

Key Points
A 6-year overall survival of 61% was observed in leukodystrophy patients after cord blood transplantation.
Mismatched cord blood donors, symptomatic disease, and lower PS before cord blood transplantation were predictors of lower survival.
Abstract
Leukodystrophies (LD) are devastating inherited disorders leading to rapid neurological deterioration and premature death. Hematopoietic stem cell transplantation (HSCT) can halt disease progression for selected LD. Cord blood is a common donor source for transplantation of these patients because it is rapidly available and can be used without full HLA matching. However, precise recommendations allowing care providers to identify patients who benefit from HSCT are lacking. In this study, we define risk factors and describe the early and late outcomes of 169 patients with globoid cell leukodystrophy, X-linked adrenoleukodystrophy, and metachromatic leukodystrophy undergoing cord blood transplantation (CBT) at an European Society for Blood and Marrow Transplantation center or at Duke University Medical Center from 1996 to 2013. Factors associated with higher overall survival (OS) included presymptomatic status (77% vs 49%; P = .006), well-matched (≤1 HLA mismatch) CB units (71% vs 54%; P = .009), and performance status (PS) of >80 vs <60 or 60 to 80 (69% vs 32% and 55%, respectively; P = .003). For patients with PS≤60 (n = 20) or 60 to 80 (n = 24) pre-CBT, only 4 (9%) showed improvement. Of the survivors with PS >80 pre-CBT, 50% remained stable, 20% declined to 60 to 80, and 30% to <60. Overall, an encouraging OS was found for LD patients after CBT, especially for those who are presymptomatic before CBT and received adequately dosed grafts. Early identification and fast referral to a specialized center may lead to earlier treatment and, subsequently, to improved outcomes.
Visual Abstract
Introduction
Leukodystrophies (LD) are a heterozygous group of rare inherited diseases that affect the development and maintenance of brain myelination. Although the age of onset and clinical course varies among this group of diseases, all inherited leukodystrophies are characterized by progressive neurological deterioration and premature death. They often arise from either a lysosomal storage disease (LSD), such as metachromatic leukodystrophy (MLD) and globoid cell leukodystrophy–Krabbe disease (GLD), or a peroxisomal disorder such as X-linked adrenoleukodystrophy (X-ALD). Hematopoietic stem cell transplantation (HSCT) has been shown to arrest or slow disease progression for MLD, GLD, and X-ALD, particularly when performed in presymptomatic patients or patients with early-stage disease.1,2 In patients with a LSD, HSCT works through engraftment of donor cells that can cross the blood-brain barrier, providing a source of cellular enzyme replacement through cross-correction of host cells by enzyme-replete donor cells.3 Conversely, in X-ALD, in which the defected protein is not an enzyme but a transporter protein, the exact mechanism of action of HSCT is not completely understood.
Umbilical cord blood (CB), related or unrelated, provides an alternative source of hematopoietic stem cells for transplantation. After over 2 decades of experience, researchers have well described the benefits of CB. Particularly relevant to patients with LDs, a rapidly progressive disease, CB is readily available, allowing for shorter time to transplant. In patients with LDs, the early outcomes of umbilical cord blood transplantation (CBT) have been described.4-10 These studies suggest that CBT is most beneficial when performed early, preferably before the onset of symptoms. Although it would be ideal to compare the early and late outcomes on the basis of cell source such as those performed for Hurler’s disease,11 this was not possible because of the very limited numbers of patients receiving other cell sources, leading us to focus on cord blood only. In this report, we describe the results of a collaboration among Eurocord, the European Society for Blood and Marrow Transplantation (EBMT), and Duke University to define risk factors and describe the early and late outcomes in a large cohort of patients with LDs (MLD, GLD, and X-ALD) treated after CBT.
Patients and methods
Data collection and patients
In this retrospective, multicenter study, patients (children and adults) with leukodystrophies (MLD, GLD, or X-ALD) who received related or unrelated donor CBT between September 1996 and August 2013 were included. Data were collected from the Eurocord Registry from EBMT centers through standardized questionnaires that included information about the patients, donors, diseases, and transplant outcomes. Data on patients from Duke University were collected through similar questionnaires as those used by the Eurocord-EBMT. Missing data were completed by institutional data managers. An additional follow-up questionnaire was developed for long-term outcomes and sent out to participating centers. Symptomatic patients were categorized into disease subtype on the basis of age of onset of symptoms; presymptomatic patients were categorized on the basis of age of onset of the index case in the family. MLD patients were classified as late infantile (0-4 years), early juvenile (4-6 years), late juvenile (6-16 years), or adult (>16 years). Patients with GLD were classified as early infantile (<6 months), late infantile (6-11 months), juvenile (1-16 years), or adult (>16 years). Patients with ALD were classified as childhood (0-10 years), adolescent (10-18 years), or adult (>18 years). All patients with ALD showed evident cerebral disease on magnetic resonance imaging (MRI) at time of transplantation. Part of this cohort (n = 70) has been reported in previous studies.5,12,13 This study was performed in agreement with the Helsinki Declaration of 1975, revised in 2008. All patients or their legal representatives gave informed consent. Eurocord and the Working Party for Inborn Errors of EBMT approved the European part of this study. In the United States, approval was given by the institutional review board of Duke University.
Outcomes of interest
Outcomes of interest were overall survival (OS) and event-free survival (EFS). OS was defined as the time from transplantation to death. EFS was defined as time from transplantation to last assessment without an event. Events were considered to be autologous reconstitution (documentation of <5% donor-derived engraftment), graft failure (lack of neutrophil recovery or transient engraftment of donor cells after transplantation, requirement for a second transplant, or both), or death. All surviving patients were censored at date of last contact.
