

Graphical abstract
Keywords: PMCA, Calcium pump, Calcium overload, Necrosis, Apoptosis, ATP
Highlights
-
•
The PMCA is an ATP-driven Ca2+ pump critical for the maintenance of low cytosolic calcium.
-
•
The PMCA has an important but paradoxical role in cell death and survival.
-
•
The PMCA can be differentially regulated by caspase/calpain cleavage.
-
•
Glycolytic ATP supply may be sufficient to fuel the PMCA during metabolic stress.
-
•
The ATP sensitivity of the PMCA can be regulated by acidic phospholipids.
Abstract
The plasma membrane Ca2+-ATPase (PMCA) is a ubiquitously expressed, ATP-driven Ca2+ pump that is critical for maintaining low resting cytosolic Ca2+ ([Ca2+]i) in all eukaryotic cells. Since cytotoxic Ca2+ overload has such a central role in cell death, the PMCA represents an essential “linchpin” for the delicate balance between cell survival and cell death. In general, impaired PMCA activity and reduced PMCA expression leads to cytotoxic Ca2+ overload and Ca2+ dependent cell death, both apoptosis and necrosis, whereas maintenance of PMCA activity or PMCA overexpression is generally accepted as being cytoprotective. However, the PMCA has a paradoxical role in cell death depending on the cell type and cellular context. The PMCA can be differentially regulated by Ca2+-dependent proteolysis, can be maintained by a localised glycolytic ATP supply, even in the face of global ATP depletion, and can be profoundly affected by the specific phospholipid environment that it sits within the membrane. The major focus of this review is to highlight some of the controversies surrounding the paradoxical role of the PMCA in cell death and survival, challenging the conventional view of ATP-dependent regulation of the PMCA and how this might influence cell fate.
1. Introduction
The plasma membrane Ca2+-ATPase (PMCA) is an ATP-driven Ca2+ pump ubiquitously expressed in the plasma membrane of all eukaryotic cells. PMCA is encoded by four separate genes (PMCA1-4) and numerous splice variants that give rise to specific tissue distribution, cellular localisation and functional diversity [1], [2]. PMCA1 and PMCA4 are ubiquitously expressed whereas PMCA2 and PMCA3 have a more tissue specific expression, mainly in excitable cells [3]. The PMCA is critical for maintaining cytosolic Ca2+ concentration ([Ca2+]i) below 300 nM (∼100 nM), due to its high affinity for Ca2+ (Kd, ∼0.2 μM) [4], [5], [6] and is the major Ca2+ efflux pathway in non-excitable cells [7]. For many years the PMCA was thought to have a “minor” house-keeping role in maintaining low resting [Ca2+]i [8]. However, the importance of PMCA in the spatiotemporal shaping of cytosolic Ca2+ signalling has steadily increased. PMCA exhibits memory of past [Ca2+]i increases, suggesting an important role in regulating the frequency of Ca2+ oscillations [9]. Moreover, the different PMCA isoforms, and numerous splice variants of PMCA, can be differentially expressed in specific regions of cells and can also be differentially regulated by a sophisticated repertoire of additional signalling pathways [10], [11], [12], [13].
Despite the emerging role of the PMCA in dynamic Ca2+ signalling, the importance of the house-keeping role of the PMCA should not be under-estimated, especially when one considers how important maintaining low resting [Ca2+]i is for cell survival and the prevention of Ca2+-dependent cell death. In this regard the PMCA can be regarded as the “last gatekeeper” for the maintenance of low resting [Ca2+]i; an essential “linchpin” for the delicate balance between cell survival and cell death [14], [15], [16], [17]. Moreover, the PMCA is inextricably linked to the specific nature of cell death. Not only does PMCA prevent Ca2+ overload associated apoptosis, but the PMCA is an ATP-driven pump and since ATP depletion induces necrosis, a decline in PMCA activity will accompany and exacerbate necrosis [17], [18], [19], [20]. Therefore, PMCA activity may act as an important switch between apoptosis and necrotic cell death, a key determinant of numerous disease processes. Thus the maintenance of PMCA activity is critical for cell survival, particularly in the face of modest-to-severe global ATP depletion, whereas inhibition of PMCA even when global ATP is maintained will facilitate Ca2+-dependent apoptosis.
2. Physiological regulation and key structural features of the PMCA
In order to understand the role of the PMCA in cell death and survival it is necessary to highlight some of the key structural features and regulatory mechanisms. Structurally, PMCA consists of ten transmembrane domains, two cytosolic loops with both N- and C-terminal cytosolic tails (Fig. 1) [1], [2]. Arguably the most functionally important structural domain is the C-terminal tail which contains the autoinhibitory calmodulin (CaM)-binding motif [21]. At low resting [Ca2+]i, the autoinhibitory CaM-binding motif interacts with the catalytic site (first and second cytosolic loops) thereby inhibiting the PMCA (Fig. 1A). When [Ca2+]i is elevated, binding of Ca2+/CaM to this autoinhibitory motif induces a conformational change which reduces its affinity for the catalytic site thereby increasing the Ca2+ transporting activity of the PMCA (Fig. 1B) [5]. The C-terminus also contains additional high affinity allosteric Ca2+ binding sites [22] and an acidic phospholipid binding site [23], [24]. Binding of acidic phospholipids such as phosphatidylinositol (PI) and phosphatidylserine (PS) increases the Ca2+ and ATP affinity of the PMCA. Phosphoinositide 4,5-bisphosphate (PIP2) is also a major activator of PMCA and is thought to account for ∼50% of the activity of PMCA at rest [4], [5]. The last few amino acid residues of the C-terminus of the PMCA contain PDZ-binding motif, which facilitate PMCA dimerization [25] which increases PMCA activity [26]. In addition, the PDZ-binding motif also facilitates the recruitment of the actin cytoskeleton [27], numerous scaffolding proteins and signalling complexes [28], [29], [30], [31], [32], [33]. Such targeting only occurs for the full-length b-variants, suggesting specialised signalling roles for different PMCA isoforms. Specifically, PMCA4b functionally interacts with neuronal nitric oxide synthase (nNOS) [34], [35], calcineurin [36] and the pro-apoptotic tumour suppressor Ras-associated factor 1 (RASSF1) [37], thereby regulating their downstream signalling.
Fig. 1.

Two-dimensional topological model of the structure of the PMCA at low resting [Ca2+]i and following activation at elevated [Ca2+]i. A., at low resting [Ca2+]i the autoinhibitory CaM binding motif within the C-terminal tail of the PMCA associates with the catalytic motif, thereby preventing Ca2+ binding and thus Ca2+ transport. B., when [Ca2+]i is elevated, Ca2+/CaM binds to the autoinhibitory CaM binding motif inducing a conformational change that causes dissociation from the catalytic motif, thereby allowing access to Ca2+ and thus the transport of Ca2+. Additional regulatory motifs include an inhibitory 14-3-3-binding site in the N-terminal region, a stimulatory phospholipid-binding motif in the first cytosolic loop and PKA/PKC phosphorylation consensus sites and a PDZ binding motif in the C-terminal tail. Splice sites A and C can generate splice variants with specific tissue-specific distribution, cellular localization and differential regulation.
The N-terminal tail exhibits the greatest diversity between the different isoforms [38] and contains an inhibitory 14-3-3-binding motif [39], [40]. In addition to part of the binding site for the autoinhibitory calmodulin (CaM)-binding motif [41] (see Fig. 1B), the first cytosolic loop of the PMCA, which spans between the second and third transmembrane domains, contains a stimulatory acidic phospholipid-binding site [42], [43] and splice site A important for the apical membrane targeting in epithelial cells [44], [45], [46]. The second cytosolic loop between the fourth and fifth transmembrane domains contains the major catalytic site (including the critical aspartate residue that becomes phosphorylated during the reaction cycle and ATP binding) and the second part of the binding site for the autoinhibitory CaM-binding motif within the C-terminal tail [47].
