

Abstract
Copy number mutations implicate excess production of α-synuclein as a possibly causative factor in Parkinson’s disease (PD). Using an unbiased screen targeting endogenous gene expression, we discovered that the β2-adrenoreceptor (β2AR) is a regulator of the α-synuclein gene (SNCA). β2AR ligands modulate SNCA transcription through histone 3 lysine 27 acetylation of its promoter and enhancers. Over 11 years of follow-up in 4 million Norwegians, the β2AR agonist salbutamol, a brain-penetrant asthma medication, was associated with reduced risk of developing PD (rate ratio, 0.66; 95% confidence interval, 0.58 to 0.76). Conversely, a β2AR antagonist correlated with increased risk. β2AR activation protected model mice and patient-derived cells. Thus, β2AR is linked to transcription of α-synuclein and risk of PD in a ligand-specific fashion and constitutes a potential target for therapies.
The brains of most patients with Parkinson’s disease (PD) are riddled with intracellular accumulations of α-synuclein protein known as Lewy bodies. Triplication or duplication of the wild-type α-synuclein gene (SNCA) locus is sufficient to cause familial PD (1, 2). In these patients, copies of functionally normal SNCA mRNA and α-synuclein protein are increased by about 50 to 100% (2, 3). Even smaller increases in α-synuclein transcription may play an analogous role in patients with sporadic disease carrying potential regulatory variants in this gene (4).
Traditionally, drug development in PD has focused on clearance of α-synuclein protein, blockade of its transformation into toxic species, or amelioration of its downstream consequences. In contrast, we hypothesized that chemical compounds designed to reduce the transcription of the SNCA gene could make it possible to prevent or slow down the disease process in selected patients, but this idea lacked a druggable target. Regulation of SNCA expression appears to include GATA transcription factor occupancy of evolutionarily conserved enhancers in intronic regions of SNCA (5) and, possibly, the NGF (nerve growth factor) and bFGF (basic fibroblast growth factor) pathways (6), methylation (7), and micro RNAs (8). However, none of these candidates can be easily targeted by available medicines.
Drug screen targeting endogenous SNCA expression identifies β2AR agonists
We developed a high-throughput gene expression assay for endogenous human SNCA expression in situ in neuronal cells. This is an alternative approach to construct-based reporter assays, which typically do not fully represent the integrated microcircuit of promoters, enhancers, and histone marks that naturally regulate gene expression in a human cell. Human SK-N-MC neuroblastoma cells were cultured and drug-treated in 384-well plates, and relative endogenous SNCA mRNA expression was assayed.
SNCA expression–lowering compounds were identified in a four-stage study design (Fig. 1) consisting of screening, replication, and confirmation of transcript expression, followed by an enzyme-linked immunosorbent assay (ELISA) stage for quantification of protein expression. We screened 1126 compounds, including drugs approved by the U.S. Food and Drug Administration (FDA) and a diverse set of natural products, vitamins, health supplements, and alkaloids (data S1 and fig. S1). SK-N-MC cells were treated with each compound for 48 hours. Forty-one compounds were included in the replication stage: Thirty-five compounds, including the selective β2-adrenoreceptor (β2AR) agonist metaproterenol, lowered SNCA expression by more than 35% in the screening stage; six related drugs, including the selective β2AR agonists clenbuterol and salbutamol, were added at the replication stage (“hit expansion”). Four compounds had P values ≤ 0.005 (two-tailed Student’s t test) in the confirmation stage and also lowered α-synuclein protein abundance (determined by ELISA) in SK-N-MC cells (P ≤ 0.05; two-tailed Student’s t test, comparing with vehicle) (Fig. 1A). Unexpectedly, three of these hits were β2AR agonists (Fig. 1B), and these were prioritized for further investigation.
Fig. 1.
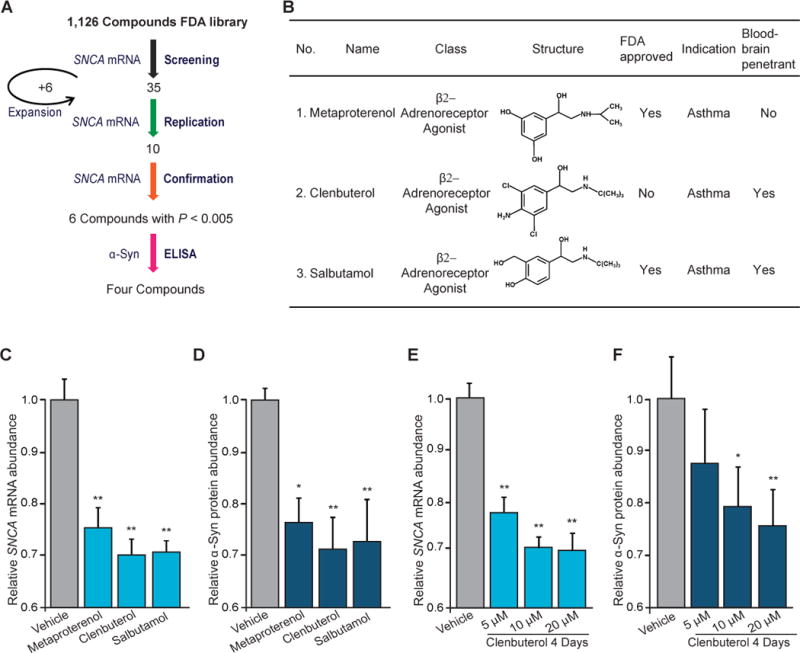
A screen of endogenous neuronal gene expression reveals β2AR as a regulator of SNCA. (A) Four out of a total of 1126 FDA-approved drugs and other compounds lowered the relative abundance of endogenous SNCA mRNA and α-synuclein protein (α-Syn) in SK-N-MC cells. (B) These included three selective β2AR compounds, whose chemical and clinical characteristics are shown. (C and D) The β2AR agonists metaproterenol (5 μM), clenbuterol (20 μM), and salbutamol (10 μM) also reduced the relative abundance of endogenous SNCA mRNA (C) and α-Syn protein (D) in rat primary cortical neurons (n = 4). (E and F) β2AR agonists lowered the expression of SNCA mRNA (E) and α-Syn protein (F) in a dose-dependent manner in neuroblastoma cells (n = 6 to 8). Means ± SEM are shown. * P < 0.05; ** P < 0.005; one-way ANOVA with Tukey’s.
