

Key Points
Clonal heterogeneity detected by iFISH is common in newly diagnosed MM.
Treatment with bortezomib overcomes the negative impact of high-risk cytogenetic abnormalities if no further subclones are detected.
Abstract
We investigated subclonal cytogenetic aberrations (CA) detected by interphase fluorescence in situ hybridization (iFISH) in patients with newly diagnosed multiple myeloma (MM) enrolled in the Haemato Oncology Foundation for Adults in the Netherlands (HOVON)–65/German-Speaking MM Group (GMMG)–HD4 phase 3 trial. Patients were either treated with 3 cycles of vincristine, Adriamycin, and dexamethasone or bortezomib, Adriamycin, and dexamethasone and then thalidomide or bortezomib maintenance after tandem autologous transplantation. Subclones were defined either by presence of different copy numbers of the same chromosome loci and/or CA present in at least 30% less and maximally 2/3 of cells compared with the main clone CA. Patients with subclones harbored more frequently high risk (31.0%) or hyperdiploid main clone aberrations (24.8%) than patients with t(11;14) in the main clone (10.1%). Gains and deletions of c-MYC were the only CA that occurred more frequently as subclone (8.1%/20.5%) than main clone (6.2%/3.9%, respectively). Treatment with bortezomib completely overcame the negative prognosis of high-risk CA in patients without subclones, but not in patients with additional subclonal CA. High-risk patients treated without bortezomib showed dismal outcome whether subclones were present or not. Cytogenetic heterogeneity defined by subclonal CA is of major prognostic significance in newly diagnosed MM patients treated with bortezomib within the HOVON-65/GMMG-HD4 trial.
Visual Abstract
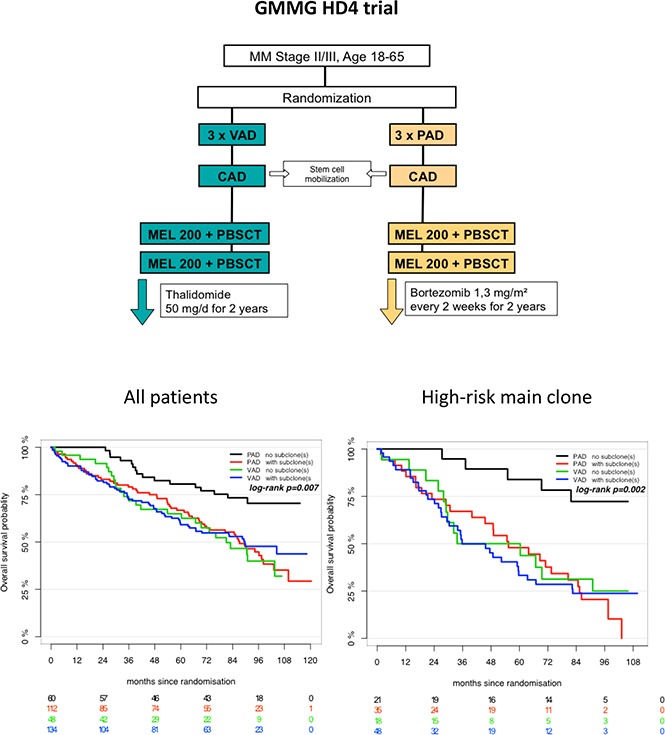
Introduction
Clonal heterogeneity is a hallmark of solid tumors and hematological malignancies. In patients with acute myeloid leukemia, the presence of different cytogenetically defined subclones in the same patient has been associated with adverse outcome.1 Recent studies demonstrated that multiple myeloma (MM) is a genetically complex and heterogeneous disease.2 It remains to be clarified whether genetic complexity of MM might be the reason for treatment resistance and, ultimately, refractoriness. Second- and third-generation novel agents improved outcome in patients with MM.3-5 However, treatment might select refractory clones, present only in a subset of cells at primary diagnosis. Furthermore, different genetically defined subclones spatially and temporarily evolve during the course of the disease and might result in disease progression.6-8 So far, there are only limited data available on the occurrence and prognostic significance of clonal heterogeneity in newly diagnosed MM patients treated with upfront autologous stem cell transplantation (ASCT).