3. The controversial role of PMCA in cell death
3.1. Intrinsic Ca2+-dependent cell death
Since the PMCA is critical for the regulation of low resting [Ca2+]i and the prevention of cytotoxic Ca2+ overload, in order to understand the role of PMCA in cell death and survival it is important to explore the mechanisms of Ca2+-dependent cell death. Ca2+ has a critical yet paradoxical role in regulating cell death [48] and both ER Ca2+ and mitochondrial Ca2+ are key contributors (Fig. 2). Cells adopt strategies to avoid cell death by activating pro-survival pathways and suppressing cell death machinery, of which the main players include the pro- and anti-apoptotic B-cell lymphoma 2 (Bcl-2) family of proteins of which there are around 20 different members [49]. At the mitochondria, tBid binds to and promotes Bax and Bak oligomerisation [50] which form pores through which cytochrome C can be released into the cytosol. Cytochrome C binds to and forms part of the apoptosome complex which activates the executioner caspases, such as caspase-3, leading to the “point-of-no-return” apoptotic cascade [48], [51]. The balance between pro-apoptotic (Bax, Bak and Bad) and anti-apoptotic proteins (Bcl-2/Bcl-xL) determines whether a cell is sensitive or resistant to apoptosis [52] (Fig. 2). The phosphorylation status of Bad is a critical checkpoint for apoptosis (Fig. 2). When phosphorylated by protein kinase-A (PKA), protein kinase-B (PKB) or Ras-mitogen activated kinase (MAPK) Bad dissociates from the mitochondria to the cytosol where it binds to 14-3-3 protein. This prevents Bad from engaging and inhibiting Bcl-2/Bcl-xL, thereby inhibiting apoptosis [53] (Fig. 2). Moreover, the Ca2+-dependent activation of calcineurin leads to the dephosphorylation of Bad, allowing it to bind to and inhibit Bcl-2/Bcl-xL and thus promote apoptosis [54]. Of relevance to the PMCA, 14-3-3 proteins directly bind to and inhibit the PMCA [39], thereby accentuating the Ca2+/calcineurin-mediated dephosphorylation of Bad and further potentiating this Ca2+-mediated apoptosis.
Fig. 2.

The mechanisms of intrinsic Ca2+-dependent cell death. Cytotoxic Ca2+ overload can mediate intrinsic cell death at both the mitochondria and ER. At the mitochondria, tBid binds to and promotes Bax and Bak oligomerisation [50] which form pores through which cytochrome C can be released into the cytosol. Cytochrome C binds to and forms part of the apoptosome complex which activates the executioner caspases, such as caspase-3, leading to the “point-of-no-return” apoptotic cascade [48], [51]. The anti-apoptotic proteins, Bcl-2 and Bcl-xL, can prevent the t-Bid/Bax/Bak interaction, thereby preventing apoptosis. The pro-apoptotic protein Bad binds to Bcl-2/Bcl-xL [53], [129], thereby preventing their interaction with the t-Bid/Bax/Bak complex and thus promoting apoptosis [130]. Phosphorylation of Bad by the protein kinases, PKB [131], PKA [132] and ERK [133], causes Bad to dissociate from the mitochondria and bind to 14-3-3 protein. Sustained Ca2+ overload can activate calcineurin which dephosphorylates Bad allowing it to sequester the anti-apoptotic proteins, Bcl-2/Bcl-xL [54]. Ca2+ uptake into the mitochondria, via the mitochondrial Ca2+ uniporter (MCU), and its associated proteins (MICU, MCUb, EMRE), can lead to the production of reactive oxygen species (ROS). Ca2+ and ROS can activate the permeability transition pore (mPTP), loss of the mitochondrial membrane potential (ΔYm), ATP depletion and necrosis. Ca2+ overload can also activate calpain and cleavage of the PMCA.
Ca2+ has a more prominent role during cell stress-induced cell death [48] (Fig. 3). Mitochondria take up Ca2+, via the mitochondrial Ca2+ uniporter (MCU) [55], [56], which was originally believed to have a low affinity for Ca2+ and thus facilitate Ca2+ uptake into the mitochondrial matrix only when cytosolic Ca2+ is very high. However, the molecular identity of MCU, and its various pore-forming subunits (MCUb and “essential MCU regulator” (EMRE)) and accessory proteins (MICU1 and MICU2), has revealed that this process is a complex molecular machine [57]. Depending on the relative expression of each of these molecular components and cellular context this process exhibits a more complex gating and Ca2+-dependency than originally thought [57]. Nevertheless, MCU can facilitate the physiological Ca2+ dependent activation of key metabolic enzymes [58], in a process referred to as stimulus-metabolism coupling. Alternatively during excessive cytosolic Ca2+ overload, which can occur when the PMCA is inhibited, MCU can lead to excessive mitochondrial Ca2+ overload and the production of reactive oxygen species (ROS) [59]. Both ROS and Ca2+ can activate the mitochondrial permeability transition pore (mPTP) [59], a molecular machine which consists a growing list of regulatory and accessory proteins, but with core members including the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT) and cyclophilin-D [60] (Fig. 3). The molecular composition and regulatory mechanism of the mPTP is hotly debated and a fast growing field and thus beyond the scope of this review. However, in simple terms the mPTP is responsible for coupling mitochondrial volume and ion homeostasis with metabolism and cellular stress. Excessive mPTP activation leads to mitochondrial swelling, outer mitochondrial membrane rupture and cytochrome C release [48]. For many years the mPTP was thought to be important for Ca2+-mediated apoptosis [61], [62], however knockout studies of VDAC [63] and cyclophilin-D [64], [65] had no effect on apoptosis but attenuated necrotic cell death. This suggests that the mPTP may be more important during necrotic cell death than for apoptosis. Nevertheless, it is clear that the mPTP can functionally interact with Bcl-2/Bcl-xL and Bax/Bak [61], [62], [66], [67], suggesting that necrosis and apoptosis may share some of the same molecular machinery. The defining feature of necrosis is that this is usually accompanied by the collapse of the mitochondrial membrane potential, due to excessive activation of the mPTP and the consequent inhibition of mitochondrial ATP synthesis. This can have a knock-on effect on the ATP-dependent PMCA, as well as SERCA and the Na+/K+-ATPase.
Fig. 3.

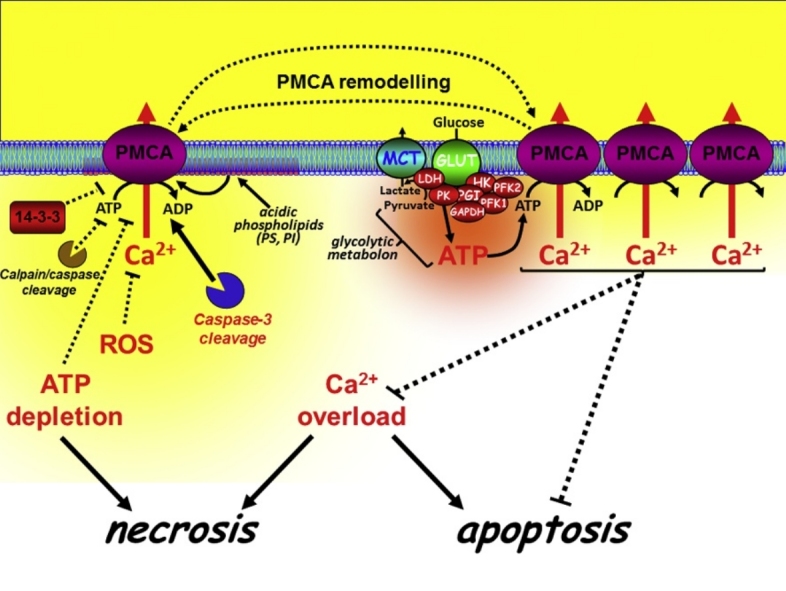
Differential regulation and paradoxical role of PMCA in cell death. The PMCA is a high affinity Ca2+ clearance pathway critical for maintaining low resting [Ca2+]i and thus prevention of cytotoxic Ca overload-induced cell death (both necrosis and apoptosis). Inhibition or reduced expression of the PMCA generally increases cell death, whereas activation or overexpression of the PMCA generally inhibits cell death. The PMCA can be directly inhibited by 14-3-3 binding, calpain/caspase cleavage, oxidation by reactive oxygen species (ROS) or by ATP depletion during metabolic stress. However, specific caspase-3 cleavage of the PMCA can remove the autoinihitory CaM-binding domain, resulting in a truncated constitutively active PMCA. Moreover, acidic phospholipids, such as phosphatidylserine (PS) and phosphatidylinositol (PI) can markedly increase the affinity of the PMCA for ATP and Ca2+/CaM, making the PMCA largely insensitive to moderate ATP depletion. Furthermore, glycolytic enzymes can associate with the plasma membrane to form a metabolic complex (metabolon) which provides the PMCA with a privileged ATP supply to the PMCA, even during global ATP depletion. These mechanisms may be important for maintaining survival during severe metabolic stress.
