

Abstract
Spontaneous brain activity is required for the development and maintenance of normal brain function. Many disease processes disrupt the organization of intrinsic brain activity but few pervasively reduce the amplitude of resting state blood oxygen level dependent (BOLD) fMRI fluctuations. We report the case of a female with anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis, longitudinally studied during the course of her illness in order to determine the contribution of NMDAR signaling to spontaneous brain activity. Resting state BOLD fMRI was measured at the height of her illness and 18 weeks following discharge from hospital. Conventional resting state networks were defined using established methods. Correlation and covariance matrices were calculated by extracting the BOLD time-series from regions of interest and calculating either the correlation or covariance quantity. The intrinsic activity was compared between visits, and to expected activity from 45 similarly-aged healthy individuals. Near the height of the illness, the patient exhibited profound loss of consciousness, high-amplitude slowing of the electroencephalogram, and a severe reduction in the amplitude of spontaneous BOLD fMRI fluctuations. The patient’s neurological status and measures of intrinsic activity improved following treatment. We conclude that NMDAR-mediated signaling plays a critical role in the mechanisms that give rise to organized spontaneous brain activity. Loss of intrinsic activity is associated with profound disruptions of consciousness and cognition.
Introduction
Intrinsic neural activity accounts for the greatest part of the brain’s high metabolic cost,[46] and is critical for both the development of normal brain organization,[27, 49] and the maintenance of homeostatic functions.[32, 57] This activity can be measured by quantifying spontaneous fluctuations in the blood oxygen level dependent (BOLD) signal utilizing resting state functional magnetic resonance imaging (rs-fMRI). BOLD signals normally exhibit a well-ordered temporal correlation structure that reflects functional organization (widely referred to as functional connectivity [FC]),[20] and defines interconnected resting-state networks (RSNs). The importance of spontaneous activity to brain development and function is exemplified by the persistence of organized spontaneous activity (i.e., FC) in healthy individuals during task performance and sleep,[46] the preservation of RSN structure in substantially altered brain states (e.g., sedation,[22] surgical anesthesia,[33, 40] and prolonged vegetative states [7]), and the observation that early changes in FC may be seen in individuals with neurodegenerative dementing illnesses (e.g. Alzheimer [9] and frontotemporal dementia [14]). Despite its importance, few opportunities exist to study the effects of focal (e.g., well-circumscribed lesions) or specific (e.g., inhibition of a single receptor system) insults on spontaneous activity. As a result, the mechanisms that give rise to FC remain largely unknown.
We present serial clinical and rs-fMRI findings from a previously healthy young adult with anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis. NMDAR encephalitis results from the production of IgG autoantibodies directed against the GluN1-subunit of central nervous system NMDARs, and classically manifests with profound changes in personality, psychiatric symptoms, memory loss, seizures and autonomic dysfunction.[53] Although most patients dramatically improve following treatment,[53] short-term mortality [15] and long-term morbidity—including residual cognitive impairment[17]—are increasingly recognized complications of NMDAR encephalitis. Accordingly, there is growing interest in identifying acute clinical and paraclinical measures that may predict longer-term individual outcomes. Description of a treatment-responsive disease associated with selective NMDAR dysfunction provides a unique opportunity to measure the within-patient effects of altered NMDAR-mediated signaling on spontaneous activity and FC. These results inform the potential association between changes in spontaneous activity, FC, and clinical outcomes.
Methods
All participants or a legal guardian provided written informed consent in accordance with the Washington University in St. Louis Institutional Review Board. Control data were obtained from ongoing imaging studies of similarly-aged healthy individuals scanned using a 3T Siemens Trio scanner (Erlangen, Germany) equipped with a standard 12-channel head coil. The present rs-fMRI results were obtained using previously described procedures.[10] Atlas registration was computed using a T1-weighted magnetization-prepared rapid gradient echo image. rs-fMRI was acquired using a gradient spin-echo sequence (TE=30 msec, TR=2200 msec, field of view=256 mm, flip angle=90°, 4mm isotropic voxels) sensitive to the BOLD contrast. The rs-fMRI scanning protocol was identical for patients and controls, except that control participants were instructed to fixate on a visual cross-hair and not fall asleep. All participants contributed two six-minute resting state runs (164 volumes each).
Initial preprocessing of rs-fMRI data followed conventional methods.[10, 45] Volumes contaminated by head movement were excluded. Frame censoring was computed using the derivative of the frame-to-frame variance (DVARS) measure, which identifies large changes in image intensity induced by head motion.[51] Signals of non-interest were extracted from white matter, ventricles, and the global signal averaged over the whole brain. Signals of non-interest along with movement time-series and their first temporal derivatives were regressed from the voxelwise BOLD time-series. The residual BOLD time-series was then low-pass filtered to retain frequencies below 0.1Hz and spatially smoothed with a Gaussian blur (6mm FWHM in each direction). Data quality with respect to motion artifact was assessed by comparing root mean square (RMS) movement to the control group and expressing this comparison as Z-scores. RMS movement was below the group mean at Visit1 (Z=−1.05) and near the mean at Visit2 (Z=−0.08). Similarly, the number of frames retained was similar to the group mean at Visit1 (Z=0.36) and Visit2 (Z=0.05).
