaine abuse disrupts dopamine system function, and reduces cocaine inhibition of the dopamine transporter (DAT), which results in tolerance. Although tolerance is a hallmark of cocaine addiction and a DSM-V criterion for substance abuse disorders, the molecular adaptations producing tolerance are unknown, and testing the impact of DAT changes on drug taking behaviors has proven difficult. In regard to treatment, amphetamine has shown efficacy in reducing cocaine intake; however, the mechanisms underlying these effects have not been explored. The goals of this study were twofold; we sought to (1) identify the molecular mechanisms by which cocaine exposure produces tolerance and (2) determine whether amphetamine-induced reductions in cocaine intake are connected to these mechanisms. Using cocaine self-administration and fast-scan cyclic voltammetry in male rats, we show that low-dose, continuous amphetamine treatment, during self-administration or abstinence, completely reversed cocaine tolerance. Amphetamine treatment also reversed escalated cocaine intake and decreased motivation to obtain cocaine as measured in a behavioral economics task, thereby linking tolerance to multiple facets of cocaine use. Finally, using fluorescence resonance energy transfer imaging, we found that cocaine tolerance is associated with the formation of DAT-DAT complexes, and that amphetamine disperses these complexes. In addition to extending our basic understanding of DATs and their role in cocaine reinforcement, we serendipitously identified a novel therapeutic target: DAT oligomer complexes. We show that dispersion of oligomers is concomitant with reduced cocaine intake, and propose that pharmacotherapeutics aimed at these complexes may have potential for cocaine addiction treatment.
SIGNIFICANCE STATEMENT Tolerance to cocaine's subjective effects is a cardinal symptom of cocaine addiction and a DSM-V criterion for substance abuse disorders. However, elucidating the molecular adaptions that produce tolerance and determining its behavioral impact have proven difficult. Using cocaine self-administration in rats, we link tolerance to cocaine effects at the dopamine transporter (DAT) with aberrant cocaine-taking behaviors. Further, tolerance was associated with multi-DAT complexes, which formed after cocaine exposure. Treatment with amphetamine deconstructed DAT complexes, reversed tolerance, and decreased cocaine seeking. These data describe the behavioral consequence of cocaine tolerance, provide a putative mechanism for its development, and suggest that compounds that disperse DAT complexes may be efficacious treatments for cocaine addiction.
Keywords: agonist therapy, behavioral economics, nucleus accumbens, self-administration, tolerance, voltammetry
Introduction
Drug addiction is one of the most damaging public health problems, resulting in close to 600,000 deaths per year and a societal cost of over $500 billion in the United States alone (McGinnis et al., 1999; O'Connor et al., 2014). Cocaine use results in disrupted neuronal function, especially of dopaminergic systems (Volkow et al., 1996, 1997; Dackis and O'Brien, 2001), which can lead to escalated and uncontrolled drug intake. In particular, studies in cocaine-dependent humans have revealed that cocaine addiction is associated with a reduction in the ability of cocaine to inhibit the dopamine transporter (DAT), increase dopamine levels, and produce euphoric effects (Volkow et al., 1996, 1997, 2014). Preclinical literature has identified the DAT as the main site of action for cocaine, necessary for both the euphoric effects and self-administration behavior (Roberts et al., 1977; Ritz et al., 1987; Chen et al., 2006; Thomsen et al., 2009); thus, changes in cocaine effects at the DAT have been hypothesized to be a major component in the development of addiction. Despite the implications of cocaine tolerance and DAT alterations in addiction, the molecular changes that underlie tolerance have remained elusive, and directly testing the effects of tolerance on drug taking and seeking behavior has been a long-standing challenge.
Amphetamine (AMPH) has shown efficacy in reducing cocaine intake in human addicts in multiple clinical trials (Grabowski et al., 2001, 2004; Rush and Stoops, 2012; Nuijten et al., 2016). Currently, AMPH is categorized as an agonist replacement therapy (i.e., a medication that acts on the same primary target as the abused compound), which typically must be administered indefinitely on a daily maintenance schedule to be effective (Rush and Stoops, 2012); however, there have been no investigations of the cellular mechanisms underlying AMPH's efficacy in reducing cocaine intake. Here we hypothesized that AMPH's therapeutic effects may be due to interactions with the DAT that result in changes in cocaine potency and dopamine neurotransmission (Ferris et al., 2015). Thus, we sought to (1) identify the molecular alterations to DATs that underlie cocaine tolerance, (2) determine the behavioral impact of cocaine tolerance, and (3) determine whether AMPH interferes with these mechanisms. To address these questions, we used the long- and short-access cocaine self-administration procedure in which animals are given daily access to cocaine for 1 h (short-access; ShA) or 6 h (long-access; LgA) per day. LgA conditions result in cardinal sequelae of addiction, including escalation of cocaine intake, compulsive cocaine seeking, and increased motivation to administer cocaine (Ahmed and Koob, 1998; Grimm et al., 2001; Paterson and Markou, 2003). This is in contrast to animals given access to cocaine under ShA conditions, which do not develop aberrant cocaine-taking behaviors over time. Comparisons of these two conditions provides a powerful model for determining the molecular underpinnings of cocaine addiction pathology. By combining drug self-administration with fast-scan cyclic voltammetry (FSCV), and fluorescence resonance energy transfer (FRET) imaging, we systematically identify the molecular alterations in the DAT that may underlie drug seeking.
We found that LgA cocaine self-administration produces neurochemical tolerance to cocaine effects concomitant with dysregulated cocaine intake. AMPH, when administered via mini-pump at a low dose during or after cocaine self-administration, ameliorated cocaine tolerance, prevented escalation of cocaine intake, and reduced maladaptive drug seeking even after cessation of treatment. Using behavioral economic analyses, we found that the increased motivation to administer cocaine after LgA was also prevented by AMPH treatment, thus linking DAT alterations to multiple aberrant cocaine-taking behaviors. Importantly, we show that cocaine exposure produced tolerance to cocaine effects concomitantly with the formation of DAT oligomer complexes, and that AMPH reversed cocaine tolerance as well as returning DATs to a monomer configuration. Thus, AMPH's ability to reduce cocaine intake likely results from more complex mechanisms than previously hypothesized. These findings provide a putative molecular mechanism underlying cocaine tolerance and provide a link between dopamine system dysfunction and aberrant cocaine-taking behaviors.
Materials and Methods
Animals.
Male Sprague Dawley rats (350–400 g at the start of experiments; Harlan Laboratories) were maintained on a 12:12 h reverse light/dark cycle (3:00 A.M. lights off; 3:00 P.M. lights on) with food and water ad libitum. All animals were maintained according to the National Institutes of Health guidelines in Association for Assessment and Accreditation of Laboratory Animal Care accredited facilities. The experimental protocol was approved by the Institutional Animal Care and Use Committee at Wake Forest School of Medicine.
Self-administration surgery and training.
Rats were anesthetized and implanted with chronic indwelling jugular catheters as previously described (Siciliano et al., 2015). Animals were singly housed, and all sessions took place in the home cage during the active/dark cycle (9:00 A.M. to 3:00 P.M.). After a 2 day recovery period, animals underwent a training paradigm within which animals were given access on a fixed ratio 1 (FR1) schedule to a cocaine-paired lever, which, upon responding, initiated an intravenous injection of cocaine (0.75 mg/kg, infused over 4 s). After each response/infusion, the lever was retracted and a stimulus light was illuminated for a 20 s timeout period. Training sessions were terminated after a maximum of 20 infusions or 6 h, whichever occurred first. Acquisition occurred when an animal responded for 20 injections for 2 consecutive days and a stable pattern of infusion intervals was present.
Osmotic mini-pumps.
Following completion of the final acquisition session, animals were anesthetized briefly with isoflurane and osmotic mini-pumps (Alzet) containing AMPH (5 mg/kg/d) or saline were implanted subcutaneously just posterior to the shoulder blades. This dose is well below doses shown to have neurotoxic effects on dopamine neurons (Jonsson and Nwanze, 1982).
Extended access cocaine self-administration.
Following acquisition, animals were allowed to administer cocaine on an FR1 schedule of reinforcement (0.75 mg/kg, infused over 4 s) with unlimited injections for a total of 6 h (LgA) or 1 h (ShA) per day for a total of 14 d. Animals were treated with mini-pump delivered AMPH or saline throughout the 14 d of self-administration.
A separate group animals were allowed to self-administer under LgA or ShA conditions for 14 d before undergoing a 7 d abstinence period. In this group, animals were treated with mini-pump delivered AMPH or saline throughout the 7 d abstinence period. In all groups, pumps were removed following the final self-administration session, ∼18 h before being euthanized for voltammetric recordings.
Threshold procedure.
In a separate group of animals, the threshold procedure was used to determine LgA-induced changes in cocaine self-administration. The threshold procedure was performed following acquisition, again following 14 d of LgA with saline or AMPH mini-pump treatment, and a final assessment was made 7 d following cessation of LgA and treatment. The threshold procedure is a behavioral economics approach to assessing drug taking and reinforcing efficacy. Using behavioral economic analyses first requires generating a demand function, or in other words, measuring the consumption of a reinforcer across varying prices. Here, price was defined as responses required to obtain 1 mg of cocaine; thus, price can be manipulated in two ways: (1) by changing the response requirement per infusion or (2) by changing the amount of cocaine received with each infusion. Both approaches have been used to generate demand functions for drugs (Wade-Galuska et al., 2007; Oleson and Roberts, 2012). The threshold procedure, used here, consists of generating a demand curve by giving rats access to a descending series of 11 unit doses of cocaine (421, 237, 133, 75, 41, 24, 13, 7.5, 4.1, 2.4, and 1.3 μg/injection) available on an FR1 schedule of reinforcement. Each dose is available for 10 min, with each bin presented consecutively across the 110 min session. Animals performed this procedure for 3 consecutive days, and responding was averaged to derive the values used. Completion of the procedure produces a within-session dose–response curve, depicted in Figure 2B. Shifts in responding across the dose–response curve can be analyzed using behavioral economics principles, as described below.