Treatment with metaproterenol reduced SNCA mRNA abundance in SK-N-MC cells compared with that in control cells (P = 0.005; two-tailed Student’s t test) in the confirmation stage (fig. S2A) and was further verified (fig. S2B). Treatment with clenbuterol (fig. S2C) and salbutamol (fig. S2D) also had similar effects on relative SNCA mRNA abundance. Thus, we concluded that β2AR activation may regulate endogenous SNCA expression in SK-N-MC cells. Interestingly, the screen highlighted riluzole hydrochloride (fig. S1E) as a fourth hit. This compound is FDA-approved for modification of amyotrophic lateral sclerosis and has been shown to attenuate dopaminergic neurodegeneration in a 6-hydroxydopamine rat model of PD (9).
β2AR activation selectively modulated the expression of SNCA without adversely affecting neuronal cell viability or housekeeping gene expression (fig. S3) (10). As expected, the effects of β2AR agonists on SNCA expression were dependent on cellular context (fig. S4). For example, in human erythroleukemia cells, which express SNCA mRNA but lack β2AR (fig. S4A), and in neuronal SH-SY5Y cells, which transcribe β2AR but express low levels of SNCA mRNA (fig. S4B), agonists did not influence SNCA expression (fig. S4, C and D). These results are consistent with the specificity of our observations.
We used a sensitive ELISA and antibodies against α-synuclein (11) to determine whether the modulation of SNCA mRNA expression by β2AR translates into changes in α-synuclein protein abundance. In rat primary cortical neurons, endogenous SNCA mRNA (Fig. 1C) and α-synuclein protein (Fig. 1D) levels were significantly, but modestly, reduced in response to β2AR activation by metaproterenol (P < 0.005 and 0.05, respectively), clenbuterol (P < 0.005), or salbutamol (P < 0.005), compared with controls [analysis of variance (ANOVA) with Tukey’s].
β2AR agonists lowered SNCA expression in a dose- and time-dependent manner (10) (fig. S5). Increasing concentrations of clenbuterol (5, 10, and 20 μM) correlated with a decrease in SNCA mRNA (Fig. 1E) and α-synuclein protein (Fig. 1F) levels in SK-N-MC cells. Similarly, metaproterenol and salbutamol lowered SNCA mRNA expression in a dose-dependent manner (P < 0.005; ANOVA with Tukey’s) (fig. S6).
β2AR activation reduces Snca expression in mouse substantia nigra
PD preferentially affects dopaminergic neurons in the substantia nigra. We examined the effects of the selective β2AR agonist clenbuterol (which can be efficiently administered intraperitoneally) to probe the effects of β2AR activation on Snca expression in the substantia nigra of wild-type C57BL/6J mice. As expected (12, 13), clenbuterol crossed the blood-brain barrier, and its brain/plasma ratio increased with doses of 1, 5, or 10 mg of drug per kilogram of body weight (Fig. 2A).
Fig. 2.