Ca2+ can also amplify cell death by activating the proteolytic enzymes, calpains [68], which can directly activate caspases [69] and can inactivate the anti-apoptotic Bcl-2 [70] and the PMCA [20], [71], [72], [73], [74] (Fig. 2). Paradoxically, some of the pro-apoptotic and anti-apoptotic proteins have been reported to directly regulate key Ca2+ transport proteins [75], [76], [77], including the PMCA [78] resulting in a complex reciprocal regulation between Ca2+ signalling and cell death.
Enhanced Ca2+ signalling and cytosolic Ca2+ overload clearly promotes both apoptosis and necrosis. Intuitively therefore, impaired PMCA activity or reduced PMCA expression would be expected to promote Ca2+-mediated cell death, whereas maintenance of PMCA activity or PMCA overexpression is generally regarded to be cytoprotective. Although such remodelling of PMCA function and expression may have evolved to attenuate Ca2+-mediated cell death, paradoxically, this may also result in apoptosis resistance which is a major hallmark of cancer.
3.2. The PMCA paradox
Although the physiological role of the PMCA has been debated for several years, the importance of the PMCA during cellular stress and under pathological conditions is undeniable. The nature of cell death (i.e. necrosis vs apoptosis) will largely depend on the extent of metabolic stress and cytosolic Ca2+ overload. However, ATP depletion during extensive metabolic stress has been suggested to be the determining factor whether a cell undergoes apoptosis or necrosis [79], [80], [81], regardless of whether this is accompanied by Ca2+ overload. This is largely because ATP is critical for many of the ATP-dependent apoptotic processes, but is not required for necrosis [81]. During pancreatitis, apoptosis is generally regarded as protective, as this involves the safe dismantling of the cell constituents [82], [83]. Necrosis, on the other hand, is an uncontrolled form of cell death characterised by cell lysis, and the subsequent release of activated proteases (zymogens) from the effected pancreatic acinar cells, which trigger the spiral of self-perpetuating tissue damage characteristic of acute pancreatitis [82], [83].
Interestingly, the anti-apoptotic factor Bcl-2/xL has been recently shown to inhibit PMCA activity in pancreatic acinar cells [78]. Although the pathophysiological relevance of this phenomenon remains to be determined, this observation highlights the critical importance of the PMCA in controlling cell fate, which may be very different depending on cell type or cellular context.
Paradoxically, the impairment of PMCA function and the subsequent dysregulation of cytosolic Ca2+ homeostasis can, in some cases, be cytoprotective [84]. During oxidative stress or tumour necrosis factor (TNF)-induced cell death, the accumulation and damage of lysosomes has been suggested to be important in determining cell fate [85], [86]. In TNF-resistant cell lines, in which PMCA4 is mutated, the resulting enhanced Ca2+ signalling has been shown to promote the exocytotic loss of lysosomes, resulting in protection against TNF-induced cell death [84]. This therefore suggests, somewhat counter-intuitively, that PMCA4 promotes TNF-induced cell death.
In MDA-MB-231 breast cancer cells the expression of different PMCA isoforms differentially regulate cell fate by protecting against either Ca2+-dependent necrosis (PMCA1) or caspase-dependent apoptosis (PMCA4) [19]. Specific siRNA-mediated knockdown of PMCA1 potentiated ionomycin-induced necrosis, whereas siRNA-mediated knockdown of PMCA4 potentiated Bcl-2 inhibitor (ABT-263)-mediated apoptosis and reduced NFkB nuclear translocation [19]. However, in colon cancer cells (HT-29), PMCA4 expression is downregulated, which appears to promote cell proliferation without any effect on the sensitivity of mitochondrial uncoupler (CCCP)-induced or TNFα-induced cell death pathways [87].
In breast epithelial cells, the PMCA has an important role in the normal physiology and in tumorigenesis. During lactation PMCA2 is up-regulated at the apical membrane where it contributes to apical Ca2+ transport and the majority of the Ca2+ composition within milk produced by normal mammary epithelial cells. During weaning, PMCA2 is down-regulated when the secretory cells die from apoptosis [88]. This loss of PMCA2 expression is thought to elevate resting Ca2+ thereby increasing the sensitivity of Ca2+-dependent apoptosis [89]. This is a form of “physiological” apoptosis known as mammary gland involution and remodelling of PMCA2 expression is central to this process. However, in breast cancer PMCA2 overexpression correlates with poor outcome due to resistance to apoptosis which is a major cancer hallmark [89].
Insulin-secreting β-cells (BRIN-BD11) also exhibit a functional paradox. Overexpression of PMCA reduces resting [Ca2+]i and mitochondrial Ca2+ overload, which intuitively one might expect would protect against Ca2+-dependent apoptosis. However, this also leads to ER Ca2+ depletion, leading to ER-stress (activation of inositol-requiring enzyme 1α/X-box binding protein 1 pathway (IRE1α-XBP1) and inhibition of Activating Transcription Factor 6 (ATF6)) and the consequent activation of caspase-mediated apoptosis [90]. Therefore, in this context PMCA overexpression may induce apoptosis.
4. Metabolism, ATP supply and ATP dependency of PMCA
To better understand the paradoxical role of the PMCA in cell death, it is necessary to explore the mechanisms by which the PMCA is regulated, the ATP dependency and the supply of ATP to the PMCA. Intuitively, one might predict that ATP depletion would inhibit PMCA leading to Ca2+ overload and necrotic cell death [17], [18], [91], [92]. However, this is likely to be an over simplification because there are numerous factors that can influence the ATP sensitivity of the PMCA.
The PMCA has a high affinity catalytic ATP binding site (Km ∼ 3 μM) and lower affinity regulatory binding site (Km ∼ 145 μM) [93]. However, more recent studies suggest a more complex ATP dependency [94]. Most normal healthy cells have a resting ATP concentration in the mM range, suggesting that ATP has to drop 100–1000 fold before the PMCA is inhibited, which might only occur under severe prolonged metabolic stress?
Studies in HeLa cells showed that high, necrosis-inducing, ATP-depleting concentrations of H2O2 (32 mM) caused a Ca2+ overload response that was due to inhibition of the PMCA but that was also dependent on functional Na+-K+-ATPase activity [18]. The authors concluded that this was due to a thermodynamic shift of free energies of the pumps in favour of the Na+-K+-ATPase [18]. It was argued that he H2O2 also induced a Na+ influx that reduced the free energy for the Na+ pump, due to the dissipation of the Na+ gradient, without any appreciable effect on the free energy for the PMCA (Ca2+ gradient maintained). Therefore, ATP reaches a critical concentration where it becomes more energetically favourable to pump Na+ than Ca2+ and so the PMCA is inhibited in favour of a fully functional Na+-K+-ATPase. In other words the Na+-pump “steals” ATP from the PMCA [18]. This study suggests that the PMCA may be inhibited by sub-maximal ATP depletion
The extent of ATP depletion will largely depend on whether mitochondria or glycolysis is inhibited, whether ATP is being rapidly consumed and the extent of PMCA inhibition will depend on whether there is a localised ATP supply. Moreover, would inhibition of mitochondrial metabolism alone deplete ATP levels sufficiently to inhibit the PMCA, especially if glycolytic ATP production remains active? The classic textbook view is that mitochondria are much more energetically efficient than glycolysis in metabolising glucose. For example, mitochondrial oxidative phosphorylation produces 32 molecules of ATP, whereas glycolysis produces only 2 molecules of ATP for every glucose molecule metabolised. However, this is likely to be an over simplification as most cells exhibit metabolic plasticity and are able to adapt to their environment (i.e. as observed in hypoxic tissues). Cancer cells are an extreme example of this and often undergo a metabolic switch from mitochondrial metabolism to glycolysis, due to mutations within key mitochondrial enzymes and the up-regulation of glycolytic enzymes [95], [96]. This phenomenon is known as the Warburg effect, named after Otto Warburg, who first described the increase in glycolysis in cancer cells in the 1920s [97]. Our recent studies in pancreatic cancer cell lines were the first to show that the PMCA is critically reliant on glycolytically-derived ATP [14], [15]. Specifically, inhibition of glycolysis, but not mitochondrial metabolism, resulted in ATP depletion, PMCA inhibition, cytotoxic Ca2+ overload and ultimately necrotic cell death [98]. This was reversed when cells were cultured in glucose-free media supplemented with either galactose or ketoisocaproate to reduce cells’ reliance on upregulated glycolytic rate [15]. This suggests that in highly glycolytic pancreatic cancer cells exhibiting the Warburg effect, glycolytically-derived ATP, rather than mitochondrially-derived ATP is more important for maintaining PMCA function and cell survival.