Cytogenetic aberrations (CA) detected by interphase fluorescence in situ hybridization (iFISH) are of major prognostic significance in newly diagnosed and relapsed MM.9 Because iFISH is a single-cell analysis, it offers the opportunity to identify CA present only in a subset of cells. With the current study, we intended to characterize subclonal CA detected by iFISH in patients with newly diagnosed MM. Our goals were (1) to investigate distribution of subclonal CA among patients with cytogenetically defined standard- and high-risk main clones; (2) to analyze which CA occurs predominantly on subclonal or main clonal level; and (3) to determine prognostic significance in patients treated either with or without bortezomib and tandem ASCT within the German part of the prospective Haemato Oncology Foundation for Adults in the Netherlands (HOVON)–65/German-Speaking Myeloma Multicenter Group (GMMG)–HD4 trial.10
Methods
Patients and treatment
We analyzed 354 patients enrolled in the prospective, randomized HOVON-65/GMMG-HD4 trial (EudraCT no. 2004-000944-26) with complete iFISH datasets. The primary end points and subgroup analyses of the HOVON-65/GMMG-HD4 trial were reported previously.10-12 In brief, after written informed consent, GMMG patients were randomly assigned to either 3 cycles of vincristine, Adriamycin, and dexamethasone followed by tandem ASCT and thalidomide maintenance therapy for 2 years (arm A) or 3 cycles of bortezomib, Adriamycin, dexamethasone (PAD) followed by tandem ASCT and bortezomib maintenance therapy for 2 years (arm B; supplemental Figure 1). The study was conducted in accordance with the Declaration of Helsinki and the protocol was approved by local ethics committees of all participating institutions. Last follow-up of the GMMG-HD4 trial was performed in November 2015. The trial met its primary end point and showed superiority of arm B with regard to prolonged progression-free survival (PFS) compared with arm A.10
Two-color iFISH was performed on CD138-purified plasma cell samples, as described previously.12 Probes for the following loci were used: 1q21, 5p15, 5q35, 6q21, 8p21, 9q34, 11q13, 11q23, 13q14, 15q22, 17p13, 19q13, 22q11, translocations t(4;14)(p16;q32), t(11;14)(q13;q32), t(14;16)(q32;q23), the c-MYC locus localized on chromosome 8q24, and an immunoglobulin H (IgH) break-apart probe. Threshold for all CA was 10%. At least 100 nuclei per patient were analyzed. Patients were grouped according to the main clone defined by the largest CA into high risk and standard risk. Presence of deletion 17p13, gain 1q21, and translocation t(4;14) were considered high-risk CA. The group with standard-risk CA was subdivided into patients with a hyperdiploid karyotype (HD) or t(11;14) in the main clone. The score by Wuilleme et al was used to define hyperdiploidy.13
Patients were considered to harbor subclones, either if they had different aberrant copy numbers of a single chromosome loci or if they had additional CA present in at least 30% less and maximally 2/3 of cells compared with the largest detected CA. Hybridization accuracy ranges from 10% to 15% based on our experience from more than 5500 FISH analyses in patients with different stages of plasma cell diseases. Therefore, we chose 30% as threshold for subclonal cytogenetic aberrations to rule out an overestimation of subclonal aberrations.
Statistical analysis
Differences between the groups were analyzed with Fisher exact test for categorical variables and Mann-Whitney Wilcoxon test for continuous variables. Overall survival (OS) was defined by time from randomization to death, PFS by the occurrence of serological or symptomatic progressive disease, or death, whichever occurred first, but was censored in case of allogeneic transplantation. Patients alive or without progression at last follow-up were censored. Kaplan-Meier method was used to estimate distribution of survival times and compared using log-rank test. P values <.05 were considered statistically significant. Multivariable Cox regression analysis was performed accounting for International Staging System (ISS) stage, age, elevated lactate dehydrogenase (LDH), and high-risk main clone CA to assess the effect of subclones on survival. Analyses were performed with statistical software R 3.3.