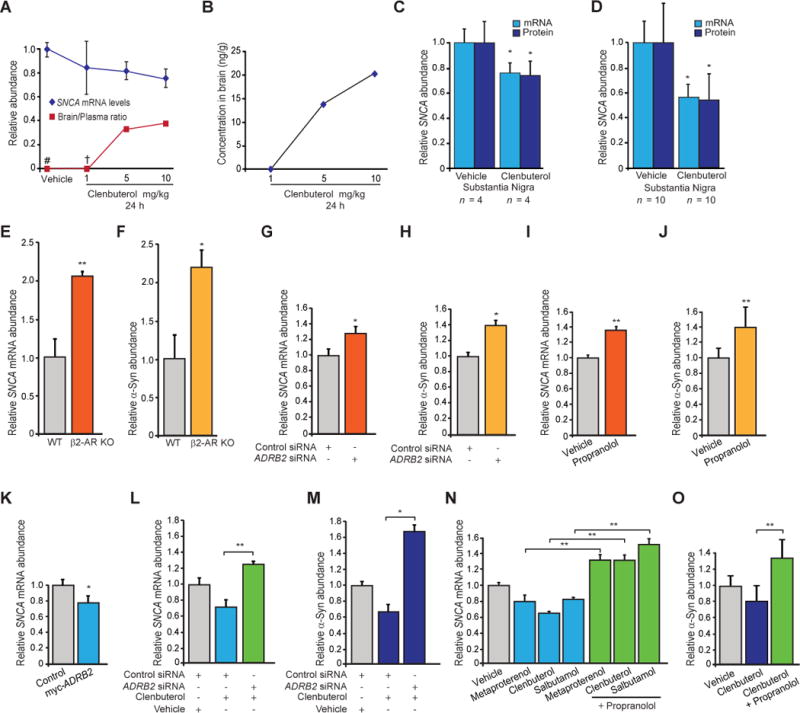
Bidirectional regulation of endogenous SNCA expression by β2AR modulation in vivo and in vitro. (A) Clenbuterol brain/plasma ratio in mice (red) and corresponding SNCA mRNA levels in the PD-vulnerable substantia nigra (blue). #Drug concentration below the quantifiable limit in brain and plasma. †Drug concentration below the quantifiable limit in brain. (B) Clenbuterol concentration in mouse brains. (C and D) β2AR activation lowered the expression of endogenous SNCA in the substantia nigra of mice in the dose-finding (C) and controlled (D) trials for 24 hours. (E to J) Knockout of the β2AR gene (Adrb2) in mouse primary neurons [(E) and (F); n = 6 to 9], silencing of β2ARs with RNA interference in human SK-N-MC cells [(G) and (H); n = 3], or chemical inhibition of β2ARs by the β-blocker propranolol in SK-N-MC cells [(I) and (J); n = 8 to 12] consistently increased the expression of SNCA mRNA [orange bars in (E), (G), and (I)] and α-Syn protein [yellow bars in (F), (H), and (J)]. (K) Transient transfection of SK-N-MC cells with ADRB2 constructs resulted in a reduction in endogenous SNCA mRNA levels, compared with those in cells transfected with empty vector (n = 6). (L to O) β2AR is necessary for mediating the effects of β2AR ligands on endogenous SNCA expression. Silencing of the β2AR gene abrogated the clenbuterol-induced reduction in SNCA mRNA and α-Syn protein expression [(L) and (M); n = 3]. Cotreatment with the β2AR antagonist propranolol abrogated the SNCA mRNA–lowering effects of metaproterenol, clenbuterol, and salbutamol [(N); n = 5 to 6]. Cotreatment with propranolol also abrogated the β2AR agonist–induced change in α-Syn protein levels [(O); n = 8 to 12]. siRNA, small interfering RNA. Means ± SEM. * P < 0.05; ** P < 0.005; two-tailed Student’s t test [(C) to (K)] or one-way ANOVA with Tukey’s [(L) to (O)].
Intraperitoneal injection of 10 mg/kg, administered for 24 hours, resulted in the highest brain/plasma ratio (Fig. 2A) and brain concentration (Fig. 2B) and induced a significant reduction in nigral α-synuclein protein and mRNA levels (P < 0.05; two-tailed Student’s t test) (Fig. 2C). We then performed a larger, randomized, placebo-controlled trial in mice to determine whether clenbuterol is efficacious in lowering α-synuclein expression in the substantia nigra of wild-type mice. Mice were euthanized after 24 hours of acute drug treatment. β2AR activation lowered the expression of endogenous α-synuclein protein and mRNA levels in the PD vulnerable substantia nigra (P = 0.01; two-tailed Student’s t test) (Fig. 2D). This was confirmed by Western blotting with various antibodies against α-synuclein (fig. S7). Overall, β2AR agonist treatment reduced SNCA expression in rodent neurons and substantia nigra.
Bidirectional modulation of SNCA expression by β2AR
We examined Snca expression levels in primary neurons derived from mice carrying a deletion of the β2AR gene (Adrb2). Endogenous Snca mRNA and α-synuclein protein levels were increased by 100 and 120%, respectively, compared with those in controls (P = 0.004 and 0.01, respectively; Student’s t test) (Fig. 2, E and F). In accord, silencing of β2AR in human SK-N-MC cells increased SNCA mRNA and α-synuclein protein levels (Fig. 2, G and H).
Moreover, chemical antagonism of β2AR with propranolol, a well-characterized b-blocker, in SK-N-MC cells similarly increased endogenous SNCA mRNA and α-synuclein protein levels (P = 0.00001 and 0.001, respectively; two-tailed Student’s t test) (Fig. 2, I and J, and fig. S8). Conversely, transient transfection of SK-N-MC cells with ADRB2 constructs reduced endogenous SNCA mRNA levels relative to those of controls (P = 0.01) (Fig. 2K). Genetic silencing of β2AR or cotreatment with propranolol blocked clenbuterol’s SNCA expression–lowering effects (Fig. 2, L to O). Collectively, these internally consistent data suggest that β2AR modulation is sufficient for altering endogenous SNCA expression and necessary for mediating the effects of β2AR ligands on endogenous SNCA expression.