Other outcomes of interest were neutrophil recovery, defined as the first day of achieving a neutrophil count of ≥0.5 × 109/L for 3 consecutive days, incidence of acute and chronic graft- versus-host disease (GVHD), donor chimerism, enzyme level (arylsulfatase A and galactocerebrosidase tested in blood cell lysates for MLD and GLD, respectively), very long chain fatty acid (VLCFA) level tested in blood cell lysates for X-ALD, and long-term outcomes. Acute GVHD grade II-IV at day 100 was diagnosed and graded according to published criteria.14 Chronic GVHD at 5 years was graded according to standard criteria15 and evaluated in patients who survived at least 100 days with sustained engraftment. Long-term outcomes included performance score (PS), either Lansky or Karnofsky on the basis of patient age, at 12 months and at latest follow-up time point; overall disease status (symptomatic, defined as clinical disease according to the local clinician, or presymptomatic, defined as no clinical disease according to the local clinician); descriptive neurological status (presence and severity of peripheral neuropathy, presence of seizures, mental development [tested by Bayley Scales of Infant Development, Stanford-Binet Intelligence Scale, Wechsler Preschool and Primary Scale of Intelligence—Revised, Wechsler Intelligence Scale for Children—III]), vision (normal, low: <20/60; blind: <20/400), hearing (normal, <25 dB hearing loss; mild hearing loss, 26-55 dB; severe hearing loss, >56 dB), and cerebral atrophy observed by magnetic resonance imaging [MRI]); incontinence; independence in daily living; school attendance; and school performance. Furthermore, gall bladder disease was registered for patients with MLD16,17 and adrenal insufficiency for patients with X-ALD (as reported by local clinicians).
Statistical analysis
Probabilities of EFS and OS were calculated using the Kaplan-Meier estimate; the 2-sided log-rank test was used for univariate comparisons. Cumulative incidence curves were created for neutrophil recovery; acute GVHD and chronic GVHD were analyzed in a competing risk setting. In univariate analyses, we considered variables associated with the recipient (median age at transplant, median weight at time of transplantation, sex, pretransplant cytomegalovirus serology status), the disease (type of diagnosis, and median interval time from diagnosis or [in ALD only] first abnormal brain MRI to transplant), the cord blood unit (HLA disparity, and median collected and infused total nucleated cells and CD34+ cell doses), and the transplant (year of transplant, use of myeloablation, antithymocyte globulin [ATG], and the type of GVHD prophylaxis). Factors associated with P <.10 in univariate analysis and factors considered relevant risk factors were included in multivariate analyses, using Cox proportional hazards for EFS, OS, neutrophil recovery, and GVHD. Subsequently, a stepwise regression analysis was performed using a threshold of 0.05. All statistical analyses were performed using SPSS (version 19; SPSS Inc., Chicago, IL) and R (version 3.32.0; R Foundation for Statistical Computing, Vienna, Austria) software packages. Figures were created with GraphPad Prism (version 7.02; GraphPad Software, La Jolla, CA).
Results
Patient, donor, and transplant characteristics
One hundred and sixty-nine patients were included: 66 MLD, 47 GLD, and 56 X-ALD. Overall, the median age at time of CBT was 5.1 years (range: 0.1-43.3); patients with X-ALD were the oldest, with a median age of 8.2 years, followed by MLD (4.3 years) and GLD (0.6 years). The majority of the patients received myeloablative conditioning (97%) with busulfan (Bu)/cyclophosphamide (Cy) as the most frequent regimen (83.4%). Ninety-two percent received a single CB graft, with a median total nucleated cell dose (TNC) of 5.7 × 107 cells/kg and a median CD34+ dose of 2.1 × 105/kg. A cyclosporine-based regimen was used as GVHD prophylaxis in 97% of the patients, and 96% received ATG as serotherapy. Median follow-up for survivors was 76 months (range: 3-211). Patient, donor, and transplant characteristics are shown in Table 1.
Table 1.
Baseline patient, donor, and transplantation characteristics
| Baseline characteristics* | ||
|---|---|---|
| Characteristics | n | % |
| Patient | ||
| Overall | 169 | |
| MLD | 66 | 39.1 |
| GLD | 47 | 27.8 |
| X-ALD | 56 | 33.1 |
| Child (<18 y)/adult | 161/8 | 95.3/4.7 |
| Sex (male/female) | 118/51 | 69.8/30.2 |
| Cytomegalovirus serology (negative) | 127 | 77.9 |
| Median | Range | |
| Weight, kg | 19.5 | 2.74-75.0 |
| Age at SCT, y | ||
| Overall | 5.1 | 0.1-43.3 |
| MLD | 4.3 | 0.1-22.7 |
| GLD | 0.6 | 0.1-16.7 |
| X-ALD | 8.2 | 2.4-43.3 |
| Interval diagnosis-transplant, mo | ||
| Overall | 2.8 | 0.6-147.9 |
| MLD | 3.0 | 0.6-70.3 |
| GLD | 1.6 | 0.6-60.0 |
| X-ALD | 5.4 | 0.6-147.9 |
| Performance score pre-SCT | ||
| <60 | 29 | 19.6 |
| 60-80 | 29 | 19.6 |
| >80 | 90 | 60.8 |
| Donor | n | % |
| sCB | 156 | 92.3 |
| dCB | 13 | 7.7 |
| Unrelated/related | 168/1 | 99.4/0.6 |
| HLA matching | ||
| 6/6 | 27 | 18.1 |
| 5/6 | 56 | 37.6 |
| 4/6 | 65 | 43.6 |
| 3/6 | 1 | 0.7 |
| CB cell dose | Median | Range |
| Collected NC, ×107/kg | 8.0 | 2.1-67.2 |
| Collected CD34+, ×105/kg | 2.7 | 0.01-28.5 |
| Infused NC, ×107/kg | 5.7 | 1.2-50.3 |
| Infused CD34+, ×105/kg | 2.1 | 0.01-32.4 |
| Transplantation | n | % |
| Conditioning regimen | ||
| MAC | 163 | 97 |
| Bu/Cy/(TT) | 135 (1) | |
| Bu/Fluda/(Cy)(Mel)(TT) | 9 (4)(3)(3) | |
| Fluda/TT/Mel | 3 | |
| Bu/Mel/other | 1 | |
| TBI/Cy | 4 | |
| RIC | 5 | 3 |
| Fluda/Mel | 1 | |
| Mel/other | 1 | |
| Fluda/other | 2 | |
| Cy/Fluda/TBI | 1 | |
| GVHD prophylaxes | ||
| CsA based | 156 | 96.9 |
| +steroids | 34 | |
| +MMF | 29 | |
| +Mtx | 4 | |
| Tacrolimus-based | 5 | 3.1 |
| +MMF | 4 | |
| +Mtx | 1 | |
| Serotherapy (ATG before day 0) | 162 | 96.4 |
| Median | Range | |
| Year of HCT | 2006 | 1996-2013 |
| Follow-up post SCT,† mo | 76.3 | 3.3-211 |
ATG, anti-thymocyte globulin; Bu, busalfan; CsA, cyclosporine; Cy, cyclophosphamide; dCB, double cord blood; Fluda, fludarabine; MAC, myoablative conditioning; Mel, melphalan; MMF, mofetil; Mtx, methotrexate; NC, neutrophil count; RIC, reduced intensity conditioning; sCD, single cord blood; SCT, stem cell transplantation; TBI, total body irradiation; TT, thiotepa.
