

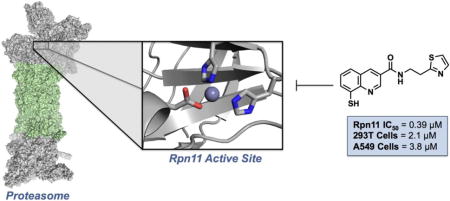
Abstract
The proteasome plays a crucial role in degradation of normal proteins that happen to be constitutively or inducibly unstable, and in this capacity it plays a regulatory role. Additionally, it degrades abnormal/damaged/mutant/misfolded proteins, which serves a quality-control function. Inhibitors of the proteasome have been validated in the treatment of multiple myeloma, with several FDA-approved therapeutics. Rpn11 is a Zn2+-dependent metalloisopeptidase that hydrolyzes ubiquitin from tagged proteins that are trafficked to the proteasome for degradation. A fragment-based drug discovery (FBDD) approach was utilized to identify fragments with activity against Rpn11. Screening of a library of metal-binding pharmacophores (MBPs) revealed that 8-thioquinoline (8TQ, IC50 value ~2.5 μM) displayed strong inhibition of Rpn11. Further synthetic elaboration of 8TQ yielded a small molecule compound (35, IC50 value ~300 nM) that is a potent and selective inhibitor of Rpn11 that blocks proliferation of tumor cells in culture.
Graphical abstract
INTRODUCTION
Multiple myeloma (MM) is a plasma cell neoplasm that affects thousands of people each year. Currently, there is no cure for MM. Even with a strong regiment of available chemotherapies, average life expectancy from time of diagnosis ranges from 2.5 to 5 years, depending upon the stage of the disease.1-2 The development of novel chemotherapeutics that inhibit components of the proteasome has proven very successful in extending progression-free and overall survival.3-4 These drugs inhibit the ubiquitin-proteasome degradation pathway through binding to one or more of the protease active sites within the proteasome.
The ubiquitin-proteasome system (UPS) plays a major role in protein quality control by degrading unwanted, damaged, or misfolded proteins within eukaryotic cells. It also controls numerous processes including cell cycle, apoptosis, transcription, and DNA repair by modulating the stability of critical regulatory proteins. Due to the UPS playing a central role in cellular metabolism, inhibition of the proteasome has emerged as a powerful strategy for anti-cancer therapy. Inhibiting this pathway was validated as a clinical target with the FDA approval of bortezomib, followed by carfilzomib, and most recently ixazomib, all approved for treatment of MM. The success of these small molecules has generated substantial interest in developing inhibitors that target other key elements of the proteasome.5-9
Degradation of proteins through the UPS occurs through a complex ATP-dependent pathway. Proteolysis is initiated with the protein destined for degradation being tagged with ubiquitin. The tagged protein undergoes several rounds of ubiquitin ligation, becoming polyubiquitinated, and is then directed to the proteasome. The constitutive 26S proteasome is composed of two subcomplexes: a catalytic barrel-shapped 20S core particle (20S CP) and a 19S regulatory particle (19S RP). The 19S RP caps one or both ends of the 20S CP to form a functional 26S proteasome. Previously reported proteasome inhibitors (bortezomib, carfilzomib, and ixazomib) inhibit the proteasome by binding preferentially to the catalytic threonine residue of the β5 subunit (also known as the chymotryptic site) within the 20S CP, which is the major site of proteolysis. The polyubiquinated protein is recognized by the 19S RP as a substrate wherein the 19S particle traps it, unfolds it, and translocates it into the 20S CP to become degraded and expelled out as oligopeptides.10-11 The Zn2+-dependent JAMM domain of the Rpn11 subunit, found within the 19S RP, cleaves ubiquitin from its substrates, thereby releasing ubiquitin for recycling. Previous reports12-14 demonstrate that mutations within the JAMM domain or addition of metal chelators to proteasome-dependent degradation reactions does not result in loss of substrate recognition, but impairs degradation of the substrate by the proteasome because it can no longer be inserted into the 20S CP due to failure to remove the bulky ubiquitin chain, the diameter of which is wider than the entry portal into the 20S CP. Inhibition of Rpn11 may lead to preferential apoptosis of neoplastic cells because these cells are thought to have a higher dependency on proteasome-dependent protein quality control compared to normal cells.15-16 Therefore, Rpn11 represents an attractive and novel therapeutic target for proteasome inhibition.
The catalytic JAMM motif of Rpn11 is found in 7 different human proteins including the Csn5 subunit of the COP9 signalosome, AMSH, AMSH-LP, the BRCC36 subunit of BRISC, MPND, and MYSM1.17-23 All of these enzymes cleave the isopeptide linkage that joins ubiquitin (or the ubiquitin-like protein Nedd8 in the case of Csn5) to a second molecule of ubiquitin or to a substrate. The conserved JAMM domain has the consensus sequence EXnHS/THX7SXXD, in which the His and Asp residues bind the Zn2+ ion and the fourth coordination site is occupied by a water molecule that is engaged in hydrogen bonding with the conserved Glu. The Zn2+ acts as a Lewis acid and increases the nucleophilic character of the bound water enough to allow hydrolytic cleavage of the isopeptide bond.20, 24
We have employed a fragment-based drug discovery (FBDD) approach to discover inhibitors of Rpn11. The use of FBDD for the discovery of biologically active compounds has become increasingly important. This strategy consists of generating small libraries of molecular fragments and screening them against the target of interest. The hits from this screen can further be elaborated into lead compounds. Here, we report the discovery, design, synthesis, and evaluation of a novel class of proteasome inhibitors that target Rpn11. Both a cell-free enzyme inhibition assay and a cellular assay identify Rpn11 as the target of inhibition. FBDD was utilized to identify a fragment, namely 8-thioquinoline (8TQ), with high affinity for the proteasome subunit Rpn11. Structure-activity relationship (SAR) experiments support the hypothesis that 8TQ inhibits Rpn11 through coordination of its catalytic Zn2+ ion. SAR of the 8TQ scaffold yielded several compounds with sub-micromolar potency, including a first-in-class Rpn11-selective inhibitor. The findings of the synthetic campaign described in this report prompted an extensive biological study that is described elsewhere.25 This work opens the door to development of a novel class of proteasome inhibitors for cancer chemotherapy.