β2AR regulates transcription of human SNCA through H3K27 acetylation
SNCA transcription appears to be finely regulated through a classical promoter spanning the non– protein-coding exon 1 and intron 1 at the 5′ end of the SNCA locus and through enhancers in the long intron 4 (Fig. 3A) (5). We clarified the endogenous SNCA promoter and putative enhancer sites by CAGE (cap analysis gene expression) in human PD relevant substantia nigra and by integrative genomics (Fig. 3A) (10). Histone 3 lysine 27 acetylation (H3K27ac) signals (indicative of active enhancer elements) were observed at the promoter and enhancer regions (Fig. 3A). Because β2AR stimulation has been implicated in regulating WNK4 transcription through histone acetylation in renal cells (14), we hypothesized that β2AR activation may regulate SNCA transcription through an analogous mechanism.
Fig. 3.
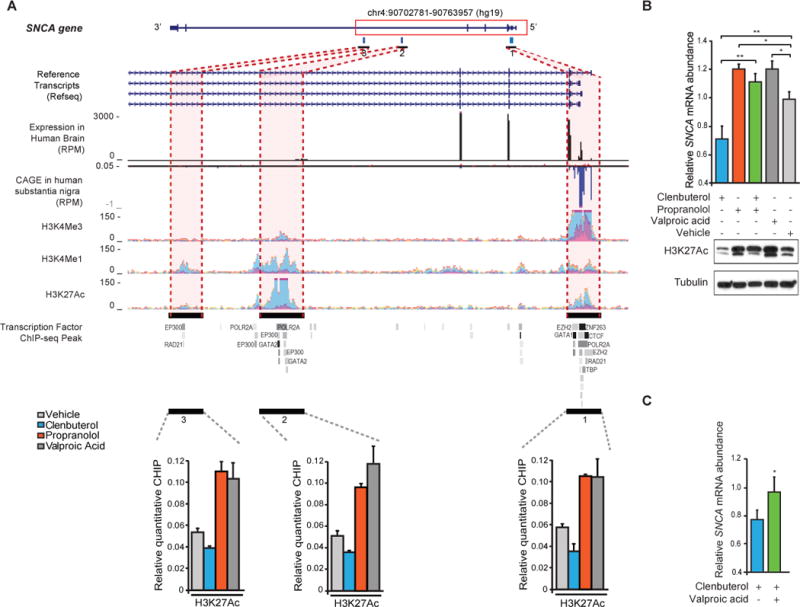
β2AR regulates the transcription of SNCA through H3K27 acetylation (H3K27ac) across the SNCA promoter and two enhancers in intron 4. (A) The SNCA gene, tracks for RefSeq transcripts, normalized read density of RNA sequencing in the human brain (34), CAGE in human substantia nigra (10), histone modifications (H3K4me3, H3K4me1, and H3K27ac), and transcription factor occupancy (35) are shown. RPM, reads per million. Vertical bar 1 corresponds to the SNCA promoter, and vertical bars 2 and 3 correspond to the two enhancers. Clenbuterol (blue) and propranolol (orange) treatments modulated H3K27ac across the three regulatory sites, as determined by quantitative chromatin immunoprecipitation (ChIP) (P < 0.05; ANOVA with Tukey’s). Dark gray, histone deacetylase inhibitor valproic acid; gray, vehicle. Means ± SEM of three independent experiments. (B) Western blotting with an antibody against H3K27ac (bottom) and relative SNCA mRNA levels (top) (n = 7). Means ± SEM. *P < 0.05; ** P < 0.005; one-way ANOVA with Tukey’s. (C) Cotreatment of clenbuterol with valproic acid abrogated the β2AR agonist’s effect on SNCA expression (green) (n = 4). Means ± SEM. * P < 0.05; two-tailed Student’s t test.
Clenbuterol treatment reduced H3K27ac across the promoter (site 1, Fig. 3A) and two putative intronic enhancers (sites 2 and 3, Fig. 3A), compared with vehicle treatment (P < 0.05; one-way ANOVA with Tukey’s). Conversely, the β-blocker propranolol increased H3K27ac across these putative regulatory sites (Fig. 3A) (P < 0.05). Consistently, the known histone deacetylase inhibitor valproic acid (15) increased H3K27ac (Fig. 3A). Western blotting with an antibody against H3K27ac confirmed our hypothesis (Fig. 3B). Clenbuterol treatment resulted in a correlated decrease in H3K27ac levels and relative SNCA mRNA abundance (Fig. 3B). Conversely, treatment with valproic acid resulted in an increase in H3K27ac levels and relative SNCA mRNA abundance, compared with vehicle treatment (Fig. 3B). Inhibition of H3K27 deacetylation (by cotreatment with valproic acid) abrogated the β2AR agonist effect on SNCA expression (Fig. 3C). Thus, β2AR regulates the transcription of α-synuclein in correlation with H3K27ac across the promoter and enhancers in the human SNCA locus.