CB transplantation was performed in the following centers: Duke (102), Utrecht (11), Marseille (6), Australia (6), Saudi Arabia (5), Madrid (4), Gent (4), Brussels (4), Paris (4), Milano (2), Murcia (2), Barcelona (2), Montréal (2), Budapest (2), Leiden (1), Leuven (1), Jerusalem (1), Porto (1), Hannover (1), Lisboa (1), Berlin (1), Manchester (1), Antalya (1), Poland (1), Nancy (1), Israel (1), and Bratislava (1).
Median follow-up of survivors.
Neutrophil and platelets recovery and chimerism
The cumulative incidence of neutrophil engraftment at day 60 was 86.3% (range: 81.2% to 91.7%; Table 2), with a median time to engraftment of 21 days (range: 11-83). In univariate analysis, factors associated with a higher probability of neutrophil recovery included shorter interval between diagnosis and CBT (≤2.84 months: 91.5%, vs >2.84 months: 80.4%; P = .01), higher infused CD34+ cell dose (≤2.05 × 105/kg: 82.7%, vs >2.05 × 105/kg: 88.6%; P = .05), and higher infused TNC (≤5.73 × 107/kg: 82.7%, vs >5.73 × 107/kg: 88.9%; P = .008). The cumulative incidence of platelet recovery was 68 ± 6% at day 180, and the median time to platelet recovery (>20 000/µL) was 52 days (range: 13-200 days). Twenty patients (12%) experienced either autologous reconstitution (n = 6; 3.5%) or secondary graft failure (n = 14; 8%) during the first 3 months after CBT. Thirteen of these patients subsequently died because of disease progression (n = 3) or graft failure (n = 10). Eight patients received a second transplant using CB (n = 5), peripheral blood stem cells (n = 1), or not specified (n = 2) as the donor source. Of these, 6 patients survived long term, and 2 died (1 because of disease progression, and 1 transplant-related mortality). One patient with autologous recovery survived long term without a second transplant. Chimerism at day 100 was available for 125 out of 148 engrafted patients. Full chimerism was achieved in 104 (83.2%) of the patients, and mixed chimerism in 21 (16.8%). Chimerism at last assessment was available for all 100 survivors (full chimerism, n = 87; mixed chimerism, n = 13). Of patients who were “alive and engrafted,” normal enzyme levels were found in 94.8% and 96.6%, for MLD and GLD, respectively.
Table 2.
Primary and secondary endpoints
| Endpoints | % | Events, n |
|---|---|---|
| Primary | ||
| 6-y OS overall | 61 | |
| MLD | 67 | |
| X-ALD | 59 | |
| GLD | 55 | |
| 6-y EFS overall | 59 | |
| MLD | 66 | |
| X-ALD | 56 | |
| GLD | 53 | |
| Secondary | ||
| Neutrophil engraftment (day 60)* | 86.3 | |
| Platelet engraftment (day 180)† | 68 | |
| Graft failure (primary/secondary) | 3.6/8.3 | 6/14 |
| Chimerism (within 100 d) | ||
| Full donor | 83.2 | 104 |
| Mixed | 16.8 | 21 |
| CIF of acute GVHD | ||
| Stage II-IV | 35.3 | 59 |
| Stage III-IV | 20.2 | 21 |
| CIF of chronic GVHD at 5 y | 30.2 | 44 |
| Limited/extended | 28/15 |
CIF, cumulative incidence function.
Median, 21; range, 11-83.
Median, 52; range, 13-200.
Acute and chronic GVHD
The cumulative incidence of acute GVHD grades II-IV and III-IV at day 100 was 35.3% ± 7%, and 20.2% ± 8% (Table 2), respectively. Univariate analysis showed that a longer interval between diagnosis and CBT was associated with a higher probability of acute GVHD grade II-IV (47.6% for ≤2.84 and 25% for >2.84; P = .002). The cumulative incidence of chronic GVHD at 5 years was 30.2% ± 5% (15 extensive and 28 limited). In univariate analysis, higher PS at the time of CBT was associated with a higher incidence of chronic GVHD (12.1% for PS ≤ 60, 20.8% for PS 60-80, and 41.6% for PS > 80; P = .009; 65% was limited chronic GVHD). Only 6 of the patients with chronic GVHD and a lower PS before CBT (n = 2 for <60, and n = 4 for 60-80) were still alive at latest follow-up.
OS, EFS, and causes of death
The 1-year and 6-year OS among all patients was 73 ± 3% and 61 ± 4% (Figure 1A), respectively, with a median follow-up of 76 months (range: 12-211). Similar OS was observed for the 3 LD types. Improved OS was observed in patients with late-onset disease in MLD and early-onset disease in patients with GLD or X-ALD (supplemental Figure 1), although this should be interpreted with caution because classification of patients might have been based on an index case. In univariate analysis, several clinical factors were associated with improved OS including recipients of 5/6 or 6/6 HLA-matched grafts (71% vs 54% for 3-4/6 HLA-matched grafts; P = .009), absence of cerebral atrophy on MRI prior to CBT (68% vs 35% for patients with cerebral atrophy; P < .001), PS before CBT > 80 (69% in comparison with 55% and 32% for patients with PS 60-80 or <60, respectively; P = .003). Presymptomatic or mildly affected patients also experienced higher OS than did symptomatic patients (77% vs 49%; P = .006). OS was not influenced by year of transplantation (median 2006; 59 ± 5% vs 63 ± 7%; P = .44). Further analysis revealed that differences in OS between presymptomatic and symptomatic patients were most notable in GLD and X-ALD patients (GLD: 78% vs 36%, P = .02; X-ALD: 88% vs 47%, P = .02; and MLD 70% vs 62%, P = .64). In multivariate analysis, recipients of 5/6 or 6/6 HLA-matched grafts (hazard ratio [HR], 1.8; 95% confidence interval [CI], 1.0-3.3; P = .04) as well as presymptomatic disease status (HR, 2.0; 95% CI, 1.0-3.7; P = .04) were significantly associated with higher OS at 6 years.