Results
A total of 833 patients were enrolled in the prospective HOVON-65/GMMG-HD4 trial. Bone marrow aspirated from 354 of 395 (89.6%) eligible GMMG patients was centrally analyzed in Heidelberg. Purity (median, 90%; interquartile range, 77%-95%) was confirmed by CD38+/CD138+ phenotypes in flow cytometry analyses.
Occurrence of subclones and correlation with baseline characteristics and treatment response
Patients with subclones had more frequently a hyperdiploid (24.8%) or high-risk main clone (31.0%) than patients with a t(11;14) in the main clone (10.1%; Table 1). Patients with high-risk main clone (n = 122) more frequently harbored subclones confined to neutral (n = 31) than other high-risk chromosome loci (n = 8).
Table 1.
Frequencies of subclonal aberrations in the analyzed cohort
| No subclone | 1 subclone | >1 subclone | ||||
|---|---|---|---|---|---|---|
| Main clone | n | % | n | % | n | % |
| High risk | 52 | 43.7 | 30 | 34.5 | 40 | 31.0 |
| Hyperdiploid | 27 | 22.7 | 31 | 35.6 | 32 | 24.8 |
| t(11;14) | 21 | 17.6 | 16 | 18.4 | 13 | 10.1 |
| Other | 19 | 16.0 | 10 | 11.5 | 44 | 34.1 |
| All | 119 | 87 | 129 | |||
| P | .0007 | |||||
Gains/deletions of the MYC locus were the only aberrations that occurred more frequently as subclone (8.1%/20.5%) than main clone (6.2%/3.9%, respectively). Main clonal gains of MYC were especially observed in patients with other high-risk CA in the main clone (14.1%) compared with patients with an HD or t(11;14) main clone (<3%; P = .0023). The second most frequently found aberrations on subclonal level besides MYC gains/deletions were gains of odd-numbered chromosomes, such as 5, 9, and 15, associated with a hyperdiploid karyotype (9.1% to 15.4% of patients).
Defined IgH translocations (t(4;14), t(11;14), t(14;16)) were almost exclusively observed as main clone aberrations and only in 1% of patients on subclonal level. High-risk CA other than t(4;14) also occurred predominantly as main clone (gain 1q21: 23.3%/del17p: 8.0%) than subclone (gain 1q21: 9.3%/del17p: 2.6%). Supplemental Table 1 summarizes frequencies of subclone and main clone aberrations in the analyzed cohort.
When comparing clinical baseline characteristics of patients with and without subclones, we did not observe any significant differences. Rates of very good partial remission (VGPR) or better after induction therapy and ASCT were higher in patients without subclones (27.8% and 47.2%) compared with patients with subclones (19.9% and 41.1%, respectively) without reaching statistical significance. Comparison of patient characteristics is depicted in Table 2.
Table 2.
Comparison of patient characteristics based on presence of subclones
| Subclone | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | Yes | All | ||||||
| Variable | Level | n | % | n | % | n | % | P |
| ISS | I | 44 | 44.0 | 87 | 38.0 | 131 | 39.8 | .60 |
| II | 33 | 33.0 | 82 | 35.8 | 115 | 35.0 | ||
| III | 23 | 23.0 | 60 | 26.2 | 83 | 25.2 | ||
| Elevated LDH | No | 91 | 86.7 | 184 | 78.6 | 275 | 81.1 | .10 |
| Yes | 14 | 13.3 | 50 | 21.4 | 64 | 18.9 | ||
| Elevated creatinine | No | 97 | 89.8 | 221 | 89.8 | 318 | 89.8 | 1.00 |
| Yes | 11 | 10.2 | 25 | 10.2 | 36 | 10.2 | ||
| Anemia | No | 95 | 88.0 | 227 | 92.3 | 322 | 91.0 | .23 |
| Yes | 13 | 12.0 | 19 | 7.7 | 32 | 9.0 | ||
| Hypercalcemia | No | 100 | 92.6 | 229 | 93.1 | 329 | 92.9 | .83 |