Regions of interest (ROIs) were defined as previously described.[9] Thirty-six 6mm radius spheres belonging to 5 RSNs, including the default mode (DMN), dorsal attention (DAN), executive control (CON), salience (SAL), and sensorimotor + visual (SMN) networks, were examined. Correlation and covariance matrices were calculated by extracting the BOLD time-series from ROIs and calculating either the pair-wise correlation or covariance quantity. These ROI-by-ROI correlation matrices then were statistically compared by calculating their eigenspectra and comparing the individual eigenvalues (at either Visit1 or Visit2) relative to the eigenvalues corresponding to Z-scores from a control group. Eigenanalysis of the control group mean covariance matrix showed several large values (corresponding to specific RSNs), followed by many near-zero values. This distribution of eigenvalues represents structured brain activity. A flat eigenspectrum, where no single eigenvalue is large compared to the others, represents a lack of structure. The eigenspectra from controls were used as a population to calculate a Z-score for each observation in the patient. These Z-scores, assuming normality, coincide with p-values indexing the probability of the null-hypothesis that the patient is indistinguishable from the controls. Correlation mapping was performed by extracting the BOLD time-series from the ROIs and calculating the Pearson correlation coefficient between that time-series and the volumetric time-series covering all voxels within the brain mask.
Results
A previously well, 20-year-old right-handed African-American female college student was transferred to our tertiary care hospital 4 weeks following the onset of psychosis, rapidly progressive encephalopathy, seizures, persistent orofacial dyskinesias and cardiac instability (Figure 1A). Circulating IgG autoantibodies against NMDAR were identified in the serum and cerebrospinal fluid (cerebrospinal fluid titers 1:20 by indirect fluorescent histochemistry; ARUP Laboratories; Salt Lake City, UT), establishing the diagnosis of NMDAR encephalitis. Structural MRI was normal and remained normal throughout the course of the illness. The EEG (Figure 1B) showed diffuse high-amplitude slowing with predominant power in the delta range and superimposed beta range activity (“extreme delta brush” pattern, a characteristic finding in NMDAR encephalitis [48]). Pelvic ultrasound and MRI were suspicious for an ovarian mass; however, no neoplasm was found following unilateral oophorectomy and histopathological review. Immunosuppressive treatment was initiated with oral prednisone (1 mg/kg) and intravenous immunoglobulin (2mg/kg divided over 5 days). Rituximab (375 mg/m2/week × 4 weeks) was provided after the patient’s condition worsened. Tracheostomy was established and an enterogastric tube was placed to maintain nutrition. The first rs-fMRI study (Visit1) was performed 8 weeks after the onset of symptoms. At that point, the patient was receiving diazepam (10 mg every 6 hours; last dose 3 hours before imaging), midazolam (2 mg IV; last dose 10 hours before imaging), and propofol (IV; 80μg/kg/min).
Figure 1.
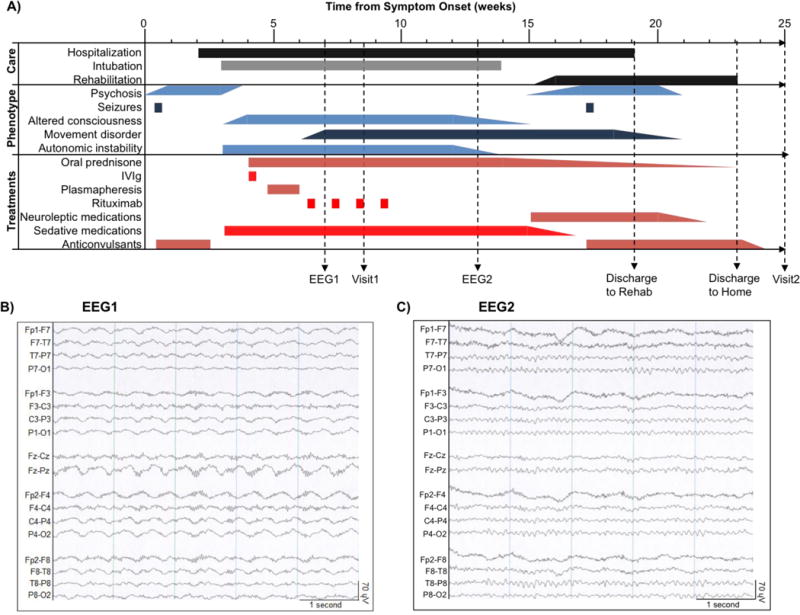
Clinical course and interventions. A: Line diagram (from time of symptom onset) depicting landmarks in clinical care, emergence of symptoms and signs (phenotype), treatments and the timing of electroencephalograms (EEG1 and EEG2) and structural / functional neuroimaging (Visit1 and Visit2). B: Abnormal EEG showing diffuse, rhythmic, delta activity at 1–3 Hz with superimposed bursts of rhythmic 20–30 Hz beta frequency activity riding on each delta wave (“extreme delta brush”[48]). C: Normal EEG with characteristic posterior-dominant alpha rhythm (8–13 Hz).
Twelve weeks following symptom onset, the patient regained spontaneous eye opening with inconsistent tracking. Over the ensuing 14 days, she began to obey 1-step commands. Autonomic instability gradually resolved. The EEG obtained at 13 weeks after symptoms onset was normal (Figure 1C). The patient was extubated at week 14 and transferred to an inpatient rehabilitation facility two weeks later. At that time, she was awake but impaired in multiple cognitive domains (Table 1). The remainder of the clinical course was marked by steady improvement in mental status. The patient was discharged home 23 weeks following the onset of symptoms. Cognitive testing at week 25 showed general improvement with persistent impairment in measures of global cognitive performance. Repeat rs-fMRI study (Visit2) was obtained without sedation at week 25. Cognitive testing at week 63 confirmed continued recovery, although delayed recall remained abnormal compared to age- and education-matched healthy individuals.