Figure 1.

Six-year overall and event-free survival. (A) OS. (B) EFS. (C) EFS per LD type. Events for EFS were considered to be autologous reconstitution, graft failure, or death.
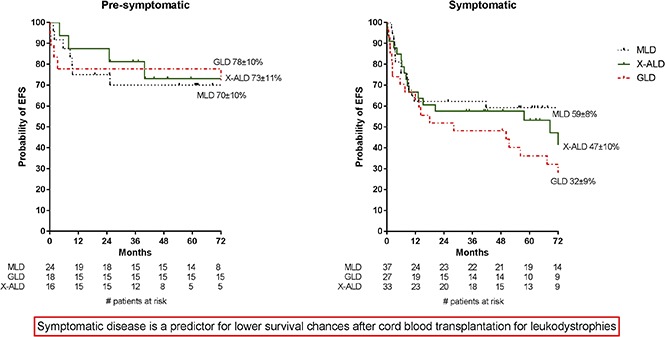
In the overall cohort, the 1-year EFS was 72 ± 4% and the 6-year EFS 59 ± 4% (Figure 1B). Although there was a trend toward improved EFS for patients with presymptomatic disease and those who received 5 or 6/6 HLA-matched grafts, no clinical factors were significantly associated with EFS in multivariate analysis (Table 3). Notably, presymptomatic GLD patients had significantly increased EFS in comparison with that of symptomatic GLD patients (78% vs 32%; P = .009; Figure 2).
Table 3.
Multivariate predictors of 6-year OS and EFS
| 6-year survival | HR | 95% CI | P |
|---|---|---|---|
| OS | |||
| HLA 0-1 vs ≥2 HLA mismatches | 1.8 | 1.0-3.3 | .04 |
| Presymptomatic vs symptomatic | 2.0 | 1.0-3.7 | .04 |
| Collected TNC ≥ 5.0 × 105 vs collected TNC < 5.0 × 105 | 1.5 | 0.7-3.2 | .34 |
| EFS | |||
| HLA 0-1 vs ≥2 HLA mismatches | 1.7 | 1.0-2.9 | .059 |
| Presymptomatic vs symptomatic | 1.8 | 1.0-3.2 | .057 |
| Collected TNC ≥ 5.0 × 105 vs collected TNC < 5.0 × 105 | 1.6 | 0.7-3.4 | .227 |
Figure 2.

EFS for patients at time of CBT. EFS is given for presymptomatic (A) and symptomatic (B) patients. Events were considered to be autologous reconstitution, graft failure, or death.
Sixty-nine patients died within 6 years after CBT (MLD n = 23; GLD n = 25; X-ALD n = 21). Forty-three patients died of transplant-related causes (infection: n = 18; GVHD: n = 9; pulmonary disease: n = 7; multisystem organ failure: n = 4; or other: n = 5) and 21 from disease progression. Additional causes of death are provided in supplemental Table 1.
Functional and disease-related outcomes
To compare functional outcomes after CBT, we used the pre- and posttransplant PS and retrospectively assigned disease status on the basis of symptoms present at the time point of interest. Pre- and posttransplant PS were available for 119 patients. Among survivors with normal pretransplant functional status (PS > 80), 50% had a PS > 80 at last follow-up (median 69 months; range: 12-211; Figure 3), and the remaining showed a decline in PS to 60-80 (20%) or <60 (30%). For patients with lower pretransplant PS (≤60, n = 20; 60-80, n = 24), only 4 patients demonstrated improved PS after CBT and at last follow-up. When decline in PS occurred after CBT, it most commonly occurred during the first year and was stable afterwards (Figures 3 and 4). Changes in performance scores according to LD type are shown in Figure 4. In addition, changes in performance score according to the different clinical phenotypes per LD can be found in supplemental Figure 2.
Figure 3.

Performance scores before and after CBT at 12 months and at most recent follow-up. Most recent follow-up consists only of patients who survived ≥12 months. Percentages show proportions of patients of corresponding pre-HCT PS. All paired bars follow the sequence of 1 year and most recent follow-up.
Figure 4.

Evolution of performance scores before and after CBT. (A) Individual PS of patients with ≥12-month survival before and at last follow-up after CBT. (B-D) The δ of PS at 12 months and at last follow-up for MLD (B), GLD (C), and X-ALD (D). A positive δ is associated with decline in PS. Color coding for pre-CBT performance score is provided within each panel. FU, follow-up, n.a., not applicable.
Data on disease-related outcomes was available for all patients pre-CBT (n = 169) and for 98 patients who survived more than 1 year post-CBT (Tables 4 and 5). In patients with enzyme testing results post-CBT, 93% and 95% of patients for MLD and GLD, respectively, achieved normal enzyme levels (arylsulfastase A or galactocerebrosidase), according to local references. In patients with LSDs (MLD and GLD), 40% and 18% of patients were presymptomatic pre- and post-CBT, respectively (Table 4). Of the presymptomatic patients, 62% remained without symptoms at last follow-up. Vision and hearing stabilized or improved in the majority of patients after CBT (94% and 96%, respectively). Seizures were reported in 24% of patients post-CBT. Neuropathy, as detected by clinical exam or symptoms, was absent or mild in 61.5% of patients pre-CBT and remained stable or improved in 72% post-CBT. Data on school performance were available for most patients alive at latest follow-up post-CBT (81%). In this primarily pediatric cohort, 28% were identified as having age-appropriate school performance. Additional educational support was provided to 71% of patients with available data. Gallbladder disease was observed in 12% (n = 3) of patients with MLD after CBT. All 3 patients required cholecystectomy. Comparatively, patients with X-ALD appear to have better late outcomes (Table 5). Of presymptomatic patients, 100% remained without symptoms at last follow-up. Neuropathy was uncommon (absent or mild in 96%) pre-CBT and remained stable in 89%. Although hearing was rarely affected (96% with normal or mild hearing loss) and remained stable or improved in 93%, vision was affected more often. Normal vision was seen in 71% pre-CBT and worsened in 20% post-CBT. Seizures were reported in 18% post-CBT. Almost all patients (92.6%) experienced stable adrenal function. Data available on school performance showed that 74% had age-appropriate school performance; educational support was provided to 32%.