Figure 2.

AMPH treatment prevented increase in motivation to administer cocaine. A, Experimental timeline showing when assessment of motivation to obtain cocaine (threshold procedure) was performed. Representative data from LgA+Saline (B) and LgA+AMPH (C) animals showing that the reinforcing efficacy of cocaine (Pmax) is decreased by AMPH treatment. D, LgA-induced increases in reinforcing efficacy are prevented by AMPH treatment during LgA. Importantly, reinforcing efficacy stays depressed for up to 7 d following cessation of treatment. Similar results were seen with Omax (E), but not Q0 (F). Error bars indicate ± SEM. *p < 0.05 versus LgA+Saline. LgA+Saline, n = 5; LgA+AMPH, n = 7.
Behavioral economic analysis was used to determine the maximal price paid (Pmax) for cocaine (Oleson et al., 2011; Oleson and Roberts, 2012). Briefly, Pmax was derived mathematically using a demand curve. Demand curves were generated by curve-fitting individual animals' intake using an equation: log(Q) = log(Q0) + k × (e − α × Q0 × C − 1)43. In this equation, Pmax was determined to be the unit price at which the first derivative point slope of the function = −1. The value k was set to 2 for all animals, whereas Q0 and α, which represent the acceleration of the function in response to changing price, were estimated to achieve best fit (Hursh and Silberberg, 2008; Bentzley et al., 2013). This measure is explained in detail below.
Q0: Q0 is a measure of the animals' preferred level of cocaine consumption. This can be measured when the dose is high and cocaine is available at low effort, or a minimally constraining price. This preferred level of consumption is established in the early bins of the threshold procedure.
Pmax: Price is expressed as the responses emitted to obtain 1 mg of cocaine; thus, as the dose is decreased in each consecutive bin of the threshold procedure, price increases. During the initial bins of the procedure, when the price is low, the animal is able to obtain a preferred level of cocaine intake with minimal responding. As the price is increased across bins, the animal must increase responding to maintain consistent intake. Pmax is the price at which the animal no longer emits enough responses to maintain intake and consumption decreases. Thus, animals with higher Pmax will increase responding to maintain cocaine levels farther into the dose–response curve; in other words, they will pay a higher price for cocaine. Here, we reported standardized Pmax which was derived by normalizing each animals' Pmax value to their Q0.
Ex vivo FCSV.
Animals were euthanized for FSCV experiments, as previously described (Siciliano et al., 2014a), the morning following the final self-administration session (∼18 h), when no drug was present. FSCV was used to characterize presynaptic dopamine system kinetics, dopamine autoreceptor sensitivity, and the ability of cocaine to inhibit dopamine uptake in the NAc core. A vibrating tissue slicer was used to prepare 400-μm-thick coronal brain sections containing the NAc core. The tissue was immersed in oxygenated aCSF containing the following (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 2.4 CaCl2, 1.2 MgCl2, 25 NaHCO3, 11 glucose, 0.4 l-ascorbic acid, pH adjusted to 7.4. Once sliced, the tissue was transferred to the testing chambers containing bath aCSF (32°C), which flowed at 1 ml/min. A carbon fiber microelectrode (100–200 μm length, 7 μm diameter) and bipolar stimulating electrode were placed into the core of the NAc. Dopamine release was evoked by a single electrical pulse (350 μA, 4 ms, monophasic) applied to the tissue every 3 min. Extracellular dopamine was recorded by applying a triangular waveform (−0.4 to 1.2 to −0.4 V vs Ag/AgCl, 400 V/s). Once the extracellular dopamine response was stable, quinpirole (3–300 nM) or cocaine (0.3–30 μm) was applied cumulatively to different brain slices.
Data analysis.
For all analysis of FSCV data, Demon Voltammetry and Analysis software was used (Yorgason et al., 2011). Recording electrodes were calibrated by recording responses (in electrical current; nA) to a known concentration of dopamine (3 μm) using a flow-injection system. To evaluate drug potency, evoked levels of dopamine were modeled using Michaelis-Menten kinetics as described previously (Siciliano et al., 2014b).
[3H]Dopamine uptake.
DAT was stably expressed in neuroblastoma N2A cells (N2A-DAT) as described previously (Chen et al., 2005). N2A-DAT cells were treated with vehicle or freshly made cocaine (10 μm) every day for 3 consecutive days. On day 4, culture medium was replaced with KRH buffer composed of the following (in mm): 25 HEPES, 125 NaCl, 4.8 KCl, 1.2 KH2PO4, 1.3 CaCl2, 1.2 MgSO4, and 5.6 glucose. AMPH (10 μm) or vehicle was added to the cells for 1 h followed by extensively washing with cold KRH three times. Dopamine uptake was initiated by adding 10 nm [3H]dopamine (specific activity: 23.5 Ci/mmol; PerkinElmer Life and Analytical Sciences) with or without cocaine (0.01–10 μm) in KRH for 10 min at 22°C as previously described (Chen et al., 2005). The reaction was terminated by washing cells twice with cold KRH, and cells were dissolved in 0.1% Triton. The amount of [3H]dopamine accumulated in the cells was determined by scintillation fluid in a liquid scintillation counter (Topcount, Packard Instrument). All experiments were performed in triplicate. A sigmoidal curve of cocaine competition for [3H]dopamine binding was generated using three-parameter nonlinear regression analysis. The IC50 values were extrapolated from the curve.
FRET.
Parental N2A cells were plated on poly-d-lysine-coated glass-bottom dishes (Mattek no. 1.5) at ∼20%–30% confluency. When the cells reached 40%–50% confluency, they were transfected with YFP-DAT (yellow fluorescent protein tagged DAT), CFP-DAT (cerulean fluorescent protein tagged DAT), using the Calcium Phosphate transfection kit (Invitrogen, K2780–01). After 12–15 h, the cells were gently washed 3 times with warm external solution (146 mm NaCl, 5 mm KCl, 30 mm dextrose, 5 mm HEPES, 1.2 mm MgCl2·6H2O, and 2.5 mm CaCl2·2H2O, pH 7.4, osmolarity 290–300 mOsm) and then again 3 times with warm media. This was followed by cocaine (10 μm) or vehicle treatment for 3 d in the incubator with media change containing fresh cocaine daily. On the day of imaging, the cells were treated with external solution or external solution containing amphetamine (10 μm) for 30 min at 37°C. The cells were then washed once in external solution and fixed with 3.7% PFA for 5 min at room temperature followed by three 5 min washes with DPBS and imaged using the Nikon Eclipse Ti confocal microscope. A similar experimental approach was used for positive and negative control experiments where FRET8 plasmid, or empty plasmids expressing either YFP or CFP, were expressed in the cells. FRET8 is a plasmid encoding tandem YFP-CFP and a generous gift from Dr. David Piston (Washington University, St. Louis).
FRET measurements were done using the method called acceptor photobleaching as previously described (Butler et al., 2015). The “dequenching” of the donor fluorophore was evaluated in the presence of an acceptor fluorophore by measuring the intensity of the fluorescence of the donor fluorophore in the same cell, preacceptor and postacceptor photobleaching. FRET was considered as positive when there was an increase in fluorescence intensity of donor fluorophore after photobleaching of the acceptor fluorophore. The microscope was configured to detect emission from the donor CFP (at 464–499 nm after excitation at 457 nm) and emission from the acceptor YFP (at 552–617 nm after excitation at 514 nm). After an initial baseline image acquisition, the acceptor (YFP) photobleaching was obtained using the 514 nm laser at 100% power for 2 s followed by another acquisition phase. Changes in the fluorescence of the donor (CFP) were quantified by subtracting postbleach images from the prebleach images after subtraction of background intensities. Quantified FRET efficiencies were calculated using the mean fluorescent intensity in the region of interest covering the plasma membrane of the cells.
The FRET efficiency was calculated at the plasma membrane using the following formula:
 |
The Donorpost is the intensity of the fluorescence of the donor (CFP) after photobleaching of the acceptor (YFP), and Donorpre is the intensity of the fluorescence of the donor before photobleaching of the acceptor.
Statistics.
Graph Pad Prism (version 6) was used to statistically analyze datasets and create graphs. Escalation data, behavioral economics data, burst frequency response curves, and concentration response curves for cocaine and quinpirole were subjected to a two-way repeated-measures ANOVA with session, burst frequency, or concentration as the within-subjects factor and experimental group as the between-subjects factor. Dopamine uptake data and FRET efficiencies between groups were compared using one-way ANOVAs. Differences between groups were tested using a Bonferroni post hoc test. All p values of <0.05 were considered to be statistically significant.