| Yes | 8 | 7.4 | 17 | 6.9 | 25 | 7.1 | ||
| Age, y | >60 | 84 | 77.8 | 175 | 71.1 | 259 | 73.2 | .24 |
| 18-60 | 24 | 22.2 | 71 | 28.9 | 95 | 26.8 | ||
| Osteolyses | None | 22 | 21.8 | 61 | 25.6 | 83 | 24.5 | .39 |
| 1 | 10 | 9.9 | 14 | 5.9 | 24 | 7.1 | ||
| 2 | 6 | 5.9 | 9 | 3.8 | 15 | 4.4 | ||
| >2 | 63 | 62.4 | 154 | 64.7 | 217 | 64.0 | ||
| ≥VGPR after induction | No | 78 | 72.2 | 197 | 80.1 | 275 | 77.7 | .13 |
| Yes | 30 | 27.8 | 49 | 19.9 | 79 | 22.3 | ||
| ≥VGPR after ASCT | No | 57 | 52.8 | 145 | 58.9 | 202 | 57.1 | .30 |
| Yes | 51 | 47.2 | 101 | 41.1 | 152 | 42.9 | ||
Prognostic significance
High-risk CA were associated with adverse outcome, regardless of whether they were present as main or subclone (Figure 1A-B). Gains and deletions of MYC resulted in shorter OS only if they were present as main clone (Figure 1C). Analysis of the entire cohort revealed shorter survival for patients with subclonal CA (Figure 2A). This was due to shorter PFS and OS in patients with a high-risk main clone (Figure 2C), whereas no subclone effect was observed in patients with a standard-risk main clone (Figure 2B).
Figure 1.

Outcome of patients with or without high-risk aberrations. (A) +1q21: blue, no +1q21; red, subclone +1q21; green, main clone +1q21. (B) −17p: blue, no −17p; red, subclone −17p; green, main clone −17p. (C) ±MYC: blue, no ±MYC; red, subclone ± MYC; green, main clone ± MYC.
Figure 2.

Outcome of patients with (red) or without (blue) subclones. (A) Entire cohort, (B) patients with standard-risk, and (C) high-risk main clone aberration.
Subgroup analysis revealed that treatment with bortezomib completely overcame the negative prognostic impact of a high-risk main clone if no subclonal CA were detected (Figure 3; supplemental Figure 4). The positive effect of bortezomib on patients with a high-risk main clone was abrogated when patients harbored additional subclonal CA (Figure 3; supplemental Figure 4). Importantly, these negative effects were observed regardless of whether subclones were confined to high-risk or neutral chromosomes (supplemental Figure 2). Because high-risk patients more frequently harbored neutral subclones, as stated previously, the observed effects were caused by the presence of subclones and not by the mere accumulation of further high-risk CA. Figure 4 summarizes results from univariate analysis.
Figure 3.

Outcome of high-risk patients. (A) Standard- and (B) high-risk patients. Bortezomib arm: purple, no subclone; red, with subclone. Thalidomide arm: green, no subclone; blue, with subclone. Colored numbers below the plots represent uncensored patients at the respective time point.
Figure 4.

Forest plots from univariate analysis. PFS (upper panel) and OS (lower panel) for the entire cohort and standard- and high-risk patients. Green, all patients; red, PAD arm; blue, VAD arm. CI, confidence interval; HR, hazard ratio; VAD, vincristine, Adriamycin, and dexamethasone.
Multivariable analysis
Multivariable Cox regression analysis accounting for ISS, age, elevated LDH, and high-risk main clone CA revealed that the presence of subclones is an independent factor for negative outcome in patients treated with bortezomib. This effect was more pronounced in patients with a high-risk main clone. Table 3 summarizes the results from multivariable analyses.
Table 3.