Table 1.
Patient Neuropsychological Performance
| Category | Test Name | Time from symptom onset | |||||
|---|---|---|---|---|---|---|---|
| 16 weeks | 25 weeks | 63 weeks | |||||
| Raw Score | Z | Raw Score | Z | Raw Score | Z | ||
| Global Function | MoCA [38] | 8 | −8.8** | 16 | −5.2** | 26 | −0.6 |
| MMSE [13] | – | – | 25 | −3.1** | 27 | −1.5 | |
| Processing Speed | Digit Symbol Modalities Test [50] | – | – | 27 | −3.9** | 57 | −0.09 |
| Trail Making Test A [53] | – | – | 42 | −2.8** | 54 | −4.5** | |
| Trail Making Test B [53] | – | – | C | – | 140 | −7.2** | |
| Episodic Memory | Logical Memory, Immediate Recall [1] | – | – | 6 | −1.8 | 17 | 1.3 |
| Word List, Delayed Recall [34] | – | – | 5 | −4.1** | 7 | −2.3* | |
| Language | BNT [31] | – | – | 12 | −4.5** | 14 | −1.2 |
| Verbal Fluency [54] | – | – | 14 | −1.3 | 17 | −0.7 | |
| FAS [54] | – | – | 12 | −3.2** | 30 | −1.2 | |
To control for social and demographic variability in the groups, Z-scores for neuropsychological testing were calculated by comparing raw scores to age and education matched norms. Z-scores were calculated on a test-by-test basis by subtracting the reported population mean from the patient’s raw score, and dividing the difference by the population standard deviation. Negative Z-scores reflect lower-than-expected patient performance; positive Z-scores reflect better-than-expected patient performance. P-values were inferred from the standard normal distribution, where p=0.05 corresponds to Z=±1.96, and p=0.01 corresponds to Z=±2.58.
= p<0.05;
= p<0.01
MoCA = Montreal Cognitive Assessment, MMSE = Mini-Mental State Examination, BNT = Boston Naming Test (15-item), C = patient unable to complete, – = not administered.
Trail Making Test A/B are scored in seconds (higher values indicate worse performance); all others scored as number correct.
Logical Memory Scores adjusted to match formatting for Form I of normalized scores
Word List Recall tested using a 10-word list; scores adjusted to match formatting of Hopkins Verbal Learning Tests scores (12-word list)
Figure 2 shows images of BOLD signal amplitude (temporal standard deviation) measured in the patient and a cohort of 45 healthy participants with a median age of 21 (19-24) years. The temporal standard deviation of the BOLD signal across uncensored (i.e., not motion contaminated) frames was calculated as a measure of spontaneous BOLD amplitude. In such images, neural activity normally gives rise to greater amplitude in grey as opposed to white matter. This feature was attenuated at Visit1 (Figure 2B) and restored at Visit2 (Figure 2C). Figure 2D shows mean BOLD signal amplitudes evaluated within 36 canonical ROIs representing 5 RSNs.[9] The bar graph results indicate that spontaneous activity was reduced in all RSNs at Visit1 but was within the normal range at Visit2.
Figure 2.
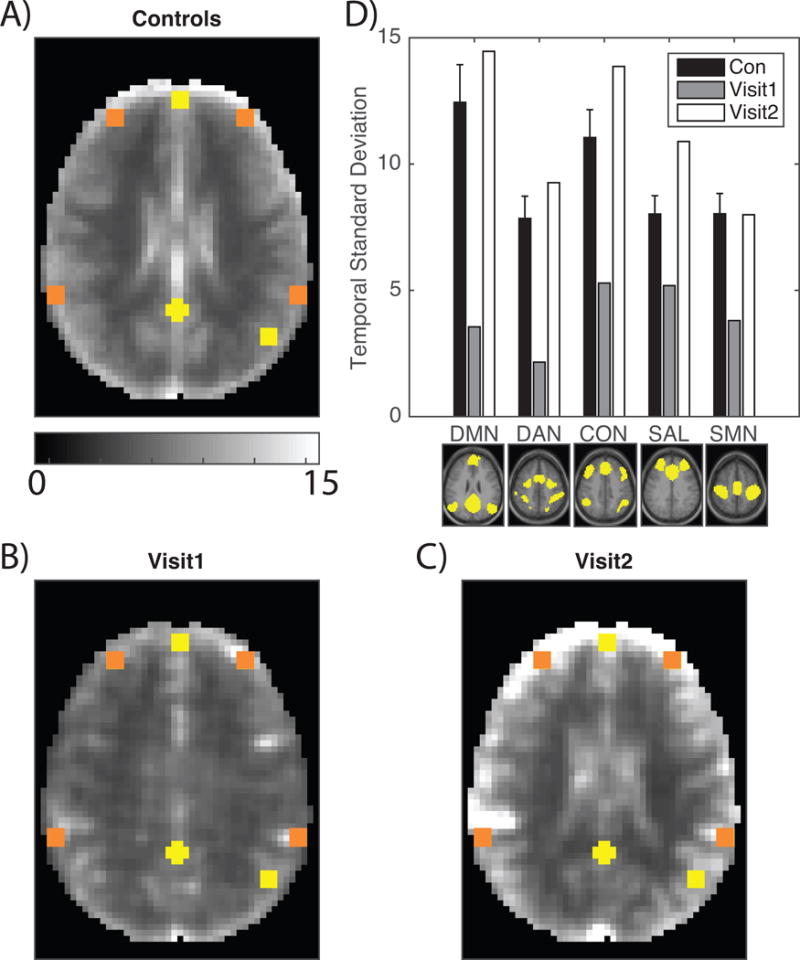
BOLD signal amplitude is diminished in a patient with NMDAR encephalitis. A: Mean BOLD temporal standard deviation derived from 45 age-matched healthy individuals. Yellow boxes correspond to DMN ROIs and orange boxes correspond to non-DMN ROIs. The control group demonstrates strong grey versus white matter contrast. B: Temporal standard deviation at Visit1 in the same style as A. Grey versus white matter contrast is markedly diminished and the overall amplitude is reduced. C: Temporal standard deviation at Visit2 in the same style as A. Grey versus white matter contrast is restored and the overall amplitude is restored. D: The temporal standard deviation of the BOLD signal measured in ROIs from each of 5 RSNs shown below the corresponding set of bars. Black bars indicate healthy individuals (±1 standard deviation); grey bars correspond to Visit1; white bars correspond to Visit2.