Results
AMPH treatment during cocaine self-administration prevents escalation of intake and augmented reinforcing efficacy of cocaine
We first assessed the effects of AMPH treatment during LgA (6 h access/d) and ShA (1 h access/d) cocaine self-administration (Fig. 1A). Increased intake over days occurred only in LgA animals treated with saline (LgA+saline), but not ShA treated with saline (ShA+saline). Intake was decreased by AMPH treatment (5 mg/kg/d, delivered continuously via subcutaneous osmotic mini-pump) in LgA animals (LgA+AMPH), but not ShA animals (ShA+AMPH) (Fig. 1B): repeated-measures two-way ANOVA, treatment group × session, group (F(3,20) = 45.31, p < 0.0001), interaction (F(39,260) = 1.572, p = 0.0216). Escalation, as defined by last session minus first session infusions, was greater than zero only in the LgA+Saline animals, demonstrating that AMPH selectively blocked escalation in LgA animals (Fig. 1C; one sample t-test, LgA+Saline vs 0, p = 0.01).
Figure 1.

AMPH treatment prevented escalation of cocaine self-administration. A, Timeline showing conditions across days (top) and within days (bottom). B, Total infusions per session were lowered by AMPH treatment during long-access, but not short-access, self-administration. C, Session 14 minus session 1 infusions reveal that LgA+Saline animals escalate intake over time, whereas all other groups do not. Error bars indicate ± SEM. *p < 0.05 versus LgA+Saline. **p < 0.01 versus LgA+Saline. ****p < 0.0001 versus LgA+Saline. #p < 0.05 versus 0. ShA+Saline, n = 6; ShA+AMPH, n = 4; LgA+Saline, n = 7; LgA+AMPH, n = 7.
We then used a behavioral economics approach to assess drug taking longitudinally across multiple points in the animal's self-administration history: at baseline, after LgA, and after an abstinence period (Fig. 2A). This approach, termed the “threshold procedure,” allows for the assessment of an animal's motivation to obtain cocaine (maximal price paid [Pmax]) (Fig. 2B,C). In LgA+saline animals, the motivation to administer cocaine was increased, which is a critical component of addiction in humans (Fig. 2D): repeated-measures two-way ANOVA (treatment group × time), group (F(1,26) = 7.762, p = 0.0098). Amphetamine treatment, delivered by osmotic mini-pump during LgA self-administration, blocked the increased motivation to administer cocaine (Fig. 2D,E). Importantly, AMPH mini-pumps were removed 18 h before the assessment of reinforcing efficacy; thus, these results are a consequence of prior AMPH treatment, rather than an acute AMPH effect. Also, in animals treated with AMPH, the reinforcing efficacy of cocaine did not rebound even 1 week following cessation of treatment (Fig. 2D), prompting us to hypothesize that AMPH effects go beyond simple agonist replacement therapy. We then aimed to determine the neurochemical mechanism underlying escalation of cocaine intake, and if amphetamine is able to reverse these deficiencies.
AMPH treatment during cocaine self-administration prevents cocaine-induced DAT alterations
To determine how the development of addictive behaviors and reversal by AMPH relate to cocaine effects at the DAT, animals were killed for ex vivo FSCV recordings in the NAc core 18 h following cessation of the final cocaine self-administration session and AMPH (or saline) treatment (Fig. 3A). Before examining cocaine effects, we first determined whether basal dopamine terminal function was altered. To probe differences in dopamine release magnitude, we used 5 pulse stimulations and varied stimulation frequency to mimic the within-burst firing rates of dopamine neurons (Schultz, 1986; Hyland et al., 2002). LgA reduced phasic dopamine release (i.e., 20 or 100 Hz stimulations) (Fig. 3B,C), and this decrease was selectively prevented by treatment with AMPH (Fig. 3B): repeated-measures two-way ANOVA (treatment group × burst frequency), burst frequency (F(4,136) = 49.46, p < 0.0001). Phasic/tonic ratios (100 Hz/5 Hz) revealed that LgA reduced the contrast between tonic and phasic signaling and that all other groups were unaffected (Fig. 3D): one-way ANOVA (F(3,34) = 2.416, p = 0.0834); Bonferroni post-test: ShA+Saline vs LgA+Saline (p < 0.05). Importantly, we saw no effect on maximal rate of dopamine uptake (Vmax) in any group (Fig. 3E), and alterations in dopamine release were not driven by changes in D2-type autoreceptor activity (Fig. 3F).
Figure 3.
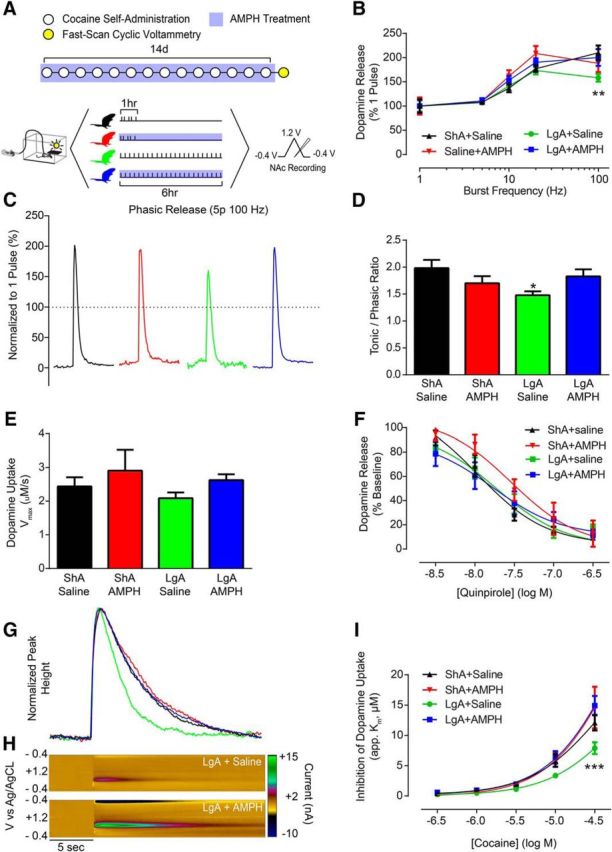
AMPH treatment prevented cocaine-evoked alterations to rapid dopamine signaling and cocaine potency. A, Timeline showing conditions across days (top) and within days (bottom). B, Dopamine release elicited by phasic-like stimulations (5 pulse, 100 Hz) is reduced in LgA animals, and this effect is prevented by treatment with AMPH during cocaine self-administration. C, Representative traces elicited by 100 Hz stimulation trains indicating that phasic dopamine release is attenuated in LgA+Saline animals, and that this effect is prevented by AMPH treatment. D, Phasic/tonic ratio (100 Hz/5 Hz) is reduced in LgA animals, and this effect is prevented by AMPH treatment. E, Maximal rate of dopamine uptake (Vmax) is unaffected in all treatment groups when AMPH treatment is given during cocaine self-administration. F, Quinpirole concentration response curves show no change in autoreceptor sensitivity across all treatment groups. G, Representative traces following bath application of 30 μm cocaine. H, Pseudo-color plots for LgA+saline (top) and LgA+AMPH (bottom) animals following bath application of 30 μm cocaine. I, Group data demonstrating that LgA cocaine self-administration results in a reduction in the ability of cocaine to inhibit the DAT, and that this effect is selectively prevented by AMPH treatment. Error bars indicate ± SEM. *p < 0.05 versus ShA+Saline. **p < 0.01 versus ShA+Saline. ***p < 0.001 versus ShA+Saline. ShA+Saline, n = 6; ShA+AMPH, n = 4; LgA+Saline, n = 6; LgA+AMPH, n = 7.
Despite basal dopamine uptake being unaltered, bath application of cocaine to brain slices (Fig. 3G,H) revealed that LgA resulted in marked tolerance to the ability of cocaine to inhibit the DAT compared with ShA animals; however, the development of tolerance was completely prevented by AMPH treatment (Fig. 3I): repeated-measures two-way ANOVA (treatment group × concentration), group (F(3,19) = 4.168, p = 0.0199), concentration (F(4,76) = 144.6, p < 0.0001), interaction (F(12,76) = 2.732, p = 0.0040). The fact that there was no effect of cocaine self-administration or AMPH treatment on dopamine uptake rate under drug-free bath conditions suggests that alterations in cocaine potency and rescue by AMPH treatment occur independently from canonical DAT function. These results prompted us to hypothesize that AMPH-induced decreases in cocaine intake are due to reversal of cocaine tolerance.
AMPH treatment during abstinence reverses the effects of chronic cocaine on the DAT
If AMPH's actions as a pharmacotherapeutic agent are indeed due to a selective amelioration of cocaine-induced dopamine deficits, it should reverse cocaine effects when given during abstinence, in addition to blocking cocaine effects when given during use. To test this hypothesis, animals were allowed 14 d of cocaine self-administration without treatment and were treated during a 7 d abstinence period (Fig. 4A). Following treatment, identical voltammetry experiments were performed to determine the ability of AMPH treatment during abstinence to reverse cocaine-induced dopaminergic deficits. Surprisingly, in contrast to decreased phasic dopamine signaling induced by LgA, following abstinence, phasic dopamine signaling was increased (Fig. 4B,C). Importantly, treatment with AMPH during abstinence selectively attenuated LgA-induced increases in phasic signaling while having no effect on ShA animals treated with AMPH (Fig. 4B): repeated-measures two-way ANOVA (treatment group × burst frequency), burst frequency (F(4,104) = 38.18, p < 0.0001). Calculation of phasic/tonic ratios revealed that LgA and withdrawal increased the dynamic range that can be encoded between tonic and phasic signaling and that all other groups were unaffected (Fig. 4D): one-way ANOVA (F(3,26) = 2.879; p = 0.0552); Bonferroni post-test: ShA+Saline vs LgA+Saline (p < 0.05). Similar to animals treated during cocaine self-administration, we found no effects on dopamine uptake rate (Fig. 4E), and no changes were observed in dopamine autoreceptor sensitivity (Fig. 4F).