These observations are particularly important when one considers that the PMCA is reported to have its own local, sub-membrane supply of glycolytically-derived ATP that may render it largely insensitive to inhibition of mitochondrial metabolism [99], [100], [101] (Fig. 3). Specifically, in isolated inside-out plasma membrane vesicles from pig stomach smooth muscle enriched with PMCA, an endogenous membrane-bound glycolytic system was observed which provided ATP to fuel PMCA-dependent Ca2+ uptake [99], [100]. Moreover, providing glycolytic substrates were present, the Ca2+ uptake (PMCA activity) persisted in the absence of an exogenously applied ATP regenerating system [99], [100]. In addition, studies in human erythrocytes which lack mitochondria have shown that several glycolytic enzymes associate with the plasma membrane, either via band 3 protein (anion-exchanger-1) [102], [103], [104], [105] or via phospholipids [106]. Moreover, the PMCA has been shown to reside within caveolae, where these phospholipids are enriched and regulate the activity of the PMCA [107]. Finally, it has been suggested that a localised pool of ATP, associated with the cytoskeleton, provides a privileged ATP supply to the PMCA [108]. Together these data implicate the possibility that a localised glycolytic ATP supply to PMCA may independently regulate PMCA activity regardless of whether global cellular ATP is maintained.
In most healthy cells under physiological conditions, when the bulk cytosolic ATP concentration is saturating for the PMCA (i.e.> 1 mM), such close functional coupling between glycolytic enzymes and the PMCA is likely to be of minimal functional significance. In other words, the PMCA does not care whether ATP comes from a mitochondrial or glycolytic source. However, in the face of impaired mitochondrial metabolism, for example under conditions of cellular stress, a glycolytic source of ATP might be critical for maintaining PMCA activity and thus restoring low resting cytosolic [Ca2+]i. Indeed, acute metabolic stress induced by pancreatitis-inducing agents markedly inhibited PMCA in pancreatic acinar cells [16], [17], [109], [110], which was attenuated by treatment with insulin [16], [17]. This insulin protection was due to an acute “cancer-like” switch from mitochondrial metabolism to glycolysis which was sufficient to preserve the ATP supply to the PMCA, thereby preventing cytotoxic Ca2+ overload and necrotic cell death [16], [17]. Under these stressed conditions, a “privileged” local glycolytic ATP supply (or more specifically an insulin-mediated “up-regulated” glycolytic supply of ATP) may be sufficient to “fuel” the PMCA, even if bulk (global) ATP is close to zero. Conversely, it is also possible that inhibition of such a localised ATP supply to the PMCA might inhibit the PMCA even when global ATP is maintained, which might be sufficient to activate Ca2+-dependent apoptosis but not necrosis.
4.1. Regulation of the PMCA by acidic phospholipids
Another important caveat when considering the ATP-sensitivity of the PMCA is that acidic phosphoplipids, such as phosphatidylinositol (PI), phosphatidylcholine (PC) and phosphatidylserine (PS) regulate the ATP sensitivity of the PMCA and mimic regulation by CaM [23], [24]. Loss of PS (or PI) from the lipid environment of the PMCA lowered the affinity of the PMCA for ATP (Kd, 5–10 mM, regulatory site) [111], [112], suggesting that disruption of the lipid environment around the PMCA may be sufficient to render the PMCA highly sensitive to ATP depletion. However, this evidence is based on in vitro cell-free assays of ATPase activity, whereby PS/PI was either absent or present in an artificial membrane, making it difficult to extrapolate these findings to intact cells. It is therefore unclear what the critical concentration of PS is to maintain “normal” ATP-sensitivity of the PMCA or whether this relationship is influenced by dynamic changes in Ca2+, Mg2+, CaM or other membrane lipids. However, functional studies in intact endothelial cells have shown that the loss of phosphatidylserine from the inner leaflet of the plasma membrane, following cholesterol depletion with β-methyl-cyclodextrin, inhibited PMCA activity [107]. This has important implications for apoptosis, since PS is known to line the inner leaflet of the plasma membrane and a proportion is thought to flip to the extracellular side of the membrane during apoptosis [113]. This provides the dying cell with an “eat me” signal detected by macrophages that then phagocytose the dying cell from the tissue [113]. Furthermore, the enzyme responsible for this PS assymetry within the plasma membrane (aminophospholipid translocase or flippase) [114] requires millimolar ATP [115], [116] and is inhibited by oxidative stress [117], [118]. Collectively these studies suggest that cellular stress may have a profound effect on the ATP sensitivity of the PMCA, and thus inhibition of the PMCA might be observed even with only mild ATP depletion. Moreover, this altered ATP sensitivity of the PMCA during apoptosis might provide a feedforward potentiation of Ca2+-dependent apoptosis before ATP declines sufficiently to trigger necrosis.
4.2. Effect of mitochondrial-derived reactive oxygen species
Severe mitochondrial stress, whatever the mechanism, often leads to the generation of reactive oxygen species (ROS) [119]. Furthermore, there is also good evidence that oxidants (H2O2) can directly oxidise PMCA and also oxidise calmodulin, which is the main activator of PMCA [120]. Hence, metabolically derived ROS may have a profound inhibitory effect on PMCA activity. In addition, H2O2 has been reported to reduce the functional expression of PMCA at the plasma membrane of cultured hippocampal neurons within 1–2 h [121]. Such rapid changes in functional expression of PMCA at the plasma membrane could lead to reduced Ca2+ efflux during metabolic stress even in the presence of continued high ATP levels.
4.3. Calpain/caspase cleavage of the PMCA
The release of cytochrome C from the mitochondria and the subsequent activation of caspases and calpain [122] have both been reported to cleave and eventually lead to the inactivation of the PMCA [20], [123], [124]. It is interesting to note that the time-frame over which cytochrome C release can occur (>2 mins) [125] coincides with the time the PMCA can be observed to be inhibited, and happens well before ATP depletion was observed [109].In fact the initial cleavage by caspase and calpain actually activates the pump, but through the subsequent internalisation and degradation of the PMCA the protease effect is manifested as inhibition of the pump [126], [127]. Interestingly, specific caspase-3 cleavage of PMCA4b produces a 120 kD fragment that is constitutively activated, due to the removal of the autoinhibitory domain [72], [73], [74]. It is also interesting to note that calpain can also be directly activated by H2O2 and Ca2+ [128].
5. Summary
In summary the PMCA represents an important regulator of cell death, both apoptosis and necrosis. Although impaired PMCA activity is generally regarded as being cytotoxic and the maintenance of PMCA activity is regarded as being cytoprotective, this rather simplistic view is by no means clear-cut. The PMCA can be differentially regulated by a vast repertoire of additional signalling pathways, proteolytic cleavage and the ATP-dependency and ATP supply is even more complex than originally thought. This raises the question as to whether the PMCA deserves greater recognition in the Pantheon of calcium signalling machinery than it currently has. Moreover, the regulation of the PMCA may offer a relatively untapped and rich tapestry of novel therapeutic targets for a variety of diseases in which Ca2+-dependent cell death is central.
Footnotes
Supported by the Medical Research Council (MR/P00251X/1) and the Pancreatic Cancer Research Fund (PCRF).