Figure 3 shows functional connectivity matrices corresponding to canonical ROIs. We present both Fisher z-transformed Pearson correlation ( ; conventional FC), as well as covariance matrices. Covariance retains sensitivity to BOLD signal amplitude whereas conventional FC does not (for additional discussion see Smyser et al. [52]). The block structure in the control cohort matrices reflects normal RSN organization. This structure is disrupted in the patient’s Visit1 results in both the correlation and covariance matrices. The near absence of structure in the Visit1 covariance matrix graphically reflects reduced amplitude of spontaneous BOLD signal fluctuations. The Visit2 results indicate substantial, but not complete, recovery of organized intrinsic activity, as evaluated in both the correlation and covariance matrices (Figure 3C).
Figure 3.
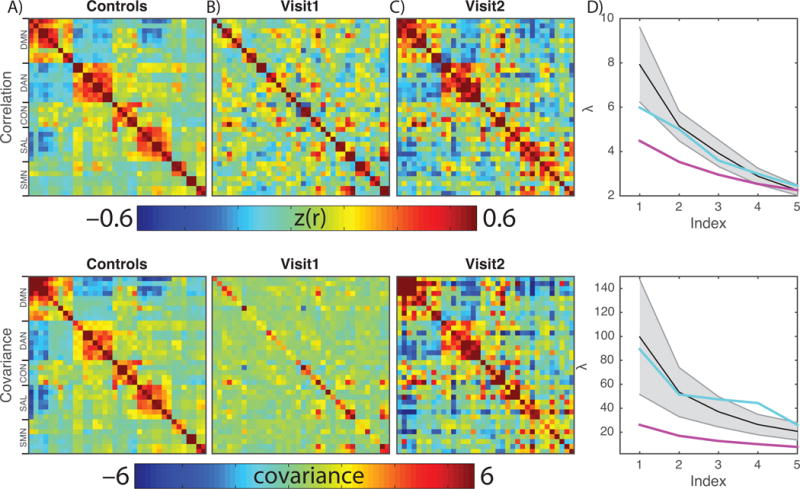
BOLD organization and amplitude is disrupted in a patient with NMDAR encephalitis. Top row shows BOLD correlation matrices; the bottom row shows BOLD covariance matrices. The salient difference in these results is that the former is normalized with respect to signal amplitude. A: BOLD correlation and covariance matrices derived from 45 age-matched healthy individuals. Rows and columns index individual regions of interest; the color indicates the correlation/covariance value of the BOLD signal extracted from ROIs. The prominent blocks represent RSNs. B: RSN structure is absent at Visit1. The covariance matrix emphasizes that the signal is both disorganized and has low amplitude. C: RSN structure and amplitude is restored at Visit2. D: Quantification of this observation is achieved by comparing the five largest eigenvalues of the matrices in healthy individuals (black line, grey area is +/− 1SD), Visit1 (magenta) and Visit2 (teal). The Visit1 result is abnormally flattened while the Visit2 result is within the normal range.
The patient’s RSN integrity, expressed as Z-scores relative to the control group, was assessed as an RSN composite score computed by averaging FC across all ROI-pairs within a given RSN.[9] At Visit1, all composite measures (DMN = −2.44; DAN = −2.89; CON = −3.04; SAL = −2.43), except for the sensorimotor network (SMN = – 0.76), were significantly abnormal (q<0.05; False Discovery Rate corrected1;[5] two-tailed) compared to matched healthy individuals. At Visit2, all composite scores were similar to controls (DMN = 0.22; DAN = 0.35; CON = −1.69; SAL = −1.03; SMN = 0.21).
Eigen-decomposition offers a means of estimating the dimensionality of rs-fMRI data.[52] Well-ordered structure in the correlation and covariance matrices manifests as large initial values in the eigenspectra (scree plots; Fig. 3D). Lack of structure manifests as flat scree plots. Normal eigenspectra are shown in black (±95% CI). The patient’s Visit1 eigenspectra (magenta traces) are abnormally flattened. The Visit2 results (teal traces) fall within the normal range.
Figure 4 shows the rs-fMRI RSN topographies. In healthy individuals, the DMN exhibits a characteristic topography in which the posterior cingulate/precuneus cortex is positively correlated with lateral parietal and medial frontal cortices, and negatively correlated with the DAN, CON, and SAL networks.[21] Complementary results are obtained in healthy controls with seeds in the DAN and CON and SAL networks. All RSNs (including the motor and visual networks) normally exhibit highly symmetric homotopic functional connectivity, as illustrated in the left columns of Figure 4. Almost all organized RSN structure was lost in the patient at Visit1; the only clear exception was continued organization of the SMN. RSN topography recovered at Visit2, except possibly in the VIS network.