Figure 4.
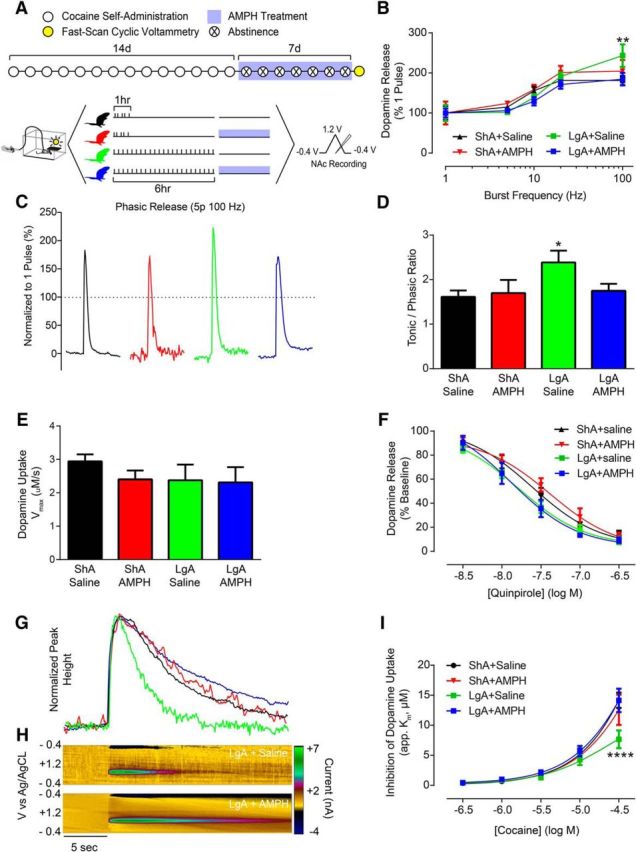
AMPH treatment reversed cocaine-induced changes to dopamine signaling. A, Experimental timeline depicting 14 d of cocaine self-administration (LgA or ShA) followed by a 7 d abstinence period. During the abstinence period, animals were treated with either saline or AMPH delivered via osmotic mini-pump. B, Dopamine release elicited by tonic-like frequency stimulations (5 pulse, 5 or 10 Hz) is unchanged in any treatment group. Dopamine release elicited by phasic-like frequency stimulations (5 pulse, 100 Hz) is increased in LgA animals after a 7 d abstinence period, and this effect is reversed by treatment with AMPH for 7 d following LgA. C, Representative traces indicating that phasic dopamine release is augmented in LgA animals after an abstinence period, and that this effect is reversed by AMPH treatment. D, Phasic/tonic ratio (100 Hz/5 Hz) is increased in LgA animals after a 7 d abstinence period, and this effect is reversed by AMPH treatment. E, Maximal rate of dopamine uptake (Vmax) is unaffected in all treatment groups when AMPH treatment is given during abstinence. F, Quinpirole concentration response curves show no change in autoreceptor sensitivity across all treatment groups. G, Representative traces following bath application of 30 μm cocaine indicating that cocaine is less effective at inhibiting dopamine uptake following LgA cocaine self-administration and abstinence. H, Pseudo-color plots for LgA+Saline (top) and LgA+AMPH (bottom) animals following bath application of 30 μm cocaine. I, Group data demonstrating that LgA cocaine self-administration animals remain tolerant to cocaine's effects following a 7 d abstinence period, and that this effect is reversed by AMPH treatment during abstinence. Error bars indicate ± SEM. *p < 0.05 versus ShA+Saline. **p < 0.01 versus ShA+Saline. ****p < 0.0001 versus ShA+Saline. ShA+Saline, n = 6; ShA+AMPH, n = 5; LgA+Saline, n = 4; LgA+AMPH, n = 5.
We then sought to determine whether AMPH treatment during abstinence reversed cocaine tolerance at the DAT. Following cocaine self-administration and abstinence (Fig. 4A), concentration-response curves for cocaine (Fig. 4G,H) revealed that LgA animals remained tolerant to cocaine's effects during abstinence. In support of our hypothesis, this effect was reversed by AMPH treatment (Fig. 4I): repeated-measures two-way ANOVA (treatment group × concentration), concentration (F(4,64) = 152.5; p < 0.0001), interaction (F(12,64) = 2.472, p = 0.0101). Together, these results demonstrate that AMPH selectively prevents or reverses cocaine-induced plasticity of DATs and ameliorates aberrant cocaine-taking behaviors.
Cocaine tolerance is concomitant with formation of DAT oligomers
Our data demonstrate a surprising effect of AMPH whereby both the neurochemical and behavioral deficits induced by escalated cocaine self-administration are prevented or reversed by AMPH treatment. We next sought to determine the molecular mechanisms underlying this amelioration. Previous studies have shown that DAT can be present at the membrane in complexes containing multiple DAT molecules, or DAT oligomers (Hastrup et al., 2001; Torres et al., 2003). Interestingly, these complexes may display altered inhibition by uptake inhibitors compared with DAT monomers (Chen and Reith, 2007), and thus may underlie the cocaine tolerance observed in the current study. Given that AMPH interacts with multiple signaling cascades that affect DAT and could alter its conformation (Fog et al., 2006; Moritz et al., 2015), we hypothesized that the development of cocaine tolerance and restoration by AMPH may be due to the formation/dispersion of DAT oligomer complexes.
To test this hypothesis, we first confirmed that the phenomenon of cocaine tolerance and reversal by AMPH could be observed in a reduced preparation where it is possible to detect DAT-DAT interactions. We exposed the N2A-DAT cells to cocaine (10 μm) for 3 d before measuring cocaine-induced inhibition of [3H]dopamine uptake. We found that, indeed, pretreatment with cocaine markedly reduced the ability of cocaine to inhibit DAT uptake activity without changing baseline uptake rates (Fig. 5A), mirroring our findings following self-administration of cocaine. Further, the addition of AMPH to the buffer fully reversed this effect back to control levels: repeated-measures two-way ANOVA (treatment group × concentration), concentration (F(5,30) = 664.1; p < 0.0001), group (F(2,6) = 11.42; p = 0.009), interaction (F(10,30) = 9.453; p < 0.0001). Calculation of IC50 concentrations for effects of cocaine on uptake revealed large cocaine pretreatment-induced increases (i.e., decreased potency) and restoration by AMPH (Fig. 5B): one-way ANOVA (F(2,6) = 104.6, p < 0.0001).
Figure 5.
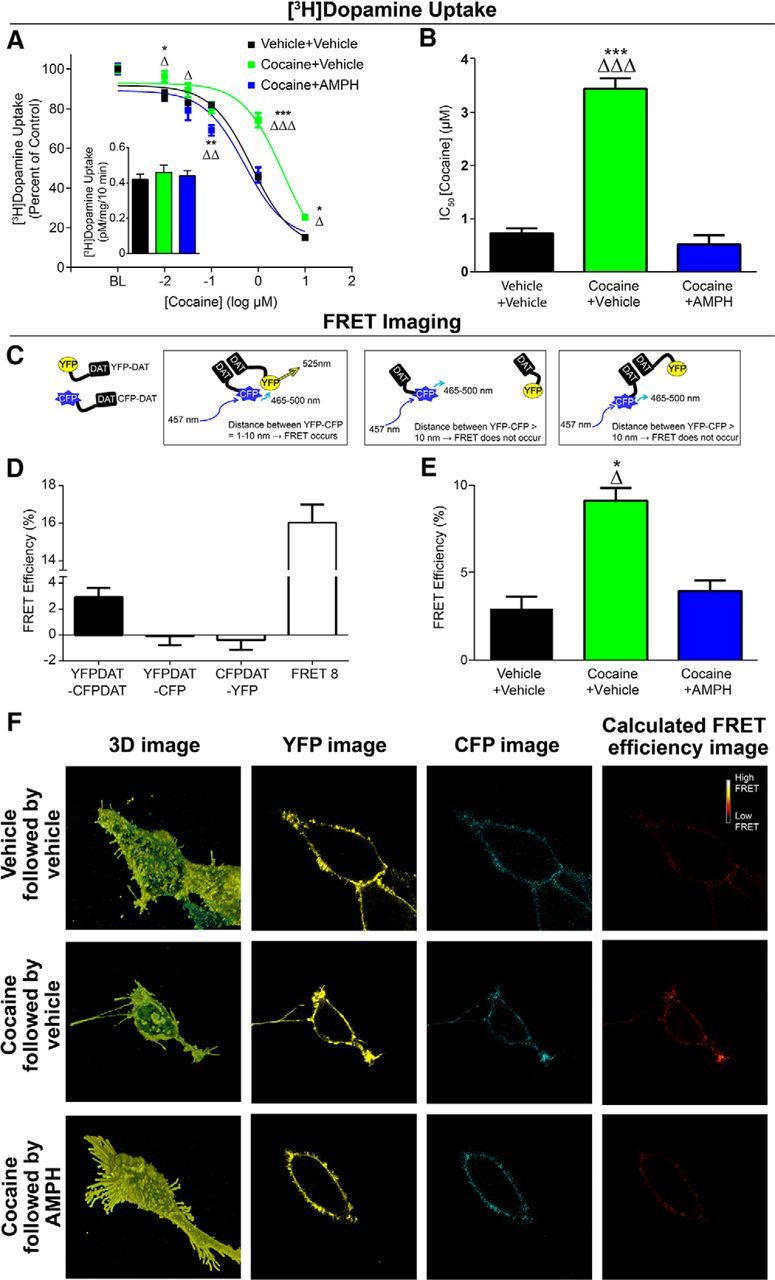
DAT oligomers are concomitant with cocaine tolerance. A, N2A cells were treated for 3 d with cocaine (10 μm) followed by AMPH (10 μm) on the fourth day before assessing cocaine-induced inhibition of [3H]dopamine uptake. We found that cocaine exposure produced marked tolerance to cocaine effects, that this effect was ameliorated by AMPH. Inset, Baseline uptake rate for 10 nm [3H]dopamine was unchanged in any group. B, IC50 values for cocaine effects on dopamine uptake across groups. C, Schematic describing the use of FRET imaging to measure DAT-DAT interactions. Cells were transfected with YFP-DAT and CFP-DAT or appropriate control constructs as described in Materials and Methods. D, Control conditions for FRET measurements. E, Group data showing that cocaine exposure increases FRET efficiency, and this was decreased by AMPH treatment. F, Representative computed FRET images generated by Nikon NIS Elements software using the FRET efficiency equation described above, showing effects of cocaine exposure and AMPH treatment on FRET efficiency between YFPDAT/CFPDAT. BL, baseline. Error bars indicate ± SEM. *p < 0.05 versus Vehicle+Vehicle. **p < 0.01 versus Vehicle+Vehicle. ***p < 0.001 versus Vehicle+Vehicle. ▵p < 0.05 versus Cocaine+AMPH. ▵▵p < 0.01 versus Cocaine+AMPH. ▵▵▵p < 0.001 versus Cocaine+AMPH. A, B, n = 3 per group. C–F, YFPDAT-CFP, n = 15; CFPDAT-YFP, n = 15; FRET8, n = 29; Vehicle+Vehicle, n = 26; Cocaine+Vehicle, n = 28; Cocaine+AMPH, n = 25.