References
- 1.Monteith G.R., Wanigasekara Y., Roufogalis B.D. The plasma membrane calcium pump, its role and regulation: new complexities and possibilities. J. Pharmacol. Toxicol. Methods. 1998;40:183–190. doi: 10.1016/s1056-8719(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 2.Strehler E.E., Zacharias D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 3.Greeb J., Shull G.E. Molecular cloning of a third isoform of the calmodulin-sensitive plasma membrane Ca2+-transporting ATPase that is expressed predominantly in brain and skeletal muscle. J. Biol. Chem. 1989;264:18569–18576. [PubMed] [Google Scholar]
- 4.Carafoli E. Calcium pump of the plasma membrane. Physiol. Rev. 1991;71:129–153. doi: 10.1152/physrev.1991.71.1.129. [DOI] [PubMed] [Google Scholar]
- 5.Carafoli E. Biogenesis: plasma membrane calcium ATPase: 15 years of work on the purified enzyme. FASEB J. 1994;8:993–1002. [PubMed] [Google Scholar]
- 6.Szasz I., Sarkadi B., Schubert A., Gardos G. Effects of lanthanum on calcium-dependent phenomena in human red cells. Biochim. Biophys. Acta. 1978;512:331–340. doi: 10.1016/0005-2736(78)90257-2. [DOI] [PubMed] [Google Scholar]
- 7.Muallem S., Beeker T., Pandol S.J. Role of Na+/Ca2+ exchange and the plasma membrane Ca2+ pump in hormone-mediated Ca2+ efflux from pancreatic acini. J. Membr. Biol. 1988;102:153–162. doi: 10.1007/BF01870453. [DOI] [PubMed] [Google Scholar]
- 8.Lopreiato R., Giacomello M., Carafoli E. The plasma membrane calcium pump: new ways to look at an old enzyme. J. Biol. Chem. 2014;289:10261–10268. doi: 10.1074/jbc.O114.555565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caride A.J., Penheiter A.R., Filoteo A.G., Bajzer Z., Enyedi A., Penniston J.T. The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J. Biol. Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa K., Tawada Y., Shigekawa M. Regulation of the plasma membrane Ca2+ pump by cyclic nucleotides in cultured vascular smooth muscle cells. J. Biol. Chem. 1988;263:8058–8065. [PubMed] [Google Scholar]
- 11.Neyses L., Reinlib L., Carafoli E. Phosphorylation of the Ca2+-pumping ATPase of heart sarcolemma and erythrocyte plasma membrane by the cAMP-dependent protein kinase. J. Biol. Chem. 1985;260:10283–10287. [PubMed] [Google Scholar]
- 12.Smallwood J.I., Gugi B., Rasmussen H. Regulation of erythrocyte Ca2+ pump activity by protein kinase C. J. Biol. Chem. 1988;263:2195–2202. [PubMed] [Google Scholar]
- 13.Zylinska L., Guerini D., Gromadzinska E., Lachowicz L. Protein kinases A and C phosphorylate purified Ca2+-ATPase from rat cortex, cerebellum and hippocampus. Biochim. Biophys. Acta. 1998;1448:99–108. doi: 10.1016/s0167-4889(98)00128-1. [DOI] [PubMed] [Google Scholar]
- 14.James A.D., Chan A., Erice O., Siriwardena A.K., Bruce J.I. Glycolytic ATP fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 2013;288:36007–36019. doi: 10.1074/jbc.M113.502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James A.D., Patel W., Butt Z., Adiamah M., Dakhel R., Latif A., Uggenti C., Swanton E., Imamura H., Siriwardena A.K., Bruce J.I. The plasma membrane calcium pump in pancreatic cancer cells exhibiting the warburg effect relies on glycolytic ATP. J. Biol. Chem. 2015;290:24760–24771. doi: 10.1074/jbc.M115.668707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mankad P., James A., Siriwardena A.K., Elliott A.C., Bruce J.I. Insulin protects pancreatic acinar cells from cytosolic calcium overload and inhibition of the plasma membrane calcium pump. J. Biol. Chem. 2012;287:1823–1836. doi: 10.1074/jbc.M111.326272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samad A., James A., Wong J., Mankad P., Whitehouse J., Patel W., Alves-Simoes M., Siriwardena A.K., Bruce J.I. Insulin protects pancreatic acinar cells from palmitoleic acid-induced cellular injury. J. Biol. Chem. 2014;289:23582–23595. doi: 10.1074/jbc.M114.589440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castro J., Ruminot I., Porras O.H., Flores C.M., Hermosilla T., Verdugo E., Venegas F., Hartel S., Michea L., Barros L.F. ATP steal between cation pumps: a mechanism linking Na+ influx to the onset of necrotic Ca2+ overload. Cell Death Differ. 2006;13:1675–1685. doi: 10.1038/sj.cdd.4401852. [DOI] [PubMed] [Google Scholar]
- 19.Curry M.C., Luk N.A., Kenny P.A., Roberts-Thomson S.J., Monteith G.R. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2012;287:28598–28608. doi: 10.1074/jbc.M112.364737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwab B.L., Guerini D., Didszun C., Bano D., Ferrando-May E., Fava E., Tam J., Xu D., Xanthoudakis S., Nicholson D.W., Carafoli E., Nicotera P. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell Death Differ. 2002;9:818–831. doi: 10.1038/sj.cdd.4401042. [DOI] [PubMed] [Google Scholar]
- 21.James P., Maeda M., Fischer R., Verma A.K., Krebs J., Penniston J.T., Carafoli E. Identification and primary structure of a calmodulin binding domain of the Ca2+ pump of human erythrocytes. J. Biol. Chem. 1988;263:2905–2910. [PubMed] [Google Scholar]
- 22.Hofmann F., James P., Vorherr T., Carafoli E. The C-terminal domain of the plasma membrane Ca2+ pump contains three high affinity Ca2+ binding sites. J. Biol. Chem. 1993;268:10252–10259. [PubMed] [Google Scholar]
- 23.Niggli V., Adunyah E.S., Carafoli E. Acidic phospholipids, unsaturated fatty acids, and limited proteolysis mimic the effect of calmodulin on the purified erythrocyte Ca2+ − ATPase. J. Biol. Chem. 1981;256:8588–8592. [PubMed] [Google Scholar]
- 24.Niggli V., Adunyah E.S., Penniston J.T., Carafoli E. Carafoli, Purified (Ca2+-Mg2+)-ATPase of the erythrocyte membrane. Reconstitution and effect of calmodulin and phospholipids. J. Biol. Chem. 1981;256:395–401. [PubMed] [Google Scholar]
- 25.Vorherr T., Kessler T., Hofmann F., Carafoli E. The calmodulin-binding domain mediates the self-association of the plasma membrane Ca2+ pump. J. Biol. Chem. 1991;266:22–27. [PubMed] [Google Scholar]
- 26.Kosk-Kosicka D., Bzdega T. Activation of the erythrocyte Ca2+-ATPase by either self-association or interaction with calmodulin. J. Biol. Chem. 1988;263:18184–18189. [PubMed] [Google Scholar]
- 27.Zabe M., Dean W.L. Plasma membrane Ca(2+)-ATPase associates with the cytoskeleton in activated platelets through a PDZ-binding domain. J. Biol. Chem. 2001;276:14704–14709. doi: 10.1074/jbc.M009850200. [DOI] [PubMed] [Google Scholar]
- 28.DeMarco S.J., Chicka M.C., Strehler E.E. Plasma membrane Ca2+ ATPase isoform 2b interacts preferentially with Na+/H+ exchanger regulatory factor 2 in apical plasma membranes. J. Biol. Chem. 2002;277:10506–10511. doi: 10.1074/jbc.M111616200. [DOI] [PubMed] [Google Scholar]
- 29.DeMarco S.J., Strehler E.E. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J. Biol. Chem. 2001;276:21594–21600. doi: 10.1074/jbc.M101448200. [DOI] [PubMed] [Google Scholar]
- 30.Goellner G.M., DeMarco S.J., Strehler E.E. Characterization of PISP, a novel single-PDZ protein that binds to all plasma membrane Ca2+-ATPase b-splice variants. Ann. N. Y. Acad. Sci. 2003;986:461–471. doi: 10.1111/j.1749-6632.2003.tb07230.x. [DOI] [PubMed] [Google Scholar]
- 31.Kim E., DeMarco S.J., Marfatia S.M., Chishti A.H., Sheng M., Strehler E.E. Plasma membrane Ca2+ ATPase isoform 4b binds to membrane-associated guanylate kinase (MAGUK) proteins via their PDZ (PSD-95/Dlg/ZO-1) domains. J. Biol. Chem. 1998;273:1591–1595. doi: 10.1074/jbc.273.3.1591. [DOI] [PubMed] [Google Scholar]