Figure 4.
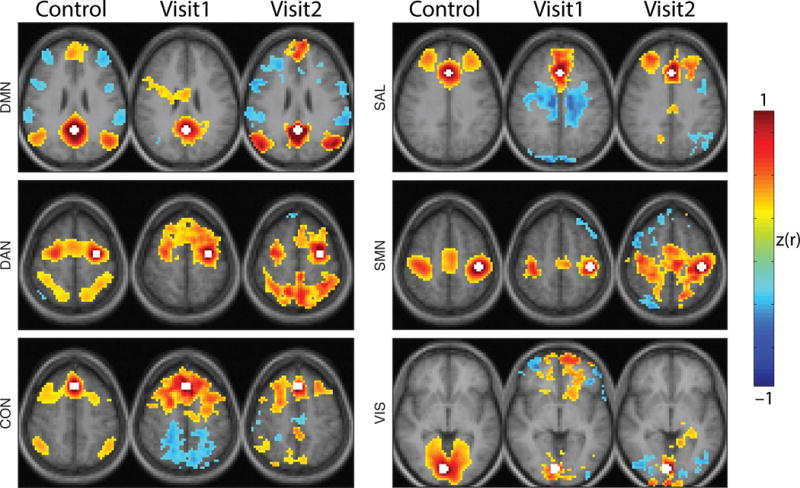
RSN topography is disrupted and recovers during NMDAR encephalitis. Seed-based functional connectivity maps for healthy individuals (first column), Visit1 and Visit2. Seed regions for each network were posterior cingulate cortex (DMN), left frontal eye field (DAN), dorsal medial prefrontal cortex (CON), anterior cingulate (SAL), left primary motor cortex (SMN) and right primary visual cortex (VIS) indicated in white. Conventional functional connectivity was calculated between the seed region and all other voxels. Resulting correlations were Fisher z-transformed. maps were thresholded voxel-wise and only suprathreshold clusters consisting of more than 100 voxels are shown. Healthy individuals demonstrate focal and defined RSN topographies that are absent at Visit1 and restored at Visit2. Images are presented in radiologic convention.
Discussion
We describe a case of NMDAR encephalitis associated with markedly reduced amplitude of spontaneous BOLD signal fluctuations and high amplitude slowing of the EEG. rs-fMRI abnormalities included reduced dimensionality and pervasive disorganization of FC, except in the SMN. These physiological abnormalities resolved, albeit incompletely, concurrent with improvement in clinical status. The present case bears substantial similarities to a previously reported child with epileptic encephalopathy and high amplitude slowing of the electroencephalogram (EEG) in whom a profound reduction in the amplitude of spontaneous BOLD fMRI fluctuations was documented.[43] These abnormalities largely normalized following successful treatment. The pathophysiological mechanism leading to encephalopathy is more strongly characterized in this report: antibody-mediated disruption of NMDAR-mediated neuronal signaling.
NMDAR encephalitis is caused by autoantibodies to the GluN1-subunit of central nervous system NMDARs. Major features of the clinical syndrome reflect the anatomical distribution of NMDARs, which are ubiquitously expressed throughout the brain but are especially prevalent in the hippocampus and cerebellum.[12, 42] In experimental models, antibody binding leads to IgG-mediated cross-linking and internalization of synaptic NMDARs, which decreases NMDAR cluster density and reduces NMDAR-associated synaptic currents without altering the number or structure of chemical synapses.[25, 37] In the absence of circulating NMDAR autoantibodies, NMDARs repopulate, thereby restoring synaptic currents.[25] Most patients recover following appropriate treatment, including immunotherapy.[53] However, cognitive deficits may across extended periods of time, and are presumed to reflect alterations in functional connectivity and white matter integrity that may be overlooked with standard clinical MR imaging.[16, 18] Our experience further advances this hypothesis, and emphasizes the potential application of longitudinal fMRI in appraising intrinsic activity and predicting meaningful clinical outcomes in a single case.
The biophysical properties of the NMDAR render it uniquely suited to play a central role in learning, memory and synaptic homeostasis (i.e., balancing long-term potentiation versus long-term depression, and excitation versus inhibition).[28, 36, 56] Dysregulation of these processes leads to cell death either owing to an excess of excitation (excitotoxicity) or withdrawal of trophic influences.[4, 58] Critically, these homeostatic processes depend on ongoing signaling, with the possibility that disruption may contribute to cognitive impairment reported in our patient and others.[16, 18]
Insight regarding the pathophysiology of NMDAR encephalitis has been obtained from animal studies. Infusion of human NMDAR autoantibodies into the ventricular cavities of mice severely disrupts learning and memory,[44] while loss of NMDARs on hippocampal parvalbumin interneurons disrupts theta oscillations, contributing to impaired spatial working memory.[11] Pharmacological blockade of NMDARs using MK801 reduces inhibition, leading to disorganized firing of pyramidal neurons,[24] and reductions in the amplitude of spontaneous BOLD fluctuations.[6] The magnitude of this effect (about 10% amplitude reduction) is quantitatively modest in comparison to the results presented in our patient with NMDAR encephalitis.
Ketamine, another NMDAR antagonist, has been reported to induce complex effects in humans, possibly owing to non-NMDA related pharmacology involving opioid receptors and monoamine transporters.[26] Immediately (minutes to hours) after administration of ketamine, the amplitude of BOLD signal fluctuations[29] and FC[23] was observed to increase, although the amplitude of task-evoked responses was reduced.[39] Chronic administration (greater than 24 hours) of ketamine focally reduced FC in selected RSNs.[47] Again, these results are quantitatively minor in comparison to the present findings.