To test the hypothesis that cocaine tolerance and its reversal by AMPH are associated with an oligomeric form of DAT complexes, we used a similar tolerance-inducing protocol in DAT-expressing cells. Here, however, we used FRET microscopy, a quantitative microscopy approach that allows examination of interactions between proteins that exist in close proximity to each other (Fig. 5C) (Schmid et al., 2001; Sorkina et al., 2003; Biener et al., 2005). We investigated the FRET efficiency between CFP-DAT, the donor molecule, and YFP-DAT, the acceptor molecule at the stacked membrane. This FRET pair has been extensively used and characterized in the literature (Oliveria et al., 2003; Rizzo et al., 2006; Grabowska et al., 2014; Butler et al., 2015). As shown in Figure 5D, we measured a positive FRET signal between YFP-DAT and CFP-DAT at or near the membrane. No FRET signal was detected in control experiments (Fig. 4D). The control experiments were performed in cells that: (1) coexpressed YFP empty vector and CFP-DAT or (2) coexpressed CFP empty vector and YFP-DAT. Furthermore, consistent with the literature (Rizzo et al., 2006; Grabowska et al., 2014), we measured a large FRET signal in cells expressing a tandem YFP-CFP (FRET8) (Fig. 5D), which served as a positive control group.
YFP-DAT/CFP-DAT-expressing cells were subjected to cocaine exposure followed by AMPH treatment as described in the [3H]dopamine uptake assay. We found that 3 d of cocaine exposure significantly increased the FRET efficiency between YFP-DAT and CFP-DAT (Fig. 5E,F), which suggests increased YFP-DAT/CFP-DAT oligomers. AMPH treatment following the 3 d cocaine exposure decreased cocaine-induced increase in FRET efficiency back to control levels (Fig. 5E,F): one-way ANOVA (F(2,76) = 24.16, p < 0.0001). These data are consistent with the interpretation that AMPH disperses cocaine-induced DAT oligomers and reverses cocaine-induced tolerance.
AMPH treatment reverses escalated cocaine use
Thus far, our data suggest that AMPH effects on self-administration of cocaine are due to AMPH-induced reversal of complex formations of the DAT, which produce functional alterations in signaling and resulting behavior. Further, these results are in opposition to proposed hypotheses stating that AMPH's efficacy as a pharmacotherapeutic agent are due to simple elevations in extracellular dopamine (Rush and Stoops, 2012). If AMPH effects are indeed due to reversal of plasticity, AMPH treatment should (1) reverse escalated cocaine consumption back to initial levels and (2) have lasting benefit beyond the treatment period. We tested for these qualities by allowing animals to escalate their cocaine intake over a 14 d self-administration period (initial escalation) before receiving AMPH treatment during an additional 7 d of self-administration (treatment period). Finally, animals were allowed to self-administer cocaine for 7 more days following the cessation of treatment (post-treatment period) (Fig. 6A). We found that AMPH treatment was able to reverse escalated cocaine intake back to pre-escalation levels. Remarkably, intake remained low during the 7 d post-treatment period (Fig. 6B,C): two-way ANOVA (treatment group × epoch), treatment group (F(1,23) = 16.41, p = 0.0005). Together, these data show AMPH-induced reversal of cocaine-evoked plasticity on a molecular, physiological, and behavioral level (Fig. 7).
Figure 6.

Cocaine reverses escalated cocaine intake. A, Experimental timeline of 28 d of cocaine self-administration split into three epochs: (1) 14 d of self-administration with no treatment (initial escalation period), (2) 7 d of AMPH treatment during cocaine self-administration (treatment period), and (3) 7 d of self-administration after cessation of treatment (post-treatment period). B, Average infusions per day over the 28 d of self-administration. Dotted line indicates average intake across groups on day 1. Shaded area represents the AMPH treatment period. C, Cocaine intake was decreased by AMPH treatment during the treatment and post-treatment periods. Error bars indicate ± SEM. *p < 0.05 versus LgA+Saline. **p < 0.01 versus LgA+Saline. LgA+Saline, n = 5; LgA+AMPH, n = 5.
Figure 7.

Summary of findings. Top, Model of the molecular underpinnings of the development of cocaine tolerance and restoration by AMPH. Bottom, Model of cocaine-induced changes to phasic dopamine signaling, cocaine potency, and reinforcing efficacy of cocaine over the course of LgA and abstinence. All three measures are normalized to control levels by AMPH.
Discussion
Here we outline molecular adaptations to the DAT produced by chronic cocaine exposure, and define their physiological impact on dopamine signaling and behavioral actions of cocaine. We show that escalated cocaine self-administration produces neurochemical tolerance to cocaine effects that lasts at least 7 d. Further, using AMPH to reverse tolerance, we show that preventing or reversing cocaine tolerance prevents or reverses escalation of cocaine intake, respectively. We provide a putative molecular mechanism for tolerance to cocaine effects, which has broad implications for the cocaine addiction field. Finally, we outline an unexpected mechanism for AMPH's pharmacotherapeutic actions, which may lead to the development of more targeted therapeutics, potentially avoiding the abuse liability associated with AMPH.
Specificity of AMPH effects and methodological considerations
One important finding of the current study is that the LgA model of cocaine self-administration accurately models the dopaminergic profile of human cocaine addicts. Indeed, PET imaging studies of cocaine-dependent individuals have revealed that they exhibit decreased dopaminergic responsiveness and tolerance to the dopamine-elevating effects of cocaine (Volkow et al., 1996, 1997, 2014), effects that were both observed following LgA in the current study (but see Ahmed et al., 2003). It is hypothesized that decreased cocaine potency is an integral component of the neurochemical consequences of cocaine use, which underlies maladaptive behaviors of cocaine addicts, such as increased intake to compensate for decreased subjective effects (Dackis and O'Brien, 2001). Our data support these hypotheses by providing a mechanistic link between cocaine potency at the DAT and the motivation to administer cocaine. We show that AMPH regimens that ameliorate cocaine-induced changes to the DAT also prevented escalation of intake, and suppressed LgA-induced increases in the reinforcing efficacy of cocaine. It is important to note that these changes are likely specific to cocaine reinforcement, as responding for food has been shown to be unchanged by AMPH using the same dose as in the current study (Chiodo et al., 2008). Additionally, studies in nonhuman primates have shown that AMPH treatment either does not affect, or transiently decreases, food consumption, whereas decreases in responding for cocaine are consistent and long-lasting (Negus et al., 2003a, b; Czoty et al., 2010, 2011). In the current study, neither cocaine self-administration nor AMPH treatment produced changes in basal dopamine uptake rate, demonstrating that changes in cocaine potency are independent of alterations to orthosteric function of the transporter (i.e., interactions with native substrates are unchanged). This is supported by previous work demonstrating that genetically increasing expression of the DAT has no effect on cocaine potency (Salahpour et al., 2008; Calipari et al., 2013), and is particularly important as it demonstrates that AMPH can modulate cocaine potency without disrupting basal DAT function. Further, this led us to hypothesize that cocaine tolerance induced by cocaine self-administration, and reversal by AMPH, were acting through an allosteric mechanism.