Table 4.
Functional outcome descriptives pre-HCT and at most recent follow-up in MLD and GLD patients
| Pre-HCT (n = 113) | Most recent follow-up (median = 87 mo, range: 13-206; n = 63) | ||||
|---|---|---|---|---|---|
| Patient characteristics | n (%) | Missing | Patient characteristics | n (%) | Missing |
| Enzyme levels | Enzyme levels | ||||
| MLD (arylsulfatase A) | 12 | MLD (arylsulfatase A) | 11 | ||
| Low | 54 (100) | Normal | 28 (93.3) | ||
| Normal | 0 (0) | ||||
| GLD (galactocerebrosidase) | 4 | GLD (galactocerebrosidase) | 3 | ||
| Low | 43 (100) | Normal | 18 (94.7) | ||
| Normal | 0 (0) | ||||
| Presymptomatic | 42 (39.6) | 7 | Presymptomatic | 10 (18.2) | 8 |
| Mental development | Mental development | ||||
| Cognitive | 40 | Cognitive | 25 | ||
| Very low | 16 (21.9) | Very low | 15 (39.5) | ||
| Low | 18 (24.7) | Low | 8 (21.0) | ||
| Average | 34 (46.6) | Average | 15 (39.5) | ||
| Above average | 5 (6.8) | ||||
| Motor | 39 | Motor | 52 | ||
| Very low | 16 (21.6) | Very low | 10 (90.9) | ||
| Low | 19 (25.7) | Low | 0 (0) | ||
| Average | 34 (45.9) | Average | 1 (9.1) | ||
| Above average | 5 (6.8) | Above average | 0 (0) | ||
| Seizures (present) | 8 (7.1) | 7 | Seizures (present) | 13 (23.6) | 8 |
| Neuropathy | 9 | Neuropathy | 9 | ||
| Absent | 36 (34.6) | Stable | 37 (68.5) | ||
| Mild | 28 (26.9) | Improved | 2 (3.7) | ||
| Severe | 40 (38.5) | Worsened | 15 (27.8) | ||
| Vision | 15 | Vision | 11 | ||
| Low | 10 (10.2) | Stable | 47 (90.4) | ||
| Blind | 2 (2.0) | Improved | 2 (3.8) | ||
| Normal | 86 (87.8) | Worsened | 3 (5.8) | ||
| Hearing | 14 | Hearing | 11 | ||
| Mildly affected | 6 (6.1) | Stable | 48 (92.3) | ||
| Severely affected | 2 (2.0) | Improved | 1 (1.9) | ||
| Normal | 91 (91.9) | Worsened | 3 (5.9) | ||
| School attendance* | 9 | School attendance* | 6 | ||
| Regular | 18 (47.4) | Regular | 11 (21.6) | ||
| Additional support | 14 (36.8) | Additional support | 36 (70.6) | ||
| No attendance | 6 (15.8) | No attendance | 4 (7.8) | ||
| School performance* | 15 | School performance* | 10 | ||
| Appropriate for age | 25 (75.8) | Appropriate for age | 15 (35.8) | ||
| Lower | 8 (24.2) | Lower | 27 (64.2) | ||
| Independence | 16 | Independence | 9 | ||
| Appropriate for age | 52 (53.6) | Appropriate for age | 15 (27.8) | ||
| Needs assistance | 45 (46.4) | Needs assistance | 39 (72.2) | ||
| Continence (incontinent) | 59 (65.6) | 23 | Continence (incontinent) | 21 (38.9) | 9 |
| Gallbladder disease† (present) | 9 (27.3) | 33 | Gallbladder disease† (present) | 3 (11.5) | 17 |
Only for pediatric patients.
Only for MLD.
Table 5.
Functional outcome descriptives pre-HCT and at most recent follow-up in X-ALD patients
| Pre-HCT (n = 56) | Most recent follow-up (median = 49 mo, range: 25-211; n = 35) | ||||
|---|---|---|---|---|---|
| Patient characteristics | n (%) | Missing | Patient characteristics | n (%) | Missing |
| Enzyme levels | Enzyme levels | ||||
| X-ALD (free fatty) | 17 | X-ALD (free fatty) | 18 | ||
| Normal | 0 (0) | Normal | 1 (5.9) | ||
| High/elevated | 39 (100) | High/elevated | 16 (94.1) | ||
| Presymptomatic | 16 (32.7) | 7 | Presymptomatic | 15 (51.7) | 6 |
| Mental development | Mental development | ||||
| Cognitive | 18 | Cognitive | 22 | ||
| Very low | 0 (0) | Very low | 1 (7.7) | ||
| Low | 10 (26.3) | Low | 2 (15.4) | ||
| Average | 22 (57.9) | Average | 7 (53.8) | ||
| Above average | 6 (15.8) | Above average | 3 (23.1) | ||
| Motor | 19 | Motor | 27 | ||
| Very low | 0 (0) | Very low | 5 (62.5) | ||
| Low | 10 (27.0) | Low | 1 (12.5) | ||
| Average | 21 (56.8) | Average | 1 (12.5) | ||
| Above average | 6 (16.2) | Above average | 1 (12.5) | ||
| Seizures (present) | 5 (10.2) | 7 | Seizures (present) | 5 (17.9) | 7 |
| Neuropathy | 9 | Neuropathy | 8 | ||
| Absent | 31(66.0) | Stable | 23 (85.2) | ||
| Mild | 14 (29.8) | Improved | 1 (3.7) | ||
| Severe | 2 (4.2) | Worsened | 3 (11.1) | ||
| Vision | 7 | Vision | 5 | ||
| Low | 12 (24.5) | Stable | 23 (76.7) | ||
| Blind | 2 (4.1) | Improved | 1 (3.3) | ||
| Normal | 35 (71.4) | Worsened | 6 (20.0) | ||
| Hearing | 8 | Hearing | 5 | ||
| Mildly affected | 5 (10.4) | Stable | 27 (90.0) | ||
| Severely affected | 2 (4.2) | Improved | 1 (3.3) | ||
| Normal | 41 (85.4) | Worsened | 2 (6.7) | ||
| School attendance* | 5 | School attendance* | 6 | ||
| Regular | 35 (79.5) | Regular | 15 (53.6) | ||
| Additional support | 7 (15.9) | Additional support | 9 (32.1) | ||
| No attendance | 2 (4.6) | No attendance | 4 (14.3) | ||
| School performance* | 6 | School performance* | 6 | ||
| Appropriate for age | 33 (82.5) | Appropriate for age | 17 (73.9) | ||
| Lower | 7 (17.5) | Lower | 6 (26.1) | ||
| Independence | 14 | Independence | 10 | ||
| Appropriate for age | 31 (73.8) | Appropriate for age | 16 (64.0) | ||
| Needs assistance | 11 (26.2) | Needs assistance | 9 (36.0) | ||
| Continence (incontinent) | 4 (10.0) | 16 | Continence (incontinent) | 3 (12.5) | 11 |
| Adrenal insufficiency | 32 | Adrenal insufficiency | 8 | ||
| Absent | 4 (16.6) | Stable | 25 (92.6) | ||
| Glucocorticoids | 18 (75) | Worsened | 2 (7.4) | ||
| Mineralcorticoids | 1 (4.2) | ||||
| Combined | 1 (4.2) | ||||
Only for pediatric patients.