- 32.Schuh K., Uldrijan S., Gambaryan S., Roethlein N., Neyses L. Interaction of the plasma membrane Ca2+ pump 4b/CI with the Ca2+/calmodulin-dependent membrane-associated kinase CASK. J. Biol. Chem. 2003;278:9778–9783. doi: 10.1074/jbc.M212507200. [DOI] [PubMed] [Google Scholar]
- 33.Sgambato-Faure V., Xiong Y., Berke J.D., Hyman S.E., Strehler E.E. The Homer-1 protein Ania-3 interacts with the plasma membrane calcium pump. Biochem. Biophys. Res. Commun. 2006;343:630–637. doi: 10.1016/j.bbrc.2006.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oceandy D., Cartwright E.J., Emerson M., Prehar S., Baudoin F.M., Zi M., Alatwi N., Venetucci L., Schuh K., Williams J.C., Armesilla A.L., Neyses L. Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation. 2007;115:483–492. doi: 10.1161/CIRCULATIONAHA.106.643791. [DOI] [PubMed] [Google Scholar]
- 35.Schuh K., Uldrijan S., Telkamp M., Rothlein N., Neyses L. The plasmamembrane calmodulin-dependent calcium pump: a major regulator of nitric oxide synthase I. J. Cell Biol. 2001;155:201–205. doi: 10.1083/jcb.200104131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buch M.H., Pickard A., Rodriguez A., Gillies S., Maass A.H., Emerson M., Cartwright E.J., Williams J.C., Oceandy D., Redondo J.M., Neyses L., Armesilla A.L. The sarcolemmal calcium pump inhibits the calcineurin/nuclear factor of activated T-cell pathway via interaction with the calcineurin A catalytic subunit. J. Biol. Chem. 2005;280:29479–29487. doi: 10.1074/jbc.M501326200. [DOI] [PubMed] [Google Scholar]
- 37.Armesilla A.L., Williams J.C., Buch M.H., Pickard A., Emerson M., Cartwright E.J., Oceandy D., Vos M.D., Gillies S., Clark G.J., Neyses L. Novel functional interaction between the plasma membrane Ca2+ pump 4b and the proapoptotic tumor suppressor Ras-associated factor 1 (RASSF1) J. Biol. Chem. 2004;279:31318–31328. doi: 10.1074/jbc.M307557200. [DOI] [PubMed] [Google Scholar]
- 38.Stauffer T.P., Guerini D., Carafoli E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J. Biol. Chem. 1995;270:12184–12190. doi: 10.1074/jbc.270.20.12184. [DOI] [PubMed] [Google Scholar]
- 39.Linde C.I., Di Leva F., Domi T., Tosatto S.C., Brini M., Carafoli E. Inhibitory interaction of the 14-3-3 proteins with ubiquitous (PMCA1) and tissue-specific (PMCA3) isoforms of the plasma membrane Ca(2+) pump. Cell Calcium. 2008;43(6):550–561. doi: 10.1016/j.ceca.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Rimessi A., Coletto L., Pinton P., Rizzuto R., Brini M., Carafoli E. Inhibitory interaction of the 14-3-3{epsilon} protein with isoform 4 of the plasma membrane Ca(2+)-ATPase pump. J. Biol. Chem. 2005;280:37195–37203. doi: 10.1074/jbc.M504921200. [DOI] [PubMed] [Google Scholar]
- 41.Falchetto R., Vorherr T., Carafoli E. The calmodulin-binding site of the plasma membrane Ca2+ pump interacts with the transduction domain of the enzyme. Protein Sci. 1992;1:1613–1621. doi: 10.1002/pro.5560011209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brodin P., Falchetto R., Vorherr T., Carafoli E. Identification of two domains which mediate the binding of activating phospholipids to the plasma-membrane Ca2+ pump. Eur. J. Biochem. 1992;204:939–946. doi: 10.1111/j.1432-1033.1992.tb16715.x. [DOI] [PubMed] [Google Scholar]
- 43.Zvaritch E., James P., Vorherr T., Falchetto R., Modyanov N., Carafoli E. Mapping of functional domains in the plasma membrane Ca2+ pump using trypsin proteolysis. Biochemistry. 1990;29:8070–8076. doi: 10.1021/bi00487a012. [DOI] [PubMed] [Google Scholar]
- 44.Chicka M.C., Strehler E.E. Alternative splicing of the first intracellular loop of plasma membrane Ca2+-ATPase isoform 2 alters its membrane targeting. J. Biol. Chem. 2003;278:18464–18470. doi: 10.1074/jbc.M301482200. [DOI] [PubMed] [Google Scholar]
- 45.Grati M., Aggarwal N., Strehler E.E., Wenthold R.J. Molecular determinants for differential membrane trafficking of PMCA1 and PMCA2 in mammalian hair cells. J. Cell Sci. 2006;119:2995–3007. doi: 10.1242/jcs.03030. [DOI] [PubMed] [Google Scholar]
- 46.Hill J.K., Williams D.E., LeMasurier M., Dumont R.A., Strehler E.E., Gillespie P.G. Splice-site A choice targets plasma-membrane Ca2+-ATPase isoform 2 to hair bundles. J. Neurosci. 2006;26:6172–6180. doi: 10.1523/JNEUROSCI.0447-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falchetto R., Vorherr T., Brunner J., Carafoli E. The plasma membrane Ca2+ pump contains a site that interacts with its calmodulin-binding domain. J. Biol. Chem. 1991;266:2930–2936. [PubMed] [Google Scholar]
- 48.Orrenius S., Zhivotovsky B., Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 49.Hardwick J.M., Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harbor Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a008722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eskes R., Desagher S., Antonsson B., Martinou J.C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cory S., Adams J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 52.Datta S.R., Ranger A.M., Lin M.Z., Sturgill J.F., Ma Y.C., Cowan C.W., Dikkes P., Korsmeyer S.J., Greenberg M.E. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell. 2002;3:631–643. doi: 10.1016/s1534-5807(02)00326-x. [DOI] [PubMed] [Google Scholar]
- 53.Zha J., Harada H., Yang E., Jockel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 54.Wang H.G., Pathan N., Ethell I.M., Krajewski S., Yamaguchi Y., Shibasaki F., McKeon F., Bobo T., Franke T.F., Reed J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 55.Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L., Koteliansky V., Mootha V.K. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Stefani D., Raffaello A., Teardo E., Szabo I., Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Stefani D., Rizzuto R., Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu. Rev. Biochem. 2016;85:161–192. doi: 10.1146/annurev-biochem-060614-034216. [DOI] [PubMed] [Google Scholar]
- 58.McCormack J.G., Denton R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993;21(3):793–799. doi: 10.1042/bst0210793. [DOI] [PubMed] [Google Scholar]
- 59.Ott M., Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 60.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341(2):233–249. [PMC free article] [PubMed] [Google Scholar]
- 61.Shimizu S., Ide T., Yanagida T., Tsujimoto Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000;275:12321–12325. doi: 10.1074/jbc.275.16.12321. [DOI] [PubMed] [Google Scholar]
- 62.Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 63.Baines C.P., Kaiser R.A., Sheiko T., Craigen W.J., Molkentin J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baines C.P., Kaiser R.A., Purcell N.H., Blair N.S., Osinska H., Hambleton M.A., Brunskill E.W., Sayen M.R., Gottlieb R.A., Dorn G.W., Robbins J., Molkentin J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 65.Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 66.Gogvadze V., Robertson J.D., Zhivotovsky B., Orrenius S. Cytochrome c release occurs via Ca2+-dependent and Ca2+-independent mechanisms that are regulated by Bax. J. Biol. Chem. 2001;276:19066–19071. doi: 10.1074/jbc.M100614200. [DOI] [PubMed] [Google Scholar]
- 67.Marzo I., Brenner C., Zamzami N., Jurgensmeier J.M., Susin S.A., Vieira H.L., Prevost M.C., Xie Z., Matsuyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 68.Storr S.J., Carragher N.O., Frame M.C., Parr T., Martin S.G. The calpain system and cancer. Nat. Rev. Cancer. 2011;11:364–374. doi: 10.1038/nrc3050. [DOI] [PubMed] [Google Scholar]