The observation that changes in cocaine potency are independent of orthosteric DAT function led us to explore possible conformational changes that could affect cocaine binding without altering basal uptake. Previous literature in cultured cells has suggested that the formation of DAT complexes can alter the effects of DAT blockers on uptake (Chen and Reith, 2007). Based on these studies, we explored the possibility that cocaine may affect the preferred conformation of the DAT, and found that cocaine exposure increased formation of oligomeric DAT complexes as indicated by increased FRET efficiency between YFP and CFP-tagged DATs. It is important to note that, although FRET microscopy reliably identifies close association of FRET pairs (∼100 Å), it does not distinguish the number of molecules within the oligomers and cannot tell us whether dimers, trimers, tetramers, or other groupings are formed, or whether there are additional proteins complexed with the DATs. In addition, our reported cocaine-induced increase in YFP-DAT/CFP-DAT FRET efficiency likely underestimates cocaine-induced DAT oligomerization because FRET does not occur between other combinations of oligomers, such as YFP-DAT/YFP-DAT or CFP-DAT/CFP-DAT oligomers. Unfavorable orientations of the two fluorophores will not result in FRET occurrence, thus further underestimating the actual magnitude of DAT-DAT interactions.
Mechanisms of DAT oligomerization
This is the first demonstration that chronic cocaine exposure can stimulate the formation of DAT oligomers. Importantly, these oligomers were dispersed by brief exposure to AMPH, similar to the restoration of cocaine effects observed in vivo. It is possible that AMPH is altering cocaine-induced oligomerization of DAT via mobilization of intracellular signaling cascades or proteins that could ultimately affect DAT conformation. For example, AMPH is known to interact with a number of different proteins that can also affect DAT, including α-synuclein (Mortensen et al., 2003; Wersinger et al., 2006; Butler et al., 2015) and dopamine D2 autoreceptors (Sevak et al., 2007; Chen et al., 2009, 2015; Bowton et al., 2010; Owens et al., 2012), as well as alter signaling pathways, including calmodulin kinases, phosphatidylinositol 3-kinase, Rho, and protein kinase A and Cβ (Carvelli et al., 2002; Fog et al., 2006; Wei et al., 2007; Lute et al., 2008; Saunders and Galli, 2015; Wheeler et al., 2015). Given that these pathways/proteins can result in differential phosphorylation, palmitoylation, and conformations of the DAT (e.g., inward- vs outward-facing DAT) and differential AMPH-induced DAT efflux (Khoshbouei et al., 2004; Cervinski et al., 2005; Foster et al., 2006, 2008, 2012; Robertson et al., 2009; Gaffaney et al., 2014; Moritz et al., 2015), it is possible that these alterations may result in restructuring of DAT-DAT interactions and return them to monomer conformation.
Alterations to presynaptic dopamine release
It is important to note that cocaine self-administration also resulted in changes in presynaptic dopamine release, which may play an important role in cocaine-seeking behaviors. Phasic dopamine release is involved in encoding the salience of rewards as well as encoding the probability that they will occur, given particular environmental cues (Waelti et al., 2001; Phillips et al., 2003; Tobler et al., 2005; Nicola, 2010; Steinberg et al., 2013). As such, phasic dopamine signaling is integral to selecting goal-oriented behaviors that produce advantageous outcomes, and its dysregulation by abused drugs has been hypothesized to be a critical component of maladaptive decision making in drug abusers. Here we show a time-dependent and biphasic effect of cocaine-induced alterations to phasic signaling at the level of the dopamine terminal. Initially, cocaine self-administration results in a reduction of dopamine terminal responsivity to phasic-like stimulations. This is consistent with previous results showing that phasic dopamine release decreases over the course of LgA proportionally with escalated cocaine intake (Willuhn et al., 2014). Combined with reduced postsynaptic dopamine receptors (Volkow et al., 1990; Nader et al., 2006), reductions in dopamine release are likely to result in a greatly decreased range of dopaminergic modulation of striatal outputs during decision-making in the acute withdrawal period. Conversely, following abstinence, phasic signaling was increased. Increased activation of NAc core output neurons by cocaine-associated cues is thought to underlie cocaine craving and seeking (Hollander and Carelli, 2007), which may be mediated, in part, by increased cue-elicited phasic dopamine signaling (Phillips et al., 2003). Thus, augmented phasic signaling during abstinence may contribute to the time-dependent increase in cocaine-seeking behaviors, termed “incubation,” which occurs following extended access cocaine self-administration (Grimm et al., 2001). It will be interesting to see whether the observed increase in phasic-like release potential ex vivo will translate to increased dopamine release to cocaine-associated cues in vivo. We found that low-dose continuous AMPH treatment ameliorated both decreases in phasic signaling observed immediately following LgA and increases in phasic signaling during abstinence.
The mechanisms governing cocaine-induced changes in release are currently unknown. One possible explanation is that release changes are also downstream of cocaine-induced alterations to the DAT. Because the DAT is responsible for reuptake and repacking of dopamine into vesicles, DAT function is tightly linked to vesicular dopamine content and storage, and changes in dopamine uptake often result in changes in dopamine release magnitude (Ferris et al., 2014). Although we did not see a change in uptake rate following LgA or AMPH treatment, vesicular repackaging also relies on membrane-bound formations of multiple proteins, including DAT, vesicular monoamine transporter-2, and scaffolding proteins, such as synaptogyrin-3 (Egaña et al., 2009; Cartier et al., 2010; Eriksen et al., 2010). Thus, it is possible that the formation of DAT oligomers may disrupt normal docking to vesicular monoamine transporter-2, thereby altering dopamine repacking and release. If this is the case, it would also explain why AMPH treatment rescued cocaine self-administration-induced changes in dopamine release.
In conclusion, we delineate a mechanistic link between cocaine-induced dopaminergic deficits and addictive behaviors. We show that escalation and increased motivation are associated with alterations in cocaine potency; and by reversing this tolerance, normal behavioral function is restored. A key aspect of the AMPH-induced restoration of dopamine system function is that cocaine self-administration remained attenuated even 7 d after cessation of treatment, suggesting that AMPH may have long-lasting benefits in human cocaine addicts after short treatment periods, rather than daily maintenance, which has been used with replacement therapies for heroin (Trussell and Gollance, 1970). Further, we demonstrate that the formation of DAT oligomers and subsequent restoration to monomers underlie the development of cocaine tolerance and AMPH reversal. Finally, one major reason that AMPH has not been approved for use in cocaine addicts despite extensive evidence of its efficacy (Grabowski et al., 2001, 2004; Negus et al., 2003a, b, 2007; Chiodo et al., 2008; Czoty et al., 2010, 2011; Rush and Stoops, 2012; Banks et al., 2015; Nuijten et al., 2016) is its inherent abuse liability (Negus and Henningfield, 2015). However, by elucidating the therapeutic action of AMPH, these findings may lead to the development of novel therapeutic compounds that have similar properties to AMPH in regard to stabilization of the dopamine system, but without abuse liability.
Footnotes
This work was supported by National Institutes of Health Grants R01 DA021325, R01 DA030161, and P50 DA006634 to S.R.J., R01 DA026947, NS071122, and OD020026 to H.K., T32 DA007246, F31 DA031533, and K99 DA042111 to E.S.C., and F31 DA037710, T32 AA007565, and F32 MH111216 to C.A.S., McKnight Brain Institute to K.S., and Brain and Behavior Foundation to E.S.C. and C.A.S.
The authors declare no competing financial interests.