Results from multivariable Cox regression analysis in patients treated with PAD
| All patients | High-risk patients | |||||||
|---|---|---|---|---|---|---|---|---|
| PFS | OS | PFS | OS | |||||
| HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | |
| Subclone | 1.28 (0.85-1.92) | .233 | 2.07 (1.15-3.73) | .016 | 2.11 (1.01-4.43) | .048 | 3.24 (1.19-8.83) | .022 |
| ISS II | 1.20 (0.77-1.88) | .414 | 1.85 (1.01-3.38) | .046 | 0.60 (0.26-1.37) | .227 | 0.93 (0.34-2.55) | .890 |
| ISS III | 1.41 (0.86-2.32) | .176 | 1.63 (0.82-3.26) | .167 | 0.78 (0.29-2.09) | .625 | 0.76 (0.23-2.53) | .654 |
| Age | 1.01 (0.98-1.03) | .653 | 1.04 (1.00-1.08) | .034 | 0.99 (0.94-1.03) | .553 | 1.00 (0.94-1.06) | .963 |
| High LDH | 2.28 (1.38-3.76) | .001 | 3.29 (1.79-6.06) | .0001 | 2.19 (0.73-6.55) | .160 | 4.68 (1.52-14.4) | .007 |
| HR main | 1.61 (1.06-2.46) | .025 | 2.06 (1.23-3.46) | .006 | — | — | — | — |
Bold type indicates significant P values.
Discussion
With the current analysis, we demonstrate for the first time occurrence and prognostic significance of subclonal CA in a large cohort of newly diagnosed MM patients treated within a prospective phase 3 trial. We show that cytogenetically defined heterogeneity is a frequent phenomenon.
An explanation for this finding might be that gain 1q21 and deletion 17p13 are especially associated with defective DNA damage repair mechanisms and chromosomal instability (CIN).14 Furthermore, proliferation of aneuploid cells is limited by TP53.15 Loss of function from deletion 17p might cause accumulation of additional CA during disease progression on a subclonal level. As in other human cancers, CIN in MM might facilitate tumorigenesis and drug resistance during the course of the disease. Therefore, subclones in high-risk MM patients might be a surrogate for CIN and lead ultimately to adverse outcome.
The same study that established the relationship between gain 1q21 as well as deletion 17p and CIN found no association between HD and CIN.14 Additionally, subclonal CA were not associated with adverse outcome in HD MM in our current study. Therefore, the mechanisms leading to subclones in patients with a HD karyotype might be different from high-risk patients. The presence of subclonal CA in patients with a HD karyotype is most likely because gains of odd-numbered chromosomes are highly associated with each other (supplemental Figure 3).16 The events leading to trisomies in MM are not fully understood. Preclinical studies in Wilms tumor cells demonstrated that bipolar mitosis with missegregation of chromosomes during anaphase causes trisomies. Another missegregation of the same chromosome in a trisomic cell leads to 1 tetrasomic and 1 disomic population. If the same event affects another chromosome, 2 daughter cells with different trisomies arise. This mechanism might explain the presence of subclones especially in patients with a HD karyotype beyond CIN.17
In contrast to patients with an HD or high-risk main clone, defined IgH translocations, especially t(11;14), occurred almost exclusively on a main clonal level and patients with the respective translocation less frequently harbored subclonal CA. This is in line with the current understanding that translocations are early events in myelomagenesis and MM cells with t(11;14) exhibit a rather stable karyotype.