BOLD signal fluctuations normally are tightly coupled to local field potentials[19] and slow cortical potentials.[41] These relationships are disrupted in the present case. The only other condition known to markedly suppress spontaneous BOLD fluctuations is deep (i.e., surgical) anesthesia with inhaled sevoflurane.[40] While we cannot offer a definitive explanation for the co-occurrence of high amplitude slowing of the EEG and markedly reduced BOLD fluctuations, we offer two speculative hypotheses. First, NMDAR encephalitis may disrupt hemodynamic coupling, which depends on glutamatergic signaling via NMDARs.[3] Second, the normally close link between local field potentials and the BOLD signal [30] was obtained in the presence of organized post-synaptic activity. Electrophysiological disorganization induced by NMDAR encephalitis may underlie reduced BOLD signal fluctuations.
The chief caveat in the present work is the requirement of sedation with several neuro-active medications at the time of the first imaging study in our patient—especially propofol. Propofol sedation may alter BOLD signal fluctuations and FC, potentially confounding the interpretation of results. However, the profound loss of intrinsic BOLD signal fluctuations reported here has not been described within a robust literature evaluating propofol effects in healthy individuals.[8, 33, 35] Similarly, it is unlikely that the use of sedative medication accounted for the high amplitude, diffusely slowed delta-frequency rhythm recorded via EEG near the time of imaging. On the contrary, propofol general anesthesia (associated with doses between 100–200 μg/kg/min) is more commonly associated with higher frequency oscillations (in the alpha range), with maximum power and coherence at 10 Hz.[2] Thus, although it is likely that propofol sedation contributed to changes in intrinsic activity, we conclude that pharmacological sedation does not account for the profound disruption of intrinsic activity observed in our patient with NMDAR encephalitis. These observations suggest that NMDAR signaling plays a critical role in the mechanisms that give rise to organized spontaneous brain activity.
Acknowledgments
The authors thank Dr. Jason Hassenstab for helpful discussions relating to the neuropsychological assessment of the patient. This work was supported by NIH: R01NR12657 (BMA), R01NR012907 (BMA), R01NR014449 (BMA), P30NS048056 (AZS); the Alzheimer’s Association (BMA); the Weston Brain Institute (GSD); the Neiss-Gain Family Endowment for Alzheimer Disease Research (GSD); and by generous support from the Paula and Rodger O. Riney fund.
Footnotes
Author Contribution: Conceptualization, BMA; Methodology, MRB, AZS, ABT; Formal Analysis, MRB; Investigation GSD, BMA; Writing-Original, MRB, GSD; Writing-Revision, AZS, BMA; Supervision, BMA
Conflict of Interest: The authors have no conflict of interest. GSD is the clinical director of the Anti-NMDA Receptor Encephalitis Foundation; the Foundation is supported by private donations.
The false discovery rate (FDR) approach to evaluating statistical significance corrects for multiple comparisons and applies when the tested hypotheses are not independent. This error rate control says that, for any set of comparisons, the rate of false positive is at most q. This is in contrast to the family-wise error rate where the false positive rate of any single test is p.
References
- 1.Abikoff H, Alvir J, Hong G, Sukoff R, Orazio J, Solomon S, Saravay S. Logical memory subtest of the Wechsler Memory Scale: age and education norms and alternate-form reliability of two scoring systems. Journal of clinical and experimental neuropsychology. 1987;9:435–448. doi: 10.1080/01688638708405063. [DOI] [PubMed] [Google Scholar]
- 2.Akeju O, Westover MB, Pavone KJ, Sampson AL, Hartnack KE, Brown EN, Purdon PL. Effects of sevoflurane and propofol on frontal electroencephalogram power and coherence. Anesthesiology. 2014;121:990–998. doi: 10.1097/ALN.0000000000000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–243. doi: 10.1038/nature09613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlett TE, Wang YT. The intersections of NMDAR-dependent synaptic plasticity and cell survival. Neuropharmacology. 2013;74:59–68. doi: 10.1016/j.neuropharm.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 6.Bettinardi RG, Tort-Colet N, Ruiz-Mejias M, Sanchez-Vives MV, Deco G. Gradual emergence of spontaneous correlated brain activity during fading of general anesthesia in rats: Evidences from fMRI and local field potentials. NeuroImage. 2015;114:185–198. doi: 10.1016/j.neuroimage.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boly M, Tshibanda L, Vanhaudenhuyse A, Noirhomme Q, Schnakers C, Ledoux D, Boveroux P, Garweg C, Lambermont B, Phillips C, Luxen A, Moonen G, Bassetti C, Maquet P, Laureys S. Functional connectivity in the default network during resting state is preserved in a vegetative but not in a brain dead patient. Human brain mapping. 2009;30:2393–2400. doi: 10.1002/hbm.20672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boveroux P, Vanhaudenhuyse A, Bruno MA, Noirhomme Q, Lauwick S, Luxen A, Degueldre C, Plenevaux A, Schnakers C, Phillips C, Brichant JF, Bonhomme V, Maquet P, Greicius MD, Laureys S, Boly M. Breakdown of within- and between-network resting state functional magnetic resonance imaging connectivity during propofol-induced loss of consciousness. Anesthesiology. 2010;113:1038–1053. doi: 10.1097/ALN.0b013e3181f697f5. [DOI] [PubMed] [Google Scholar]