References
- Ahmed SH, Koob GF (1998) Transition from moderate to excessive drug intake: change in hedonic set point. Science 282:298–300. 10.1126/science.282.5387.298 [DOI] [PubMed] [Google Scholar]
- Ahmed SH, Lin D, Koob GF, Parsons LH (2003) Escalation of cocaine self-administration does not depend on altered cocaine-induced nucleus accumbens dopamine levels. J Neurochem 86:102–113. 10.1046/j.1471-4159.2003.01833.x [DOI] [PubMed] [Google Scholar]
- Banks ML, Hutsell BA, Schwienteck KL, Negus SS (2015) Use of preclinical drug vs. food choice procedures to evaluate candidate medications for cocaine addiction. Curr Treat Options Psychiatry 2:136–150. 10.1007/s40501-015-0042-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzley BS, Fender KM, Aston-Jones G (2013) The behavioral economics of drug self-administration: a review and new analytical approach for within-session procedures. Psychopharmacology (Berl) 226:113–125. 10.1007/s00213-012-2899-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biener E, Charlier M, Ramanujan VK, Daniel N, Eisenberg A, Bjørbaek C, Herman B, Gertler A, Djiane J (2005) Quantitative FRET imaging of leptin receptor oligomerization kinetics in single cells. Biol Cell 97:905–919. 10.1042/BC20040511 [DOI] [PubMed] [Google Scholar]
- Bowton E, Saunders C, Erreger K, Sakrikar D, Matthies HJ, Sen N, Jessen T, Colbran RJ, Caron MG, Javitch JA, Blakely RD, Galli A (2010) Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J Neurosci 30:6048–6057. 10.1523/JNEUROSCI.5094-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler B, Saha K, Rana T, Becker JP, Sambo D, Davari P, Goodwin JS, Khoshbouei H (2015) Dopamine transporter activity is modulated by α-synuclein. J Biol Chem 290:29542–29554. 10.1074/jbc.M115.691592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Ferris MJ, Salahpour A, Caron MG, Jones SR (2013) Methylphenidate amplifies the potency and reinforcing effects of amphetamines by increasing dopamine transporter expression. Nat Commun 4:2720. 10.1038/ncomms3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier EA, Parra LA, Baust TB, Quiroz M, Salazar G, Faundez V, Egaña L, Torres GE (2010) A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J Biol Chem 285:1957–1966. 10.1074/jbc.M109.054510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvelli L, Morón JA, Kahlig KM, Ferrer JV, Sen N, Lechleiter JD, Leeb-Lundberg LM, Merrill G, Lafer EM, Ballou LM, Shippenberg TS, Javitch JA, Lin RZ, Galli A (2002) PI 3-kinase regulation of dopamine uptake. J Neurochem 81:859–869. 10.1046/j.1471-4159.2002.00892.x [DOI] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD, Vaughan RA (2005) Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J Biol Chem 280:40442–40449. 10.1074/jbc.M501969200 [DOI] [PubMed] [Google Scholar]
- Chen N, Reith ME (2007) Substrates and inhibitors display different sensitivity to expression level of the dopamine transporter in heterologously expressing cells. J Neurochem 101:377–388. 10.1111/j.1471-4159.2006.04384.x [DOI] [PubMed] [Google Scholar]
- Chen R, Han DD, Gu HH (2005) A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate. J Neurochem 94:352–359. 10.1111/j.1471-4159.2005.03199.x [DOI] [PubMed] [Google Scholar]
- Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH (2006) Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci U S A 103:9333–9338. 10.1073/pnas.0600905103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Furman CA, Zhang M, Kim MN, Gereau RW 4th, Leitges M, Gnegy ME (2009) Protein kinase Cbeta is a critical regulator of dopamine transporter trafficking and regulates the behavioral response to amphetamine in mice. J Pharmacol Exp Ther 328:912–920. 10.1124/jpet.108.147959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Daining CP, Sun H, Fraser R, Stokes SL, Leitges M, Gnegy ME (2013) Protein kinase Cβ is a modulator of the dopamine D2 autoreceptor-activated trafficking of the dopamine transporter. J Neurochem 125:663–672. 10.1111/jnc.12229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodo KA, Läck CM, Roberts DC (2008) Cocaine self-administration reinforced on a progressive ratio schedule decreases with continuous d-amphetamine treatment in rats. Psychopharmacology (Berl) 200:465–473. 10.1007/s00213-008-1222-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty PW, Martelle JL, Nader MA (2010) Effects of chronic D-amphetamine administration on the reinforcing strength of cocaine in rhesus monkeys. Psychopharmacology (Berl) 209:375–382. 10.1007/s00213-010-1807-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czoty PW, Gould RW, Martelle JL, Nader MA (2011) Prolonged attenuation of the reinforcing strength of cocaine by chronic D-amphetamine in rhesus monkeys. Neuropsychopharmacology 36:539–547. 10.1038/npp.2010.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackis CA, O'Brien CP (2001) Cocaine dependence: a disease of the brain's reward centers. J Subst Abuse Treat 21:111–117. 10.1016/S0740-5472(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Egaña LA, Cuevas RA, Baust TB, Parra LA, Leak RK, Hochendoner S, Peña K, Quiroz M, Hong WC, Dorostkar MM, Janz R, Sitte HH, Torres GE (2009) Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-3. J Neurosci 29:4592–4604. 10.1523/JNEUROSCI.4559-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen J, Jørgensen TN, Gether U (2010) Regulation of dopamine transporter function by protein-protein interactions: new discoveries and methodological challenges. J Neurochem 113:27–41. 10.1111/j.1471-4159.2010.06599.x [DOI] [PubMed] [Google Scholar]
- Ferris MJ, Calipari ES, Rose JH, Siciliano CA, Sun H, Chen R, Jones SR (2015) A Single Amphetamine Infusion Reverses Deficits in Dopamine Nerve-Terminal Function Caused by a History of Cocaine Self-Administration. Neuropsychopharmacology 40:1826–1836. 10.1038/npp.2015.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris MJ, España RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, Jones SR (2014) Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci U S A 111:E2751–E2759. 10.1073/pnas.1407935111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U (2006) Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51:417–429. 10.1016/j.neuron.2006.06.028 [DOI] [PubMed] [Google Scholar]
- Foster JD, Cervinski MA, Gorentla BK, Vaughan RA (2006) Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol 175:197–214. 10.1007/3-540-29784-7_10 [DOI] [PubMed] [Google Scholar]
- Foster JD, Adkins SD, Lever JR, Vaughan RA (2008) Phorbol ester induced trafficking-independent regulation and enhanced phosphorylation of the dopamine transporter associated with membrane rafts and cholesterol. J Neurochem 105:1683–1699. 10.1111/j.1471-4159.2008.05262.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Yang JW, Moritz AE, Challasivakanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA (2012) Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux. J Biol Chem 287:29702–29712. 10.1074/jbc.M112.367706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffaney JD, Shetty M, Felts B, Pramod AB, Foster JD, Henry LK, Vaughan RA (2014) Antagonist-induced conformational changes in dopamine transporter extracellular loop two involve residues in a potential salt bridge. Neurochem Int 73:16–26. 10.1016/j.neuint.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowska MM, Elliott AD, DeGraff DJ, Anderson PD, Anumanthan G, Yamashita H, Sun Q, Friedman DB, Hachey DL, Yu X, Sheehan JH, Ahn JM, Raj GV, Piston DW, Gronostajski RM, Matusik RJ (2014) NFI transcription factors interact with FOXA1 to regulate prostate-specific gene expression. Mol Endocrinol 28:949–964. 10.1210/me.2013-1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski J, Rhoades H, Schmitz J, Stotts A, Daruzska LA, Creson D, Moeller FG (2001) Dextroamphetamine for cocaine-dependence treatment: a double-blind randomized clinical trial. J Clin Psychopharmacol 21:522–526. 10.1097/00004714-200110000-00010 [DOI] [PubMed] [Google Scholar]
- Grabowski J, Rhoades H, Stotts A, Cowan K, Kopecky C, Dougherty A, Moeller FG, Hassan S, Schmitz J (2004) Agonist-like or antagonist-like treatment for cocaine dependence with methadone for heroin dependence: two double-blind randomized clinical trials. Neuropsychopharmacology 29:969–981. 10.1038/sj.npp.1300392 [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y (2001) Neuroadaptation: incubation of cocaine craving after withdrawal. Nature 412:141–142. 10.1038/35084134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastrup H, Karlin A, Javitch JA (2001) Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc Natl Acad Sci U S A 98:10055–10060. 10.1073/pnas.181344298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM (2007) Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci 27:3535–3539. 10.1523/JNEUROSCI.3667-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursh SR, Silberberg A (2008) Economic demand and essential value. Psychol Rev 115:186–198. 10.1037/0033-295X.115.1.186 [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R (2002) Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114:475–492. 10.1016/S0306-4522(02)00267-1 [DOI] [PubMed] [Google Scholar]
- Jonsson G, Nwanze E (1982) Selective (+)-amphetamine neurotoxicity on striatal dopamine nerve terminals in the mouse. Br J Pharmacol 77:335–345. 10.1111/j.1476-5381.1982.tb09303.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA (2004) N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol 2:E78. 10.1371/journal.pbio.0020078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lute BJ, Khoshbouei H, Saunders C, Sen N, Lin RZ, Javitch JA, Galli A (2008) PI3K signaling supports amphetamine-induced dopamine efflux. Biochem Biophys Res Commun 372:656–661. 10.1016/j.bbrc.2008.05.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis JM, Foege WH (1999) Mortality and morbidity attributable to use of addictive substances in the United States. Proc Assoc Am Physicians 111:109–118. 10.1046/j.1525-1381.1999.09256.x [DOI] [PubMed] [Google Scholar]
- Moritz AE, Rastedt DE, Stanislowski DJ, Shetty M, Smith MA, Vaughan RA, Foster JD (2015) Reciprocal phosphorylation and palmitoylation control dopamine transporter kinetics. J Biol Chem 290:29095–29105. 10.1074/jbc.M115.667055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OV, Amara SG (2003) Dynamic regulation of the dopamine transporter. Eur J Pharmacol 479:159–170. 10.1016/j.ejphar.2003.08.066 [DOI] [PubMed] [Google Scholar]