Gains and deletions of MYC were the only CA that occurred more often on a subclonal level. This confirms the hypothesis that MYC aberrations are secondary events and linked to progression from monoclonal gammopathy of undetermined significance to symptomatic MM as well as adverse outcome.2,18 However, gains and deletions of MYC were only associated with adverse outcome if they were present as main clone. An explanation for this finding might be that main clonal gains of MYC were predominantly observed in patients with other high-risk CA in the main clone. However, the analysis of MYC abnormalities is hampered by the fact that they often involve small rearrangements around the MYC locus that are underestimated when using FISH.19
When investigating the prognostic significance of subclone or main clonal high-risk CA, we observed adverse outcomes in both groups compared with patients without the respective CA. This underlines the hypothesis that high-risk clones might outgrow initially predominant standard-risk, therapy-sensitive clones under the selection pressure of chemotherapy. During the course of the disease, the different clones might gain dominance over each other, resulting in clonal tiding.7 That our results were actually more pronounced when looking at OS data compared with PFS data supports this theory. Additionally, recent studies in patients with deletion 17p demonstrated that higher cutoffs for the aberration (eg, 60% instead of 10% as in our routine practice) resulted in significantly shorter PFS, whereas results for OS were less significant.20,21 In that context, longitudinal iFISH analyses might help identify distinct clones that are susceptible to certain agents.22 In line with that hypothesis, we show that bortezomib might overcome the poor prognostic impact of gain 1q21 and deletion 17p if there are no additional subclones. Furthermore, multivariable analysis confirmed that the presence of subclones is a negative prognostic factor in patients treated with bortezomib, independent from age, ISS, or LDH levels. It has been shown in a subanalysis of the HD4 trial that bortezomib partially abrogates the negative prognostic impact of deletion 17p.12 Our current analysis provides the first evidence for clonal heterogeneity being associated with poor outcome and a possible explanation for the beneficial effect of bortezomib in patients with deletion 17p.
One of the main criticisms of the current study is the lack of whole-genome data, which might lead to an underestimation of subclones. However, iFISH is a widely available and comparably cheap method to detect subclones compared with whole-genome approaches. Future studies investigating subclones on, for example, the mutational level, to clarify the mechanisms leading to the observed effects will follow.
Taken together, we show for the first time that clonal heterogeneity detected by iFISH is a common phenomenon in MM. High-risk patients without subclones benefit from continuous bortezomib treatment.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Maria Dörner, Ewelina Nickel, Hendrike Seidt, and Marie-Louise Brygider for technical assistance in the enrichment of CD138+ plasma cells and Michaela Brough, Michelle Ebentheuer, Stephanie Pschowski-Zuck, and Annekathrin Borowski for performing iFISH analysis.
This work was supported by grants from the German Federal Ministry of Education (BMBF) CLIOMMICS (01ZX1309) and CAMPSIMM (01ES1103), the Deutsche Forschungsgemeinschaft (SFB/TRR79), the Dietmar Hopp Stiftung Heidelberger Konzept zur Optimierung der Diagnostik und Therapie des Multiplen Myeloms, the 7th EU-framework program OverMyR, and Takeda (IISR-2016-101654).
Authorship
Contribution: M.M., A.J., and T.H. planned and designed the study; H.G. is the principal investigator of the study; T.H. performed the statistical analysis; A.J. performed iFISH analysis; A.S. and D.H. prepared bone marrow samples for iFISH analysis; M.S.R., T.B., S.O.S., U.B., K.N., M.S.R., H.S., I.W.B., H.-W.L., I.G.H.S.-W., C.S., M.H., K.C.W., J.H., and H.G. provided clinical data; M.M., T.H., and A.J. interpreted the data; and all authors read, wrote, reviewed, and approved the final manuscript.
Conflict-of-interest disclosure: M.M. received travel grants from Celgene, Abbvie, Amgen, and Janssen and acts on advisory boards for Amgen. D.H. received research funding from Sanofi and EngMab, as well as personal fees outside this work from Celgene. H.S. received speakers honoraria and travel grants from Celgene, Amgen, and Sanofi. I.G.H.S.-W. received research funding from Janssen and Novartis. C.S. received funding outside this work from Novartis, Celgene, and Janssen. K.C.W. received consultancy honoraria from Amgen, Takeda, and BMS, as well as honoraria from Novartis, research funding from Janssen and Celgene, and consultancy honoraria from Onyx. H.G. received consultancy honoraria and research funding from Janssen, Celgene, and Novartis, as well as consultancy honoraria from Onyx and Millenium and fees outside this work from BMS. J.H. received speakers honoraria from Celgene, acts on advisory boards for Janssen, BMS, and Amgen, and received consultancy honoraria from Janssen, Novartis, and Amgen as well as research funding from Sanofi. The remaining authors declare no competing financial interests.
Correspondence: Maximilian Merz, Medizinische Klinik V, Universitätsklinikum Heidelberg, Im Neuenheimer Feld 410, 69120 Heidelberg, Germany; e-mail: maximilian.merz@med.uni-heidelberg.de.