The most frequent interventions and complications after CBT were G-tube placement (13 with MLD, 20 patients with GLD, and 9 with X-ALD), followed by issues with dentition (9 with MLD, 14 with GLD, and 2 with X-ALD) and surgery (9 MLD, 9 GLD, and 2 X-ALD). Late pulmonary toxicity was seen in 16 patients (3 MLD, 4 GLD, and 9 X-ALD) and cardiac toxicity in 14 patients (5 MLD, 7 GLD, 2 X-ALD; supplemental Table 2).
Discussion
To the best of our knowledge, this retrospective study, spanning over 20 years of clinical experience, is the largest to describe both early and late outcomes after CBT in patients with leukodystrophies undergoing CBT. In this joint study of Eurocord, Inborn Errors Working Party EBMT, and Duke University, we demonstrate a promising 1-year and 6-year OS (73% and 61%, respectively) and EFS (72% and 59%, respectively). Importantly, we observed an OS at 6 years of nearly 80% in patients who were presymptomatic at time of CBT. Furthermore, the majority of these patients (53%) maintained a performance score >80 after CBT at latest follow-up. This confirms previous smaller analyses6,18,19 showing that leukodystrophy patients transplanted prior to clinical symptoms experience long-term survival while maintaining cognitive and motor function, in comparison with symptomatic or nontransplanted patients who all will deteriorate or die prematurely. Our results emphasize the importance of early diagnosis and treatment.
We observed rapid and robust neutrophil and platelet engraftment, which supports other studies of CBT in LDs.4,20 Time to neutrophil recovery was predicted by higher infused CD34+ cell dose and higher infused TNC, as has been previously reported.4,11,20 In multivariate analysis of OS, patients who received grafts matched at 5-6/6 HLA loci or who were presymptomatic at time of CBT experienced improved OS. Conversely, those with poor performance status were at higher risk for morbidity (supplemental Figure 3). Although a recent report described >95% of patients as alive and engrafted at 8 years after CBT for LSD in specialized centers,21 these patients all met strict eligibility criteria and received harmonized conditioning regimens and GVHD prophylaxis. It is important to acknowledge that our report reflects transplants that occurred in nearly 30 centers over a timeframe of almost 20 years. An incompletely understood observation was the higher incidence of (mainly limited) chronic GVHD in the higher performance status group of patients. This may be due to the very low number of survivors in the lowest performance group (only 9 of 29 survived). It is likely that refinements in patient eligibility and donor selection and improvements in supportive care that have occurred over the years will translate into improved outcomes in contemporary patient cohorts.22
It has been well described that PS is an important predictor for survival and late outcomes after HSCT, including CBT.5-8,18,19,23,24 Although PS is not the ideal tool for assessing neurocognitive development, this was the best available surrogate of cognitive function for long-term follow-up. It is important to note that at most recent follow-up, there were some patients with discrepant PS and neurocognitive scores (ie, PS > 80, but a low or very low score on mental development, or vice versa). Nevertheless, PS generally correlated with disease status and was able to quantify overall well-being that, even if less specific, can include general characteristics of the disease. Our results also demonstrated that there was minimal or no decline in PS beyond 1 year after CBT. Notably, presymptomatic disease was correlated with a higher probability of overall and event-free survival. Combining the PS before CBT and presymptomatic status could be used as a tool to predict outcomes. However, prospective studies that include longitudinal assessment of cognitive and motor function along with quality-of-life measures are warranted.
Moreover, this study emphasizes the importance of presymptomatic status and short duration from diagnosis to transplant on both early and late outcomes. These results provide further support that donor cord blood should be strongly considered when a noncarrier sibling donor is lacking. Because of the rapid course of these diseases, CB has some practical advantages above unrelated donors. Although haploidentical related donors are also readily available, most of these donors will be disease carriers. Previous studies of HCT in other LSDs have demonstrated an association between lower enzyme levels and worse late outcomes.25 Therefore, haploidentical donors are not routinely recommended for patients with leukodystrophies. Previous studies have also demonstrated that patients who achieved full-donor chimerism experienced improved late outcomes.25,26 Although these studies were conducted in other LSDs, all studies with CB as a donor source in LD showed high rates of full-donor chimerism.4-6 These results also support the use of newborn screening (NBS), which allows identification of neonates eligible for CBT at a time when they still experience maximal benefit. In select US and EU member states, NBS for GLD or X-ALD has been or will be implemented in upcoming years.6,27,28 To date, NBS for MLD is not available, but our results, along with those from other studies, provide evidence that early diagnosis and transplant prior to onset of symptoms improve early and late outcomes after CBT.8,9,12,24
This study recognizes that the long-term outcomes of many children are affected to various extents by their underlying disease, the transplant procedure, or a combination of both. Although nearly half of the children in this study were able to perform at an age-appropriate level at school, many needed additional support services to achieve that level of performance, especially MLD and GLD patients. Although these results are better than those of the natural course in nontransplanted patients,29,30 more research concerning the effects of transplantation on functional outcomes and quality of life is warranted to optimize late outcomes, including the quality of life of these patients.