β2AR ligands are associated with risk of PD in Norwegians
We evaluated the effects of β2AR activation in two nationwide, longitudinal analyses of incident PD in Norway; a mouse model of MPTP (N-methyl- 4-phenyl-1, 2, 3, 6-tetrahydropyridine)–induced human parkinsonism; and an iPSC (induced pluripotent stem cell)–derived neuronal culture system from a patient with autosomal dominant PD due to a triplication of the SNCA locus. The Norwegian Prescription Database (NorPD) contains complete information on all prescribed drugs dispensed at pharmacies to individuals in Norway since 2004 (16). Given that β2ARmodulates SNCA expression, we hypothesized that use of β2AR ligands would affect PD risk. We thus tested salbutamol and propranolol, respectively the most commonly used β2AR agonist and antagonist in Norway, as time-dependent covariates in two separate Cox proportional hazard models. We adjusted for sex, age, and level of education and included the total Norwegian population alive on 1 January 2004 as the study population (n = 4.6 million). We observed a yearly incidence rate of PD similar to that found in a recent clinical incidence study in Norway (10, 17). Salbutamol was associated with decreased risk of PD, with a rate ratio of 0.66 [95% confidence interval (CI), 0.58 to 0.76] (Tables 1 and 2, Fig. 4A, and fig. S9). Propranolol was associated with a markedly increased risk of PD, with a rate ratio of 2.20 (95% CI, 1.62 to 3.00) (Table 1 and Fig. 4B).
Table 1.
Rate ratio (RR) for Parkinson’s disease in persons treated with salbutamol or propranolol during a complete 11 years follow-up of the entire population of Norway.
| RR (95% CI)
|
|||||
|---|---|---|---|---|---|
| Users
|
Cases
|
Person-years
|
Age-, sex- adjusted
|
Multivariate adjusteda
|
|
| Salbutamol | |||||
| Never user | 4,066,119 | 4,398 | 36,700,554 | 1 (ref) | 1 (ref) |
| Ever user | 619,863 | 236 | 3,135,956 | 0.65 (0.57–0.74) | 0.66 (0.58–0.76) |
| Propranolol | |||||
| Never user | 4,671,188 | 4,593 | 39,770,912 | 1 (ref) | 1 (ref) |
| Ever userb | 14,794 | 41 | 65,598 | 2.16(1.59–2.94) | 2.20 (1.62–3.00) |
Adjusted for age in 5 year periods, sex and level of education.
Use of at least 365 defined daily doses.
RR: Rate ratio; PD: Parkinson’s disease; Cl: confidence interval
Table 2.
Rate ratio (RR) for Parkinson’s disease during 2008–2014 for salbutamol prescribed during 2004–2007 in the entire population of Norway.
| RR (95% CI)
|
|||
|---|---|---|---|
| Users 2004–07
|
Cases 2008–14
|
Multivariate Adjusteda
|
|
| Salbutamol | |||
| Never user | 4,201,011 | 2,338 | 1 (ref) |
| Low (<60 DDD) | 152,965 | 68 | 0.96 (0.76–1.23) |
| Medium (60–180 DDD) | 72,911 | 23 | 0.60 (0.40–0.91) |
| High (≥180 DDD) | 69,511 | 25 | 0.45 (0.31–0.67) |
Adjusted for age in five year periods, sex and level of education.
DDD: Defined daily dose; RR: Rate ratio; CI: confidence interval
Fig. 4.
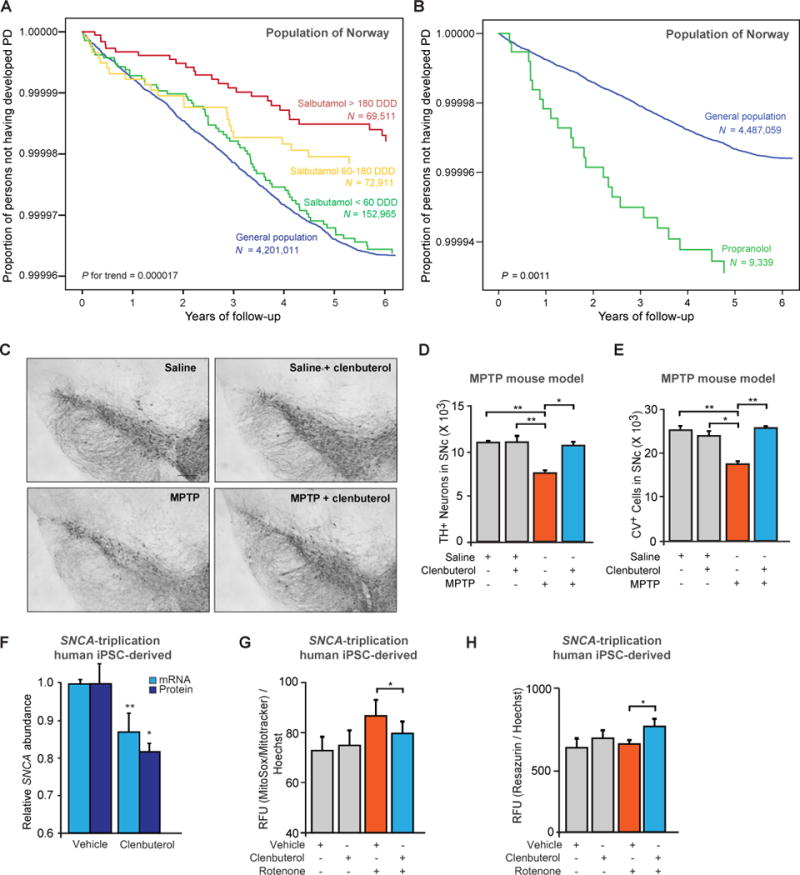
β2AR ligands are associated with risk of PD in Norway, and agonists show neuroprotective effects. (A and B) Covariate-adjusted survival curves show the proportion of individuals not developing PD from 2008 to 2014 for different exposure groups. Cox’s proportional hazard regression model adjusted for age, sex, and level of education was used for these analyses. In (A), Norwegians who never were prescribed salbutamol (“never users”) are represented by the blue survival curve. Individuals who were prescribed salbutamol at high [>180 defined daily doses (DDDs); red] or medium doses (60 to 180 DDDs; yellow) between 2004 and 2007 had lower proportions of incident PD during longitudinal follow-up. In (B), Norwegians who never were prescribed propranolol (“never users”) are represented by the blue survival curve. Individuals (n = 9339) who used at least 365 DDDs of propranolol between 2004 