References
- 1.Bochtler T, Stölzel F, Heilig CE, et al. . Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J Clin Oncol. 2013;31(31):3898-3905. [DOI] [PubMed] [Google Scholar]
- 2.Manier S, Salem KZ, Park J, et al. . Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2016;14(2):100-113. [DOI] [PubMed] [Google Scholar]
- 3.Moreau P, Masszi T, Grzasko N, et al. ; TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621-1634. [DOI] [PubMed] [Google Scholar]
- 4.Avet-Loiseau H, Fonseca R, Siegel D, et al. . Carfilzomib significantly improves the progression-free survival of high-risk patients in multiple myeloma. Blood. 2016;128(9):1174-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leleu X, Karlin L, Macro M, et al. ; Intergroupe Francophone du Myélome (IFM). Pomalidomide plus low-dose dexamethasone in multiple myeloma with deletion 17p and/or translocation (4;14): IFM 2010-02 trial results. Blood. 2015;125(9):1411-1417. [DOI] [PubMed] [Google Scholar]
- 6.Egan JB, Shi C-X, Tembe W, et al. . Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120(5):1060-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keats JJ, Chesi M, Egan JB, et al. . Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raab MS, Lehners N, Xu J, et al. . Spatially divergent clonal evolution in multiple myeloma: overcoming resistance to BRAF inhibition. Blood. 2016;127(17):2155-2157. [DOI] [PubMed] [Google Scholar]
- 9.Palumbo A, Avet-Loiseau H, Oliva S, et al. . Revised International Staging System for Multiple Myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonneveld P, Schmidt-Wolf IGH, van der Holt B, et al. . Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J Clin Oncol. 2012;30(24):2946-2955. [DOI] [PubMed] [Google Scholar]
- 11.Scheid C, Sonneveld P, Schmidt-Wolf IGH, et al. . Bortezomib before and after autologous stem cell transplantation overcomes the negative prognostic impact of renal impairment in newly diagnosed multiple myeloma: a subgroup analysis from the HOVON-65/GMMG-HD4 trial. Haematologica. 2014;99(1):148-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neben K, Lokhorst HM, Jauch A, et al. . Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119(4):940-948. [DOI] [PubMed] [Google Scholar]
- 13.Wuilleme S, Robillard N, Lodé L, et al. ; Intergroupe Francophone de Myélome. Ploidy, as detected by fluorescence in situ hybridization, defines different subgroups in multiple myeloma. Leukemia. 2005;19(2):275-278. [DOI] [PubMed] [Google Scholar]
- 14.Decaux O, Lodé L, Magrangeas F, et al. ; Intergroupe Francophone du Myélome. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the Intergroupe Francophone du Myélome. J Clin Oncol. 2008;26(29):4798-4805. [DOI] [PubMed] [Google Scholar]
- 15.Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188(3):369-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chretien M-L, Corre J, Lauwers-Cances V, et al. . Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126(25):2713-2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gisselsson D, Jin Y, Lindgren D, et al. . Generation of trisomies in cancer cells by multipolar mitosis and incomplete cytokinesis. Proc Natl Acad Sci USA. 2010;107(47):20489-20493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinhold N, Kirn D, Seckinger A, et al. . Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116-e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. J Natl Cancer Inst Monogr. 2008; (39):25-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.An G, Li Z, Tai Y-T, et al. . The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin Cancer Res. 2015;21(9):2148-2156. [DOI] [PubMed] [Google Scholar]
- 21.Merz M, Hielscher T, Seckinger A, et al. . Baseline characteristics, chromosomal alterations, and treatment affecting prognosis of deletion 17p in newly diagnosed myeloma. Am J Hematol. 2016;91(11):E473-E477. [DOI] [PubMed] [Google Scholar]
- 22.Merz M, Hielscher T, Seckinger A, et al. . Longitudinal fluorescence in situ hybridization at primary diagnosis and relapse reveals clonal evolution after autologous stem cell transplantation in multiple myeloma. Haematologica. 2017;102(8):1432-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.