- Nader MA, Morgan D, Gage HD, Nader SH, Calhoun TL, Buchheimer N, Ehrenkaufer R, Mach RH (2006) PET imaging of dopamine D2 receptors during chronic cocaine self-administration in monkeys. Nat Neurosci 9:1050–1056. 10.1038/nn1737 [DOI] [PubMed] [Google Scholar]
- Negus SS, Henningfield J (2015) Agonist medications for the treatment of cocaine use disorder. Neuropsychopharmacology 40:1815–1825. 10.1038/npp.2014.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Mello NK (2003a) Effects of chronic D-amphetamine treatment on cocaine- and food-maintained responding under a second-order schedule in rhesus monkeys. Drug Alcohol Depend 70:39–52. 10.1016/S0376-8716(02)00339-3 [DOI] [PubMed] [Google Scholar]
- Negus SS, Mello NK (2003b) Effects of chronic D-amphetamine treatment on cocaine- and food-maintained responding under a progressive-ratio schedule in rhesus monkeys. Psychopharmacology (Berl) 167:324–332. 10.1007/s00213-003-1409-y [DOI] [PubMed] [Google Scholar]
- Negus SS, Mello NK, Blough BE, Baumann MH, Rothman RB (2007) Monoamine releasers with varying selectivity for dopamine/norepinephrine versus serotonin release as candidate “agonist” medications for cocaine dependence: studies in assays of cocaine discrimination and cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther 320:627–636. 10.1124/jpet.106.107383 [DOI] [PubMed] [Google Scholar]
- Nicola SM. (2010) The flexible approach hypothesis: unification of effort and cue-responding hypotheses for the role of nucleus accumbens dopamine in the activation of reward-seeking behavior. J Neurosci 30:16585–16600. 10.1523/JNEUROSCI.3958-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuijten M, Blanken P, van de Wetering B, Nuijen B, van den Brink W, Hendriks VM (2016) Sustained-release dexamfetamine in the treatment of chronic cocaine-dependent patients on heroin-assisted treatment: a randomised, double-blind, placebo-controlled trial. Lancet. 387:2226–2234. 10.1016/S0140-6736(16)00205-1 [DOI] [PubMed] [Google Scholar]
- O'Connor PG, Sokol RJ, D'Onofrio G (2014) Addiction medicine: the birth of a new discipline. JAMA Intern Med 174:1717–1718. 10.1001/jamainternmed.2014.4211 [DOI] [PubMed] [Google Scholar]
- Oleson EB, Roberts DC (2012) Cocaine self-administration in rats: threshold procedures. Methods Mol Biol 829:303–319. 10.1007/978-1-61779-458-2_20 [DOI] [PubMed] [Google Scholar]
- Oleson EB, Richardson JM, Roberts DC (2011) A novel IV cocaine self-administration procedure in rats: differential effects of dopamine, serotonin, and GABA drug pre-treatments on cocaine consumption and maximal price paid. Psychopharmacology (Berl) 214:567–577. 10.1007/s00213-010-2058-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Gomez LL, Dell'Acqua ML (2003) Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J Cell Biol 160:101–112. 10.1083/jcb.200209127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens WA, Williams JM, Saunders C, Avison MJ, Galli A, Daws LC (2012) Rescue of dopamine transporter function in hypoinsulinemic rats by a D2 receptor-ERK-dependent mechanism. J Neurosci 32:2637–2647. 10.1523/JNEUROSCI.3759-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson NE, Markou A (2003) Increased motivation for self-administered cocaine after escalated cocaine intake. Neuroreport 14:2229–2232. 10.1097/00001756-200312020-00019 [DOI] [PubMed] [Google Scholar]
- Phillips PE, Stuber GD, Heien ML, Wightman RM, Carelli RM (2003) Subsecond dopamine release promotes cocaine seeking. Nature 422:614–618. 10.1038/nature01476 [DOI] [PubMed] [Google Scholar]
- Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ (1987) Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237:1219–1223. 10.1126/science.2820058 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer G, Segawa K, Zipfel WR, Piston DW (2006) Optimization of pairings and detection conditions for measurement of FRET between cyan and yellow fluorescent proteins. Microsc Microanal 12:238–254. 10.1017/S1431927606060235 [DOI] [PubMed] [Google Scholar]
- Roberts DC, Corcoran ME, Fibiger HC (1977) On the role of ascending catecholaminergic systems in intravenous self-administration of cocaine. Pharmacol Biochem Behav 6:615–620. 10.1016/0091-3057(77)90084-3 [DOI] [PubMed] [Google Scholar]
- Robertson SD, Matthies HJ, Galli A (2009) A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Mol Neurobiol 39:73–80. 10.1007/s12035-009-8053-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush CR, Stoops WW (2012) Agonist replacement therapy for cocaine dependence: a translational review. Future Med Chem 4:245–265. 10.4155/fmc.11.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Ramsey AJ, Medvedev IO, Kile B, Sotnikova TD, Holmstrand E, Ghisi V, Nicholls PJ, Wong L, Murphy K, Sesack SR, Wightman RM, Gainetdinov RR, Caron MG (2008) Increased amphetamine-induced hyperactivity and reward in mice overexpressing the dopamine transporter. Proc Natl Acad Sci U S A 105:4405–4410. 10.1073/pnas.0707646105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C, Galli A (2015) Insights in how amphetamine ROCKs (Rho-associated containing kinase) membrane protein trafficking. Proc Natl Acad Sci U S A 112:15538–15539. 10.1073/pnas.1520960112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid JA, Scholze P, Kudlacek O, Freissmuth M, Singer EA, Sitte HH (2001) Oligomerization of the human serotonin transporter and of the rat GABA transporter 1 visualized by fluorescence resonance energy transfer microscopy in living cells. J Biol Chem 276:3805–3810. 10.1074/jbc.M007357200 [DOI] [PubMed] [Google Scholar]
- Schultz W. (1986) Responses of midbrain dopamine neurons to behavioral trigger stimuli in the monkey. J Neurophysiol 56:1439–1461. [DOI] [PubMed] [Google Scholar]
- Sevak RJ, Owens WA, Koek W, Galli A, Daws LC, France CP (2007) Evidence for D2 receptor mediation of amphetamine-induced normalization of locomotion and dopamine transporter function in hypoinsulinemic rats. J Neurochem 101:151–159. 10.1111/j.1471-4159.2006.04358.x [DOI] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Ferris MJ, Jones SR (2014a) Biphasic mechanisms of amphetamine action at the dopamine terminal. J Neurosci 34:5575–5582. 10.1523/JNEUROSCI.4050-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Calipari ES, Jones SR (2014b) Amphetamine potency varies with dopamine uptake rate across striatal subregions. J Neurochem 131:348–355. 10.1111/jnc.12808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siciliano CA, Ferris MJ, Jones SR (2015) Cocaine self-administration disrupts mesolimbic dopamine circuit function and attenuates dopaminergic responsiveness to cocaine. Eur J Neurosci 42:2091–2096. 10.1111/ejn.12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A (2003) Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J Biol Chem 278:28274–28283. 10.1074/jbc.M210652200 [DOI] [PubMed] [Google Scholar]
- Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH (2013) A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci 16:966–973. 10.1038/nn.3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Han DD, Gu HH, Caine SB (2009) Lack of cocaine self-administration in mice expressing a cocaine-insensitive dopamine transporter. J Pharmacol Exp Ther 331:204–211. 10.1124/jpet.109.156265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler PN, Fiorillo CD, Schultz W (2005) Adaptive coding of reward value by dopamine neurons. Science 307:1642–1645. 10.1126/science.1105370 [DOI] [PubMed] [Google Scholar]
- Torres GE, Carneiro A, Seamans K, Fiorentini C, Sweeney A, Yao WD, Caron MG (2003) Oligomerization and trafficking of the human dopamine transporter: mutational analysis identifies critical domains important for the functional expression of the transporter. J Biol Chem 278:2731–2739. 10.1074/jbc.M201926200 [DOI] [PubMed] [Google Scholar]
- Trussell RE, Gollance H (1970) Methadone maintenance treatment is successful for heroin addicts. Hosp Manage 110:56–62. [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wolf AP, Schlyer D, Shiue CY, Alpert R, Dewey SL, Logan J, Bendriem B, Christman D (1990) Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am J Psychiatry 147:719–724. 10.1176/ajp.147.6.719 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Hitzemannn R, Gatley SJ, MacGregor RR, Wolf AP (1996) Cocaine uptake is decreased in the brain of detoxified cocaine abusers. Neuropsychopharmacology 14:159–168. 10.1016/0893-133X(95)00073-M [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R, Chen AD, Dewey SL, Pappas N (1997) Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386:830–833. 10.1038/386830a0 [DOI] [PubMed] [Google Scholar]
- Volkow ND, Tomasi D, Wang GJ, Logan J, Alexoff DL, Jayne M, Fowler JS, Wong C, Yin P, Du C (2014) Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol Psychiatry 19:1037–1043. 10.1038/mp.2014.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade-Galuska T, Winger G, Woods JH (2007) A behavioral economic analysis of cocaine and remifentanil self-administration in rhesus monkeys. Psychopharmacology (Berl) 194:563–572. 10.1007/s00213-007-0858-0 [DOI] [PubMed] [Google Scholar]
- Waelti P, Dickinson A, Schultz W (2001) Dopamine responses comply with basic assumptions of formal learning theory. Nature 412:43–48. 10.1038/35083500 [DOI] [PubMed] [Google Scholar]
- Wei Y, Williams JM, Dipace C, Sung U, Javitch JA, Galli A, Saunders C (2007) Dopamine transporter activity mediates amphetamine-induced inhibition of Akt through a Ca2+/calmodulin-dependent kinase II-dependent mechanism. Mol Pharmacol 71:835–842. 10.1124/mol.106.026351 [DOI] [PubMed] [Google Scholar]
- Wersinger C, Jeannotte A, Sidhu A (2006) Attenuation of the norepinephrine transporter activity and trafficking via interactions with alpha-synuclein. Eur J Neurosci 24:3141–3152. 10.1111/j.1460-9568.2006.05181.x [DOI] [PubMed] [Google Scholar]
- Wheeler DS, Underhill SM, Stolz DB, Murdoch GH, Thiels E, Romero G, Amara SG (2015) Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine. Proc Natl Acad Sci U S A 112:E7138–E7147. 10.1073/pnas.1511670112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willuhn I, Burgeno LM, Groblewski PA, Phillips PE (2014) Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci 17:704–709. 10.1038/nn.3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorgason JT, España RA, Jones SR (2011) Demon voltammetry and analysis software: analysis of cocaine-induced alterations in dopamine signaling using multiple kinetic measures. J Neurosci Methods 202:158–164. 10.1016/j.jneumeth.2011.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]