Further research is also needed to better understand the duration for which CBT can provide a therapeutic effect. It remains unclear whether the cross-correction of the deficient enzyme after CBT completely halts disease in MLD and GLD, because significant posttransplantation peripheral nerve disease has been reported by others.6,8 Lentiviral haemopoietic stem cell gene therapy that aims for supranormal enzyme levels may provide a solution for this in the future, as a recent report suggests for MLD patients. Although promising, longer follow-up is needed, and only half of patients achieved supranormal enzyme levels.31
In conclusion, CBT in patients with LD showed promising early and late outcomes, especially for those who are presymptomatic before CBT and those who received adequately dosed grafts. Early identification and treatment appear to be important predictors of outcomes, which suggests that NBS may further improve outcomes. Furthermore, fast referral to a specialized center after diagnosis is necessary. For symptomatic patients transplanted with a low performance score (<60 and 60-80), only 5% and 12%, respectively, of these patients were alive with improved performance at last known follow-up. Counseling of these patients and parents is of extreme importance. As life expectancy for patients with LSDs increases after CBT, international collaboration is of utmost importance to further optimize therapies, including CBT, to improve outcomes for these patients. Moreover, prospective trials comparing new (gene) therapies to the standard therapies are needed.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the collaborating transplant centers for sharing patient information: Hospital La Timone, Marseille, France; King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia; Niño Jesus Children’s Hospital, Madrid, Spain; University Hospital Gent, Gent, Belgium; Randwick Sydney Children’s Hospital, Sydney, Australia; Children’s Hospital at Westmead, Sydney, Australia; Necker Hospital, Paris, France; Cliniques Universitaires St. Luc, Brussels, Belgium; Monza Ospedale San Gerardo Clinica Pediatrica dell'Università di Milano Bicocca, Monza, Italy; Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain; Hospital Vall d'Hebron Barcelona, Spain; CHU Sainte-Justine, Montréal, QC, Canada; Universitair Ziekenhuis Brussel, Brussels, Belgium; St. László Hospital, Budapest, Hungary; University Medical Center, Leiden, The Netherlands; University Hospital Gasthuisberg–University of Leuven, Leuven, Belgium; Hadassah University Hospital, Jerusalem, Israel; Pitié-Salpêtrière Hospital, Paris, France; Instituto Português De Oncologia do Porto Francisco Gentil, Porto, Portugal; Department of Haematology/Oncology/Stem Cell Transplantation, Medical School, Hannover, Germany; Instituto Português De Oncologia de Lisboa Francisco Gentil, Lisbon, Portugal; Charité–CVK University Medicine, Berlin, Germany; Rambam Medical Center, Haifa, Israel; Department of Paediatric Haematology, Royal Manchester Children's Hospital, Manchester, United Kingdom; Akdeniz University Medical School, Antalya, Turkey; University of Medical Sciences Poznan, Poznan, Poland; CHU Brabois, Nancy, France; Children’s University Hospital, Bratislava, Slovakia; Wilhelmina Children’s Hospital, Utrecht, The Netherlands; and Duke University Medical Center, Durham, NC. The authors would also like to thank Mahsa Taskindoust for helping with the data collection at Duke University Medical Center.
B.T.A.v.d.B. received a grant from the Sylvia Toth Charity Foundation during the conduct of the study. K.P. was supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (K23HL104575).
B.T.A.v.d.B. is a PhD candidate at Utrecht University, and this work is submitted in partial fulfillment of the requirement for a PhD.
The sponsors of this study are public or nonprofit organizations that support science in general. They had no role in gathering, analyzing, or interpreting the data.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, Blood Institute or the National Institutes of Health.
Appendix: study group members
The members of the Eurocord study group are: E.G., A.R., A.P., F.V., C.K. (all Paris, France), and V.R. (Paris, France, and Oxford, United Kingdom).
The members of the EBMT study group are: M.A.D. (Madrid, Spain); V.B. (Gent, Belgium); T.O. and P.J.S. (both of Sydney, Australia); A.A.-S. (Riyadh, Saudi Arabia); A.R.G. (Newcastle, United Kingdom); and J.J.B. (Utrecht, The Netherlands).
The members of the Duke University Medical Center (Blood and Marrow Transplantation Program) study group are: K.P., H.A., and J.K.
Authorship
Contribution: J.J.B. and J.K. designed and supervised the study; B.T.A.v.d.B., K.P., A.P., F.V., J.K., and J.J.B. drafted and revised the manuscript; B.T.A.v.d.B., J.J.B., A.P., A.R., J.H., F.V., P.M.v.H., K.P., and J.K. performed statistical analyses and analyzed and interpreted the data; J.H., H.A., F.V., G.M., M.A.D., V.B., T.O., P.J.S., C.K., A.A.-S., P.M.v.H., A.R.G., E.G., V.R., and A.R. contributed to the critical revision of the manuscript; B.T.A.v.d.B., K.P., A.P., J.H., H.A., F.V., G.M., M.A.D., V.B., T.O., P.J.S., C.K., A.A.-S., P.M.v.H., A.R.G., E.G., V.R., A.R., J.K., and J.J.B. contributed to the acquisition of the data. All authors approved the final manuscript as submitted.
Conflict-of-interest disclosure: J.K. is Director of the Carolinas Cord Blood Bank and Medical Director of the CORD:USE Cord Blood Bank (no personal compensation). The remaining authors declare no competing financial interests.
A complete list of the members of the Eurocord, Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation, and Duke University Medical Center study groups appears in “Appendix: study group members.”