and 2007 had a higher proportion of incident PD (green) during longitudinal follow-up. (C) Representative images illustrating TH+ neurons in the substantia nigra pars compacta (SNpc). MPTP-treated animals show loss of TH+ neurons relative to control animals treated with saline or saline plus clenbuterol. Scale bar, 100 m. (D and E) Clenbuterol abrogated MPTP induced loss of nigral neurons in mice, as assayed by anti-TH immunostaining (D) or cresyl violet (CV) staining of cells (E) and stereology (n = 6 to 8 animals per group). Means ± SEM. * P < 0.05; ** P < 0.01; one-way ANOVA with Tukey’s. (F) Effect of clenbuterol treatment (20 μM) on SNCA mRNA expression (light blue; 3 days) and α-Syn protein expression (dark blue; 4 days) in PD patient iPSC– derived neuronal precursor cells (NPCs) carrying the SNCA locus triplication. Means ± SEM. * P <0.05; ** P < 0.005; two-tailed Student’s t test. (G) Clenbuterol treatment and levels of mitochondria‐associated superoxide in NPCs carrying the SNCA triplication. Cells were treated with or without 20 μM clenbuterol for four days and challenged with 20 μM rotenone during the last 18 hours (n = 6). (H) Clenbuterol treatment affects cellular viability of these NPCs, as determined by using resazurin, a fluorescent indicator dye of mitochondrial and other cellular reductive potentials. Cells were treated with or without 20 μM clenbuterol for 4 days and challenged with 20 μM rotenone during the last 18 hours (n = 6). RFU, relative fluorescence units. Means ± SD [(G) and (H)]. * P < 0.05; two-way ANOVA with Tukey’s [(G) and (H)].
The most common indication for salbutamol in our database was asthma. Smoking has been associated with decreased risk of PD (18). Tobacco exposure is also associated with early childhood asthma (19). If smoking explained the reduced risk associated with salbutamol, we would expect to see a similarly reduced risk for other asthma drugs not acting on β2AR. However, inhaled corticosteroids, which are frequently prescribed for asthma, did not reduce the PD risk (rate ratio, 0.95; 95% CI, 0.80 to 1.12) (table S1) after adjusting for salbutamol use and level of education. Further, adjusting for education, which is strongly associated with smoking habits in Norway (20), we observed only a slight change in the effect of salbutamol (Table 1). Thus, it is unlikely that smoking can fully explain the association between salbutamol and PD.
Propranolol is used to treat cardiovascular diseases and essential tremor, which might be misdiagnosed as a first sign of PD. To reduce this source of bias, we excluded all individuals with an indication of essential tremor or other neurological diseases and included only those with cardiovascular diagnoses. Moreover, we introduced a time lag between time of first exposure to propranolol and PD onset. Using time lags of 1 and 2 years only slightly reduced the effect estimates (rate ratio reduced from 2.20 to 1.82). This makes it unlikely that reverse causality explains a major part of this association.
β2AR activation protects MPTP model mice
In addition to α-synuclein, chemicals such as MPTP (21, 22) and rotenone (23, 24) are implicated in the mechanism of sporadic PD. These chemicals inhibit the flow of electrons through complex I of the electron transport chain and foster buildup of superoxide and other reactive oxygen species, particularly in dopamine neurons (22, 25, 26). We tested whether clenbuterol treatment could protect against MPTP-induced degeneration of tyrosine hydroxylase–positive (TH+) neurons in the substantia nigra pars compacta (SNpc) of a mouse model of PD (10, 22). Clenbuterol treatment abrogated the MPTP-induced loss of TH+ neurons (Fig. 4, C and D) and, importantly, also blocked the loss of cresyl violet–stained cells in the SNpc (Fig. 4E and fig. S10).