- 9.Brier MR, Thomas JB, Snyder AZ, Benzinger TL, Zhang D, Raichle ME, Holtzman DM, Morris JC, Ances BM. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32:8890–8899. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brier MR, Thomas JB, Snyder AZ, Wang L, Fagan AM, Benzinger T, Morris JC, Ances BM. Unrecognized preclinical Alzheimer disease confounds rs-fcMRI studies of normal aging. Neurology. 2014;83:1613–1619. doi: 10.1212/WNL.0000000000000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, Ruhlmann C, Jones SR, Deisseroth K, Sheng M, Moore CI, Tsai LH. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Molecular psychiatry. 2012;17:537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti F, Barbaresi P, Melone M, Ducati A. Neuronal and glial localization of NR1 and NR2A/B subunits of the NMDA receptor in the human cerebral cortex. Cereb Cortex. 1999;9:110–120. doi: 10.1093/cercor/9.2.110. [DOI] [PubMed] [Google Scholar]
- 13.Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and education level. JAMA : the journal of the American Medical Association. 1993;269:2386–2391. [PubMed] [Google Scholar]
- 14.Day GS, Farb NA, Tang-Wai DF, Masellis M, Black SE, Freedman M, Pollock BG, Chow TW. Salience network resting-state activity: prediction of frontotemporal dementia progression. JAMA neurology. 2013;70:1249–1253. doi: 10.1001/jamaneurol.2013.3258. [DOI] [PubMed] [Google Scholar]
- 15.Day GS, High SM, Cot B, Tang-Wai DF. Anti-NMDA-receptor encephalitis: case report and literature review of an under-recognized condition. Journal of general internal medicine. 2011;26:811–816. doi: 10.1007/s11606-011-1641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finke C, Kopp UA, Pajkert A, Behrens JR, Leypoldt F, Wuerfel JT, Ploner CJ, Pruss H, Paul F. Structural Hippocampal Damage Following Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Biol Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.02.024. In Press. [DOI] [PubMed] [Google Scholar]
- 17.Finke C, Kopp UA, Pruss H, Dalmau J, Wandinger KP, Ploner CJ. Cognitive deficits following anti-NMDA receptor encephalitis. Journal of Neurology Neurosurgery and Psychiatry. 2012;83:195–198. doi: 10.1136/jnnp-2011-300411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finke C, Kopp UA, Scheel M, Pech LM, Soemmer C, Schlichting J, Leypoldt F, Brandt AU, Wuerfel J, Probst C, Ploner CJ, Pruss H, Paul F. Functional and Structural Brain Changes in Anti-N-Methyl-D-Aspartate Receptor Encephalitis. Annals of Neurology. 2013;74:284–296. doi: 10.1002/ana.23932. [DOI] [PubMed] [Google Scholar]
- 19.Florin E, Watanabe M, Logothetis NK. The role of sub-second neural events in spontaneous brain activity. Curr Opin Neurobiol. 2015;32:24–30. doi: 10.1016/j.conb.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nature reviews Neuroscience. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- 21.Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci U S A. 2005;102:9673–9678. doi: 10.1073/pnas.0504136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greicius MD, Kiviniemi V, Tervonen O, Vainionpaa V, Alahuhta S, Reiss AL, Menon V. Persistent default-mode network connectivity during light sedation. Human brain mapping. 2008;29:839–847. doi: 10.1002/hbm.20537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimm O, Gass N, Weber-Fahr W, Sartorius A, Schenker E, Spedding M, Risterucci C, Schweiger JI, Bohringer A, Zang Z, Tost H, Schwarz AJ, Meyer-Lindenberg A. Acute ketamine challenge increases resting state prefrontal-hippocampal connectivity in both humans and rats. Psychopharmacology (Berl) 2015;232:4231–4241. doi: 10.1007/s00213-015-4022-y. [DOI] [PubMed] [Google Scholar]
- 24.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, Balice-Gordon RJ. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30:5866–5875. doi: 10.1523/JNEUROSCI.0167-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kapur S, Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Molecular psychiatry. 2002;7:837–844. doi: 10.1038/sj.mp.4001093. [DOI] [PubMed] [Google Scholar]
- 27.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 28.Kullmann DM, Moreau AW, Bakiri Y, Nicholson E. Plasticity of inhibition. Neuron. 2012;75:951–962. doi: 10.1016/j.neuron.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 29.Littlewood CL, Jones N, O’Neill MJ, Mitchell SN, Tricklebank M, Williams SC. Mapping the central effects of ketamine in the rat using pharmacological MRI. Psychopharmacology (Berl) 2006;186:64–81. doi: 10.1007/s00213-006-0344-0. [DOI] [PubMed] [Google Scholar]
- 30.Logothetis NK. The neural basis of the blood-oxygen-level-dependent functional magnetic resonance imaging signal. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2002;357:1003–1037. doi: 10.1098/rstb.2002.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mack WJ, Freed DM, Williams BW, Henderson VW. Boston Naming Test: shortened versions for use in Alzheimer’s disease. J Gerontol. 1992;47:P154–158. doi: 10.1093/geronj/47.3.p154. [DOI] [PubMed] [Google Scholar]