Correspondence: Jaap Jan Boelens, Department of Pediatrics, Pediatric Blood and Marrow Transplantation Program, Room KC03.063.0, University Medical Center, Utrecht, Lundlaan 6, 3584 EA Utrecht, The Netherlands; e-mail: j.j.boelens@umcutrecht.nl; and Joanne Kurtzberg, Pediatric Blood and Marrow Transplant Program, Carolinas Cord Blood Bank, Duke University Medical Center, 2400 Pratt St, Room 9026, Durham, NC 27705; e-mail: kurtz001@mc.duke.edu.
References
- 1.Boelens JJ, van Hasselt PM. Neurodevelopmental outcome after hematopoietic cell transplantation in inborn errors of metabolism: current considerations and future perspectives. Neuropediatrics. 2016;47(5):285-292. [DOI] [PubMed] [Google Scholar]
- 2.Aldenhoven M, Kurtzberg J. Cord blood is the optimal graft source for the treatment of pediatric patients with lysosomal storage diseases: clinical outcomes and future directions. Cytotherapy. 2015;17(6):765-774. [DOI] [PubMed] [Google Scholar]
- 3.Wynn R. Stem cell transplantation in inherited metabolic disorders. Hematology Am Soc Hematol Educ Program. 2011;2011:285-291. [DOI] [PubMed] [Google Scholar]
- 4.Martin PL, Carter SL, Kernan NA, et al. Results of the cord blood transplantation study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12(2):184-194. [DOI] [PubMed] [Google Scholar]
- 5.Prasad VK, Mendizabal A, Parikh SH, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112(7):2979-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352(20):2069-2081. [DOI] [PubMed] [Google Scholar]
- 7.Beam D, Poe MD, Provenzale JM, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13(6):665-674. [DOI] [PubMed] [Google Scholar]
- 8.Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A, Escolar ML. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant. 2013;19(4):616-624. [DOI] [PubMed] [Google Scholar]
- 9.Groeschel S, Kühl J-S, Bley AE, et al. Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 2016;73(9):1133-1140. [DOI] [PubMed] [Google Scholar]
- 10.Escolar ML, Poe MD, Martin HR, Kurtzberg J. A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics. 2006;118(3):e879-e889. [DOI] [PubMed] [Google Scholar]
- 11.Boelens JJ, Aldenhoven M, Purtill D, et al. ; Centre for International Blood and Marrow Research. Outcomes of transplantation using various hematopoietic cell sources in children with Hurler syndrome after myeloablative conditioning. Blood. 2013;121(19):3981-3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allewelt HB, Page K, Taskindoust M, et al. Long-term functional outcomes following hematopoietic stem cell transplantation for Krabbe disease. Biol Blood Marrow Transplant. 2016;22(3):S102-S103. [DOI] [PubMed] [Google Scholar]
- 13.Page KM, Zhang L, Mendizabal A, et al. Total colony-forming units are a strong, independent predictor of neutrophil and platelet engraftment after unrelated umbilical cord blood transplantation: a single-center analysis of 435 cord blood transplants. Biol Blood Marrow Transplant. 2011;17(9):1362-1374. [DOI] [PubMed] [Google Scholar]
- 14.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295-304. [DOI] [PubMed] [Google Scholar]
- 15.Shulman HM, Sullivan KM, Weiden PL, et al. Chronic graft-versus-host syndrome in man. A long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69(2):204-217. [DOI] [PubMed] [Google Scholar]
- 16.van Rappard DF, Bugiani M, Boelens JJ, et al. Gallbladder and the risk of polyps and carcinoma in metachromatic leukodystrophy. Neurology. 2016;87(1):103-111. [DOI] [PubMed] [Google Scholar]
- 17.Kim J, Sun Z, Ezekian B, et al. Gallbladder abnormalities in children with metachromatic leukodystrophy. J Surg Res. 2017;208:187-191. [DOI] [PubMed] [Google Scholar]
- 18.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118(7):1971-1978. [DOI] [PubMed] [Google Scholar]
- 19.van Rappard DF, Boelens JJ, van Egmond ME, et al. Efficacy of hematopoietic cell transplantation in metachromatic leukodystrophy: the Dutch experience. Blood. 2016;127(24):3098-3101. [DOI] [PubMed] [Google Scholar]
- 20.Boelens JJ, Rocha V, Aldenhoven M, et al. ; EUROCORD, Inborn Error Working Party of EBMT, and Duke University. Risk factor analysis of outcomes after unrelated cord blood transplantation in patients with hurler syndrome. Biol Blood Marrow Transplant. 2009;15(5):618-625. [DOI] [PubMed] [Google Scholar]
- 21.Aldenhoven M, Jones SA, Bonney D, et al. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant. 2015;21(6):1106-1109. [DOI] [PubMed] [Google Scholar]
- 22.Lum SH, Miller WP, Jones S, et al. Changes in the incidence, patterns and outcomes of graft failure following hematopoietic stem cell transplantation for Hurler syndrome. Bone Marrow Transplant. 2017;52(6):846-853. [DOI] [PubMed] [Google Scholar]
- 23.Musolino PL, Lund TC, Pan J, et al. Hematopoietic stem cell transplantation in the leukodystrophies: a systematic review of the literature. Neuropediatrics. 2014;45(3):169-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucher AA, Miller W, Shanley R, et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis. 2015;10(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164-2172. [DOI] [PubMed] [Google Scholar]
- 26.Lum SH, Ghosh A, Stepien K, et al. Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation in Manchester. Mol Genet Metab. 2017;120(1):S87-S88. [DOI] [PubMed] [Google Scholar]
- 27.Msall M, Duffner P, Lyon N, Shapiro E. Health, developmental, and functional outcome surveillance in preschool children with lysosomal storage diseases (LSD). Mol Genet Metab. 2009;96(2):S32. [Google Scholar]
- 28.Wasserstein MP, Andriola M, Arnold G, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med. 2016;18(12):1235-1243. [DOI] [PubMed] [Google Scholar]
- 29.Krägeloh-Mann I, Groeschel S, Kehrer C, et al. Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow Transplant. 2013;48(3):369-375. [DOI] [PubMed] [Google Scholar]
- 30.Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R. The burden of inherited leukodystrophies in children. Neurology. 2010;75(8):718-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388(10043):476-487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.