β2AR agonist in patient-derived cells carrying a SNCA triplication
Triplication of the SNCA locus causes autosomal dominant PD (1, 2), with iPSC-derived neurons constitutively overexpressing endogenous α-synuclein (27). Increased levels of wild-type α-synuclein cause mitochondrial impairment and an increase in superoxide and other reactive oxygen species (28, 29), possibly because of interference with mitochondrial protein import (30). We tested whether clenbuterol may be helpful in normalizing SNCA expression levels in human iPSC derived neuronal cells of a patient carrying the SNCA triplication. SNCA-triplication iPSC-derived neuronal precursor cells were treated with clenbuterol (20 μM), and endogenous SNCA mRNA expression and α-synuclein protein levels were significantly reduced (P < 0.005 and 0.05, respectively; two-tailed Student’s t test) (Fig. 4F). Similarly, SNCA expression was reduced in SNCA-triplication iPSC-derived neurons cultured for 8 weeks and then treated with clenbuterol (20 μM) for 3 days (fig. S11).
Furthermore, PD patient–derived neuronal precursor cells carrying the pathogenic SNCA locus triplication show increased mitochondria associated superoxide production and reduced viability under exposure to the environmental mitochondrial complex I toxin rotenone (28). Clenbuterol treatment ameliorated this increased mitochondria-associated superoxide production (Fig. 4G) and increased viability (Fig. 4H), similarly to partial SNCA knockdown (28).
Discussion
We found effects of β2AR activation in two epidemiologic analyses, in mice modeling neurotoxin induced human parkinsonism, and in iPSC-derived neuronal cultures modeling SNCA dosage and rotenone toxicity. We propose a model in which β2AR antagonists increase SNCA expression through H3K27 acetylation, resulting in α-synuclein accumulation, mitochondrial oxidative stress, dopaminergic neurodegeneration, and increased risk of PD. In contrast, we expect β2AR agonists to promote dopamine neuron health by reducing SNCA expression (through H3K27 deacetylation) and mitochondrial free radicals. This may benefit nigral dopamine neurons, which are prone to mitochondrial bioenergetics dysfunction even at early stages of Lewy body neuropathology (31) and are preferentially vulnerable to mitochondrial complex I toxins (22). There is precedent for β2AR stimulation acting as a regulator of transcription (14). β2ARs are expressed in the substantia nigra and cortex (32), regions that are progressively affected in PD. The ligand-specific regulatory mechanism that we uncovered is consistent with the clinical association in Norway, where the selective β2AR agonist salbutamol (typically prescribed for asthma) was associated with a reduced risk of PD, whereas the β2AR antagonist propranolol (commonly used for hypertension) was associated with increased risk.
We demonstrate associations of β2AR with neuronal SNCA expression and risk of PD. It is important to note that association does not imply causation. β2AR agonists are not currently FDA-approved for PD treatment. Cardiovascular disease can be exacerbated by β2AR agonists. Evaluation in additional populations and in clinical trials will be required to determine whether the insights gained in this work can be translated to patients with PD. The described regulatory pathway and the impacts of various compounds present a new view of SNCA biology and offer clues for medicinal chemistry and drug repurposing. Our screen targeted neuronal SNCA; however, β2AR may have additional beneficial effects on glia and inflammation (12, 33). A complete chart of the pathway components linking β2AR to PD pathobiology can now be realized and might inspire more potent and PD-specific interventions.
Our study presents a path to drug development that is distinct from traditional approaches. Targeting the endogenous expression of a human disease gene may be a useful strategy for other diseases attributed to copy number variation or regulatory variants. The drug development pipeline tested in this study could be more generally applicable to rapid discovery and translation of therapeutics for other brain diseases.
Supplementary Material
Acknowledgments
We thank J. B. Concannon, K. Seyb, Paul J. Lorello, K. J. Shankaran, J. B. Sanderson, R. Passas, S. Aziz, and A. J. Scherzer (Brigham and Women’s Hospital); V. Mishra (Purdue University); and P. C. Marcogliese (University of Ottawa) for invaluable technical assistance. Funding was provided by the Michael J. Fox Foundation (to C.R.S.), the National Institute of Neurological Disorders and Stroke (grants U01 NS082157 and U01NS095736 to C.R.S. and grant R01 NS083845 to D.J.S.), the U.S. Department of Defense (to C.R.S.), the M.E.M.O. Hoffman Foundation (to C.R.S.), Prinses Beatrix Spier Fonds (to P.H.), the American Parkinson’s Disease Association (to T.B.), the Parkinson’s Disease Foundation (to T.B.), the Branfman Family Foundation (to J.C.R.), the Canadian Institute of Health Research (to D.S.P.), Brain Canada/Krembil Foundation (to D.S.P.), the Heart and Stroke Foundation of Canada (to D.S.P.), the Multiple System Atrophy Coalition (to V.K.), and Harvard NeuroDiscovery Center (to V.K.). B.W.H. has applied for a related U.S. patent. C.R.S. is named as inventor on patent application 62487541 submitted by Brigham and Women’s Hospital that relates to modifications and combinations of b-adrenoreceptor agonists as potential therapeutics for Parkinson’s disease. NorPD data are accessible by application at http://norpd.no.