- 32.Maffei A, Fontanini A. Network homeostasis: a matter of coordination. Curr Opin Neurobiol. 2009;19:168–173. doi: 10.1016/j.conb.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mhuircheartaigh RN, Rosenorn-Lanng D, Wise R, Jbabdi S, Rogers R, Tracey I. Cortical and subcortical connectivity changes during decreasing levels of consciousness in humans: a functional magnetic resonance imaging study using propofol. J Neurosci. 2010;30:9095–9102. doi: 10.1523/JNEUROSCI.5516-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miotto EC, Campanholo KR, Rodrigues MM, Serrao VT, Lucia MC, Scaff M. Hopkins verbal learning test-revised and brief visuospatial memory test-revised: preliminary normative data for the Brazilian population. Arquivos de neuro-psiquiatria. 2012;70:962–965. doi: 10.1590/s0004-282x2012001200014. [DOI] [PubMed] [Google Scholar]
- 35.Monti MM, Lutkenhoff ES, Rubinov M, Boveroux P, Vanhaudenhuyse A, Gosseries O, Bruno MA, Noirhomme Q, Boly M, Laureys S. Dynamic change of global and local information processing in propofol-induced loss and recovery of consciousness. PLoS Comput Biol. 2013;9:e1003271. doi: 10.1371/journal.pcbi.1003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris RG. NMDA receptors and memory encoding. Neuropharmacology. 2013;74:32–40. doi: 10.1016/j.neuropharm.2013.04.014. [DOI] [PubMed] [Google Scholar]
- 37.Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014;76:108–119. doi: 10.1002/ana.24195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I, Cummings JL, Chertkow H. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53:695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- 39.Northoff G, Richter A, Bermpohl F, Grimm S, Martin E, Marcar VL, Wahl C, Hell D, Boeker H. NMDA hypofunction in the posterior cingulate as a model for schizophrenia: an exploratory ketamine administration study in fMRI. Schizophr Res. 2005;72:235–248. doi: 10.1016/j.schres.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Palanca BJ, Mitra A, Larson-Prior L, Snyder AZ, Avidan MS, Raichle ME. Resting-state Functional Magnetic Resonance Imaging Correlates of Sevoflurane-induced Unconsciousness. Anesthesiology. 2015;123:346–356. doi: 10.1097/ALN.0000000000000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan WJ, Thompson GJ, Magnuson ME, Jaeger D, Keilholz S. Infraslow LFP correlates to resting-state fMRI BOLD signals. NeuroImage. 2013;74:288–297. doi: 10.1016/j.neuroimage.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pizoli CE, Shah MN, Snyder AZ, Shimony JS, Limbrick DD, Raichle ME, Schlaggar BL, Smyth MD. Resting-state activity in development and maintenance of normal brain function. Proc Natl Acad Sci U S A. 2011;108:11638–11643. doi: 10.1073/pnas.1109144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, Titulaer MJ, Petit-Pedrol M, Jain A, Balice-Gordon R, Lakadamyali M, Graus F, Maldonado R, Dalmau J. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain : a journal of neurology. 2015;138:94–109. doi: 10.1093/brain/awu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. NeuroImage. 2012;59:2142–2154. doi: 10.1016/j.neuroimage.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raichle ME. The restless brain. Brain connectivity. 2011;1:3–12. doi: 10.1089/brain.2011.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H, Boesiger P, Henning A, Seifritz E. Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS One. 2012;7:e44799. doi: 10.1371/journal.pone.0044799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmitt SE, Pargeon K, Frechette ES, Hirsch LJ, Dalmau J, Friedman D. Extreme delta brush: a unique EEG pattern in adults with anti-NMDA receptor encephalitis. Neurology. 2012;79:1094–1100. doi: 10.1212/WNL.0b013e3182698cd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shatz CJ. Emergence of order in visual system development. Proc Natl Acad Sci U S A. 1996;93:602–608. doi: 10.1073/pnas.93.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheridan LK, Fitzgerald HE, Adams KM, Nigg JT, Martel MM, Puttler LI, Wong MM, Zucker RA. Normative Symbol Digit Modalities Test performance in a community-based sample. Arch Clin Neuropsychol. 2006;21:23–28. doi: 10.1016/j.acn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Smyser CD, Inder TE, Shimony JS, Hill JE, Degnan AJ, Snyder AZ, Neil JJ. Longitudinal analysis of neural network development in preterm infants. Cereb Cortex. 2010;20:2852–2862. doi: 10.1093/cercor/bhq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smyser CD, Snyder AZ, Shimony JS, Mitra A, Inder TE, Neil JJ. Resting-State Network Complexity and Magnitude Are Reduced in Prematurely Born Infants. Cereb Cortex. 2016;26:322–333. doi: 10.1093/cercor/bhu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet neurology. 2013;12:157–165. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tombaugh T. Trail Making Test A and B: Normative data stratified by age and education. Archives of Clinical Neuropsychology. 2004;19:203–214. doi: 10.1016/S0887-6177(03)00039-8. [DOI] [PubMed] [Google Scholar]
- 55.Tombaugh TN, Kozak J, Rees L. Normative data stratified by age and education for two measures of verbal fluency: FAS and animal naming. Arch Clin Neuropsychol. 1999;14:167–177. [PubMed] [Google Scholar]
- 56.Tsien JZ. Linking Hebb’s coincidence-detection to memory formation. Curr Opin Neurobiol. 2000;10:266–273. doi: 10.1016/s0959-4388(00)00070-2. [DOI] [PubMed] [Google Scholar]
- 57.Vitureira N, Letellier M, Goda Y. Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr Opin Neurobiol. 2012;22:516–521. doi: 10.1016/j.conb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Q, Sheng M. NMDA receptors in nervous system diseases. Neuropharmacology. 2013;74:69–75. doi: 10.1016/j.neuropharm.2013.03.030. [DOI] [PubMed] [Google Scholar]