- 69.Nakagawa T., Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gil-Parrado S., Fernandez-Montalvan A., Assfalg-Machleidt I., Popp O., Bestvater F., Holloschi A., Knoch T.A., Auerswald E.A., Welsh K., Reed J.C., Fritz H., Fuentes-Prior P., Spiess E., Salvesen G.S., Machleidt W. Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J. Biol. Chem. 2002;277:27217–27226. doi: 10.1074/jbc.M202945200. [DOI] [PubMed] [Google Scholar]
- 71.Bano D., Young K.W., Guerin C.J., Lefeuvre R., Rothwell N.J., Naldini L., Rizzuto R., Carafoli E., Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 72.Paszty K., Antalffy G., Hegedus L., Padanyi R., Penheiter A.R., Filoteo A.G., Penniston J.T., Enyedi A. Cleavage of the plasma membrane Ca + ATPase during apoptosis. Ann. N. Y. Acad. Sci. 2007;1099:440–450. doi: 10.1196/annals.1387.003. [DOI] [PubMed] [Google Scholar]
- 73.Paszty K., Antalffy G., Penheiter A.R., Homolya L., Padanyi R., Ilias A., Filoteo A.G., Penniston J.T., Enyedi A. The caspase-3 cleavage product of the plasma membrane Ca2+-ATPase 4b is activated and appropriately targeted. Biochem. J. 2005;391:687–692. doi: 10.1042/BJ20051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paszty K., Verma A.K., Padanyi R., Filoteo A.G., Penniston J.T., Enyedi A. Plasma membrane Ca2 + ATPase isoform 4b is cleaved and activated by caspase-3 during the early phase of apoptosis. J. Biol. Chem. 2002;277:6822–6829. doi: 10.1074/jbc.M109548200. [DOI] [PubMed] [Google Scholar]
- 75.Chen R., Valencia I., Zhong F., McColl K.S., Roderick H.L., Bootman M.D., Berridge M.J., Conway S.J., Holmes A.B., Mignery G.A., Velez P., Distelhorst C.W. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Foyouzi-Youssefi R., Arnaudeau S., Borner C., Kelley W.L., Tschopp J., Lew D.P., Demaurex N., Krause K.H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuo T.H., Kim H.R., Zhu L., Yu Y., Lin H.M., Tsang W. Modulation of endoplasmic reticulum calcium pump by Bcl-2. Oncogene. 1998;17:1903–1910. doi: 10.1038/sj.onc.1202110. [DOI] [PubMed] [Google Scholar]
- 78.Ferdek P.E., Gerasimenko J.V., Peng S., Tepikin A.V., Petersen O.H., Gerasimenko O.V. A novel role for Bcl-2 in regulation of cellular calcium extrusion. Curr. Biol. 2012;22:1241–1246. doi: 10.1016/j.cub.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Criddle D.N., Gerasimenko J.V., Baumgartner H.K., Jaffar M., Voronina S., Sutton R., Petersen O.H., Gerasimenko O.V. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death Differ. 2007;14:1285–1294. doi: 10.1038/sj.cdd.4402150. [DOI] [PubMed] [Google Scholar]
- 80.Kim J.S., Qian T., Lemasters J.J. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124:494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 81.Leist M., Single B., Castoldi A.F., Kuhnle S., Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhatia M. Apoptosis versus necrosis in acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G189–196. doi: 10.1152/ajpgi.00304.2003. [DOI] [PubMed] [Google Scholar]
- 83.Gukovskaya A.S., Mareninova O.A., Odinokova I.V., Sung K.F., Lugea A., Fischer L., Wang Y.L., Gukovsky I., Pandol S.J. Cell death in pancreatitis: effects of alcohol. J. Gastroenterol. Hepatol. 2006;21(Suppl. 3):S10–13. doi: 10.1111/j.1440-1746.2006.04571.x. [DOI] [PubMed] [Google Scholar]
- 84.Ono K., Wang X., Han J. Resistance to tumor necrosis factor-induced cell death mediated by PMCA4 deficiency. Mol. Cell. Biol. 2001;21:8276–8288. doi: 10.1128/MCB.21.24.8276-8288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brunk U.T., Svensson I. Oxidative stress, growth factor starvation and Fas activation may all cause apoptosis through lysosomal leak. Redox Rep. 1999;4:3–11. doi: 10.1179/135100099101534675. [DOI] [PubMed] [Google Scholar]
- 86.Ollinger K., Brunk U.T. Cellular injury induced by oxidative stress is mediated through lysosomal damage. Free Radic. Biol. Med. 1995;19:565–574. doi: 10.1016/0891-5849(95)00062-3. [DOI] [PubMed] [Google Scholar]
- 87.Aung C.S., Ye W., Plowman G., Peters A.A., Monteith G.R., Roberts-Thomson S.J. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis. 2009;30:1962–1969. doi: 10.1093/carcin/bgp223. [DOI] [PubMed] [Google Scholar]
- 88.Watson C.J. Involution: apoptosis and tissue remodelling that convert the mammary gland from milk factory to a quiescent organ. Breast Cancer Res. 2006;8:203. doi: 10.1186/bcr1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.VanHouten J., Sullivan C., Bazinet C., Ryoo T., Camp R., Rimm D.L., Chung G., Wysolmerski J. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 2010;107:11405–11410. doi: 10.1073/pnas.0911186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang L., Allagnat F., Nguidjoe E., Kamagate A., Pachera N., Vanderwinden J.M., Brini M., Carafoli E., Eizirik D.L., Cardozo A.K., Herchuelz A. Plasma membrane Ca2+-ATPase overexpression depletes both mitochondrial and endoplasmic reticulum Ca2+ stores and triggers apoptosis in insulin-secreting BRIN-BD11 cells. J. Biol. Chem. 2010;285:30634–30643. doi: 10.1074/jbc.M110.116681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barrow S.L., Voronina S.G., da Silva Xavier G., Chvanov M.A., Longbottom R.E., Gerasimenko O.V., Petersen O.H., Rutter G.A., Tepikin A.V. ATP depletion inhibits Ca(2+) release, influx and extrusion in pancreatic acinar cells but not pathological Ca(2+) responses induced by bile. Pflugers Arch. 2008;455:1025–1039. doi: 10.1007/s00424-007-0360-x. [DOI] [PubMed] [Google Scholar]
- 92.Criddle D.N., Murphy J., Fistetto G., Barrow S., Tepikin A.V., Neoptolemos J.P., Sutton R., Petersen O.H. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130:781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 93.Richards D.E., Rega A.F., Garrahan P.J. Two classes of site for ATP in the Ca2+-ATPase from human red cell membranes. Biochim. Biophys. Acta. 1978;511:194–201. doi: 10.1016/0005-2736(78)90313-9. [DOI] [PubMed] [Google Scholar]
- 94.Echarte M.M., Rossi R.C., Rossi J.P. Phosphorylation of the plasma membrane calcium pump at high ATP concentration. On the mechanism of ATP hydrolysis. Biochemistry. 2007;46:1034–1041. doi: 10.1021/bi061857x. [DOI] [PubMed] [Google Scholar]
- 95.Pedersen P.L. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the Warburg Effect, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- 96.Pedersen P.L. The cancer cell's power plants as promising therapeutic targets: an overview. J. Bioenerg. Biomembr. 2007;39:1–12. doi: 10.1007/s10863-007-9070-5. [DOI] [PubMed] [Google Scholar]
- 97.Warburg O., Wind F., Negelein E. The metabolism of tumors in the body. J. Gen. Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.James A., Chan A., Erice Azparren O., Siriwardena A.K., Bruce J.I.E. Glycolytic ATP fuels the plasma membrane calcium pump critical for pancreatic cancer cell survival. J. Biol. Chem. 2013;288:36007–36019. doi: 10.1074/jbc.M113.502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hardin C.D., Raeymaekers L., Paul R.J. Comparison of endogenous and exogenous sources of ATP in fueling Ca2+ uptake in smooth muscle plasma membrane vesicles. J. Gen. Physiol. 1992;99:21–40. doi: 10.1085/jgp.99.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hardin C.D., Zhang C., Kranias E.G., Steenaart N.A., Raeymaekers L., Paul R.J. Regulation of glycolytically fueled Ca2+ uptake in smooth muscle plasmalemmal vesicles by phosphorylation. Am. J. Physiol. 1993;265:H1326–1333. doi: 10.1152/ajpheart.1993.265.4.H1326. [DOI] [PubMed] [Google Scholar]