Footnotes
References
- 1.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 2.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Annals of neurology. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 3.Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62:1835–1838. doi: 10.1212/01.wnl.0000127517.33208.f4. [DOI] [PubMed] [Google Scholar]
- 4.Pihlstrom L, Toft M. Genetic variability in SNCA and Parkinson's disease. Neurogenetics. 2011;12:283–293. doi: 10.1007/s10048-011-0292-7. [DOI] [PubMed] [Google Scholar]
- 5.Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10907–10912. doi: 10.1073/pnas.0802437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clough RL, Stefanis L. A novel pathway for transcriptional regulation of alpha synuclein. Faseb J. 2007;21:596–607. doi: 10.1096/fj.06-7111com. [DOI] [PubMed] [Google Scholar]
- 7.Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. The Journal of neuroscience. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carbone M, Duty S, Rattray M. Riluzole neuroprotection in a Parkinson’s disease model involves suppression of reactive astrocytosis but not GLT-1 regulation. BMC Neurosci. 2012;13:38. doi: 10.1186/1471-2202-13-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Supplementary Results and Methods are available online.
- 11.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314. doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gleeson LC, Ryan KJ, Griffin EW, Connor TJ, Harkin A. The beta2- adrenoceptor agonist clenbuterol elicits neuroprotective, anti-inflammatory and neurotrophic actions in the kainic acid model of excitotoxicity. Brain Behav Immun. 2010;24:1354–1361. doi: 10.1016/j.bbi.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 13.O’Donnell JM. Pharmacological characterization of the discriminative stimulus effects of clenbuterol in rats. Pharmacology, biochemistry, and behavior. 1997;58:813–818. doi: 10.1016/s0091-3057(97)00038-5. [DOI] [PubMed] [Google Scholar]
- 14.Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, Fujita T. Epigenetic modulation of the renal betaadrenergic-WNK4 pathway in salt-sensitive hypertension. Nature medicine. 2011;17:573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 15.Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate induced excitotoxicity. The Journal of neuroscience. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Norwegian Prescription Database. The Norwegian Institute of Public Health. www.norpd.no.
- 17.Alves G, Muller B, Herlofson K, HogenEsch I, Telstad W, Aarsland D, Tysnes OB, Larsen JP. Incidence of Parkinson’s disease in Norway: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry. 2009;80:851–857. doi: 10.1136/jnnp.2008.168211. [DOI] [PubMed] [Google Scholar]
- 18.Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA, Ross GW, Strickland D, Van Den Eeden SK, Gorell J. Pooled analysis of tobacco use and risk of Parkinson disease. Archives of neurology. 2007;64:990–997. doi: 10.1001/archneur.64.7.990. [DOI] [PubMed] [Google Scholar]
- 19.Subbarao P, Mandhane PJ, Sears MR. Asthma: epidemiology, etiology and risk factors. CMAJ. 2009;181:E181–190. doi: 10.1503/cmaj.080612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lund M. Social Inequality in Cigarette Consumption, Cigarette Dependence, and Intention to Quit among Norwegian Smokers. Biomed Res Int. 2015;2015:835080. doi: 10.1155/2015/835080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 22.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 23.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 24.Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston JW. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasegawa E, Kang D, Sakamoto K, Mitsumoto A, Nagano T, Minakami S, Takeshige K. A dual effect of 1-methyl-4-phenylpyridinium (MPP+)-analogs on the respiratory chain of bovine heart mitochondria. Arch Biochem Biophys. 1997;337:69–74. doi: 10.1006/abbi.1996.9726. [DOI] [PubMed] [Google Scholar]
- 26.Bezard E, Dovero S, Bioulac B, Gross C. Effects of different schedules of MPTP administration on dopaminergic neurodegeneration in mice. Exp Neurol. 1997;148:288–292. doi: 10.1006/exnr.1997.6648. [DOI] [PubMed] [Google Scholar]
- 27.Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, Baru V, Lou Y, Freyzon Y, Cho S, Mungenast AE, Muffat J, Mitalipova M, Pluth MD, Jui NT, Schule B, Lippard SJ, Tsai LH, Krainc D, Buchwald SL, Jaenisch R, Lindquist S. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science. 2013;342:983–987. doi: 10.1126/science.1245296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flierl A, Oliveira LM, Falomir-Lockhart LJ, Mak SK, Hesley J, Soldner F, Arndt-Jovin DJ, Jaenisch R, Langston JW, Jovin TM, Schule B. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS One. 2014;9:e112413. doi: 10.1371/journal.pone.0112413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J, Chu CT, Burton EA, Hastings TG, Greenamyre JT. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci Transl Med. 2016;8:342ra378. doi: 10.1126/scitranslmed.aaf3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grunblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wullner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, Young AB, Vance JM, Davis RL, Hedreen JC, Adler CH, Beach TG, Graeber MB, Middleton FA, Rochet JC, Scherzer CR. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, Wilson B, Lu RB, Hong JS, Flood PM. β2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol. 2011;186:4443–4454. doi: 10.4049/jimmunol.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.GTEx Consortium, Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S, Park DS. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27:3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi H, Lassmann T, Murata M, Carninci P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nature protocols. 2012;7:542–561. doi: 10.1038/nprot.2012.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.