- 101.Paul R.J., Hardin C.D., Raeymaekers L., Wuytack F., Casteels R. Preferential support of Ca2+ uptake in smooth muscle plasma membrane vesicles by an endogenous glycolytic cascade. FASEB J. 1989;3:2298–2301. doi: 10.1096/fasebj.3.11.2528493. [DOI] [PubMed] [Google Scholar]
- 102.Campanella M.E., Chu H., Low P.S. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Campanella M.E., Chu H., Wandersee N.J., Peters L.L., Mohandas N., Gilligan D.M., Low P.S. Characterization of glycolytic enzyme interactions with murine erythrocyte membranes in wild-type and membrane protein knockout mice. Blood. 2008;112:3900–3906. doi: 10.1182/blood-2008-03-146159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chu H., Low P.S. Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochem. J. 2006;400:143–151. doi: 10.1042/BJ20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Puchulu-Campanella E., Chu H., Anstee D.J., Galan J.A., Tao W.A., Low P.S. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J. Biol. Chem. 2013;288:848–858. doi: 10.1074/jbc.M112.428573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dabrowska A., Pietkiewicz J., Dabrowska K., Czapinska E., Danielewicz R. Interaction of M1 and M2 isozymes pyruvate kinase from human tissues with phospholipids. Biochim. Biophys. Acta. 1998;1383:123–129. doi: 10.1016/s0167-4838(97)00192-1. [DOI] [PubMed] [Google Scholar]
- 107.Zhang J., Xiao P., Zhang X. Phosphatidylserine externalization in caveolae inhibits Ca2+ efflux through plasma membrane Ca2+-ATPase in ECV304. Cell Calcium. 2009;45:177–184. doi: 10.1016/j.ceca.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 108.Chu H., Puchulu-Campanella E., Galan J.A., Tao W.A., Low P.S., Hoffman J.F. Identification of cytoskeletal elements enclosing the ATP pools that fuel human red blood cell membrane cation pumps. Proc. Natl. Acad. Sci. U. S. A. 2012;109:12794–12799. doi: 10.1073/pnas.1209014109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baggaley E.M., Elliott A.C., Bruce J.I. Oxidant-induced inhibition of the plasma membrane Ca2+-ATPase in pancreatic acinar cells: role of the mitochondria. Am. J. Physiol. Cell Physiol. 2008;295:C1247–1260. doi: 10.1152/ajpcell.00083.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bruce J.I., Elliott A.C. Oxidant-impaired intracellular Ca2+ signaling in pancreatic acinar cells: role of the plasma membrane Ca2+-ATPase. Am. J. Physiol. Cell Physiol. 2007;293:C938–950. doi: 10.1152/ajpcell.00582.2006. [DOI] [PubMed] [Google Scholar]
- 111.Lehotsky J., Raeymaekers L., Missiaen L., Wuytack F., De Smedt H., Casteels R. Stimulation of the catalytic cycle of the Ca2+ pump of porcine plasma-membranes by negatively charged phospholipids. Biochim. Biophys. Acta. 1992;1105:118–124. doi: 10.1016/0005-2736(92)90169-m. [DOI] [PubMed] [Google Scholar]
- 112.Rossi J.P., Rega A.F. A study to see whether phosphatidylserine, partial proteolysis and EGTA substitute for calmodulin during activation of the Ca2+-ATPase from red cell membranes by ATP. Biochim. Biophys. Acta. 1989;996:153–159. doi: 10.1016/0167-4838(89)90241-0. [DOI] [PubMed] [Google Scholar]
- 113.Fadok V.A., Voelker D.R., Campbell P.A., Cohen J.J., Bratton D.L., Henson P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 114.Daleke D.L. Phospholipid flippases. J. Biol. Chem. 2007;282:821–825. doi: 10.1074/jbc.R600035200. [DOI] [PubMed] [Google Scholar]
- 115.Seigneuret M., Devaux P.F. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc. Natl. Acad. Sci. U. S. A. 1984;81:3751–3755. doi: 10.1073/pnas.81.12.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smriti, Nemergut E.C., Daleke D.L. ATP-dependent transport of phosphatidylserine analogues in human erythrocytes. Biochemistry. 2007;46:2249–2259. doi: 10.1021/bi061333x. [DOI] [PubMed] [Google Scholar]
- 117.Brunauer L.S., Moxness M.S., Huestis W.H. Hydrogen peroxide oxidation induces the transfer of phospholipids from the membrane into the cytosol of human erythrocytes. Biochemistry. 1994;33:4527–4532. doi: 10.1021/bi00181a013. [DOI] [PubMed] [Google Scholar]
- 118.Herrmann A., Devaux P.F. Alteration of the aminophospholipid translocase activity during in vivo and artificial aging of human erythrocytes. Biochim. Biophys. Acta. 1990;1027:41–46. doi: 10.1016/0005-2736(90)90045-p. [DOI] [PubMed] [Google Scholar]
- 119.Ricci J.E., Waterhouse N., Green D.R. Mitochondrial functions during cell death, a complex (I-V) dilemma. Cell Death Differ. 2003;10:488–492. doi: 10.1038/sj.cdd.4401225. [DOI] [PubMed] [Google Scholar]
- 120.Zaidi A., Michaelis M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free Radic. Biol. Med. 1999;27:810–821. doi: 10.1016/s0891-5849(99)00128-8. [DOI] [PubMed] [Google Scholar]
- 121.Kip S.N., Strehler E.E. Rapid downregulation of NCX and PMCA in hippocampal neurons following H2O2 oxidative stress. Ann. N. Y. Acad. Sci. 2007;1099:436–439. doi: 10.1196/annals.1387.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harwood S.M., Yaqoob M.M., Allen D.A. Caspase and calpain function in cell death: bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem. 2005;42:415–431. doi: 10.1258/000456305774538238. [DOI] [PubMed] [Google Scholar]
- 123.Brown C.S., Dean W.L. Regulation of plasma membrane Ca2+-ATPase in human platelets by calpain. Platelets. 2007;18:207–211. doi: 10.1080/09537100600954037. [DOI] [PubMed] [Google Scholar]
- 124.Guerini D., Pan B., Carafoli E. Expression, purification, and characterization of isoform 1 of the plasma membrane Ca2+ pump: focus on calpain sensitivity. J. Biol. Chem. 2003;278:38141–38148. doi: 10.1074/jbc.M302400200. [DOI] [PubMed] [Google Scholar]
- 125.Gerasimenko J.V., Gerasimenko O.V., Palejwala A., Tepikin A.V., Petersen O.H., Watson A.J. Menadione-induced apoptosis: roles of cytosolic Ca(2+) elevations and the mitochondrial permeability transition pore. J. Cell Sci. 2002;115:485–497. doi: 10.1242/jcs.115.3.485. [DOI] [PubMed] [Google Scholar]
- 126.Chami M., Ferrari D., Nicotera P., Paterlini-Brechot P., Rizzuto R. Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 2003;278:31745–31755. doi: 10.1074/jbc.M304202200. [DOI] [PubMed] [Google Scholar]
- 127.Pottorf W.J., 2nd, Johanns T.M., Derrington S.M., Strehler E.E., Enyedi A., Thayer S.A. Glutamate-induced protease-mediated loss of plasma membrane Ca2+ pump activity in rat hippocampal neurons. J. Neurochem. 2006;98:1646–1656. doi: 10.1111/j.1471-4159.2006.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weber H., Huhns S., Luthen F., Jonas L., Schuff-Werner P. Calpain activation contributes to oxidative stress-induced pancreatic acinar cell injury. Biochem. Pharmacol. 2005;70:1241–1252. doi: 10.1016/j.bcp.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 129.Yang E., Zha J., Jockel J., Boise L.H., Thompson C.B., Korsmeyer S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 130.Adams J.M., Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 131.Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 132.Lizcano J.M., Morrice N., Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 133.Fang X., Yu S., Eder A., Mao M., Bast R.C., Jr., Boyd D., Mills G.B. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene. 1999;18:6635–6640. doi: 10.1038/sj.onc.1203076. [DOI] [PubMed] [Google Scholar]