RESULTS
Screening of the MBP library
The majority of previously reported proteasome inhibitors bind to the catalytic β1, β2, or β5 subunits within the 20S CP,3, 26-28 with fewer reports of inhibitors binding subunits within the 19S RP.29-33 An alternative approach to high-throughput screening (HTS) for the development of metalloenzyme inhibitors is through use of fragment libraries of metal-binding pharmacophores (MBPs). MBPs are small molecules that have one or a set of donor atoms that are capable of forming coordinate covalent bonds to the active site metal ion of metalloenzymes. To identify potent MBPs that can serve as initial building blocks for inhibitor design, a chemical library containing 96 MBP fragments34 was screened against Rpn11. The fragments are small (<300 amu) and have a known or predictable metal-binding motif (Figure 1). This MBP library has been previously used to successfully identify scaffolds for the development of several metalloenzyme inhibitors.35-36
Figure 1.
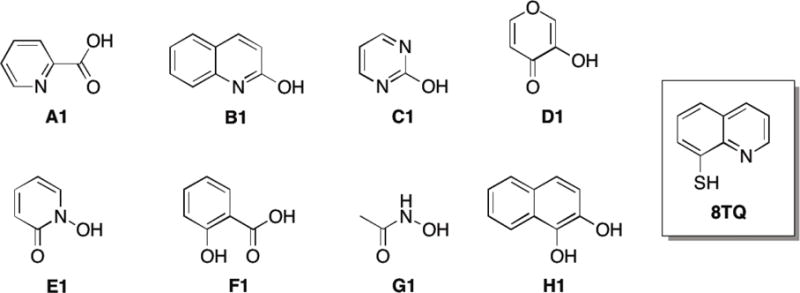
Representative fragments from the MBP library, including the lead fragment for this study 8TQ.
The MBP library was initially screened at a fragment concentration of 200 μM against Rpn11 by utilizing a fluorescence polarization assay.37-38 The fluorescence polarization assay specifically measures the deubiquitinating activity of Rpn11. The assay features a proteasome substrate with four tandem repeats of ubiquitin (Ub4) followed by a peptide labeled with Oregon Green on a unique cysteine residue. Incubation of this substrate, Ub4peptideOG, with proteasome resulted in depolarization of Oregon Green fluorescence due to release of the peptideOG from Ub4.25 Initial evaluation of the MBP library revealed three compounds with >50% inhibition, with the majority of the compounds exhibiting 0-30% inhibition (Figure 2). One fragment (8TQ, Figure 1) demonstrated essentially complete inhibition at a concentration of 200 μM. Because of the particularly strong activity of 8TQ the fragment was chosen for lead development. 8TQ was determined to have an IC50 value of 2.8±0.36 μM, which translates to an extremely high ligand efficiency of 0.69.39
Figure 2.
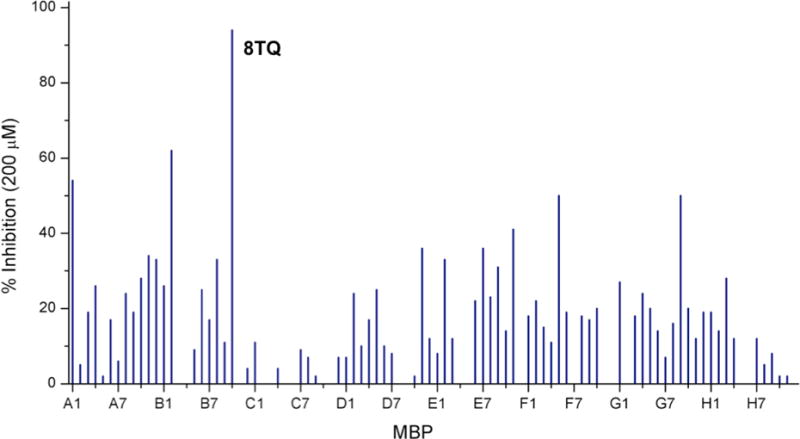
Plot of screening results of the MBP library against Rpn11 using a fluorescence polarization assay. Lines represent percent enzyme inhibition for a give MBP fragment at a concentration of 200 μM.
As described in greater detail elsewhere,25 an independent screen of >300,000 compounds produced only one hit – a thioester derivative of 8TQ – that, given the hydrolytic instability of the thioester group, is proposed to form 8TQ as the active species. Incredibly, the results from a small MBP library screen uncovered the same privileged scaffold as a large HTS campaign, which highlight the efficiency and effectiveness of our MBP library approach to drugging metalloenzymes.
Investigating the Mechanism of Inhibition
Due to the structural similarity of 8TQ to the common metal chelator 8-hydroxyquinoline, as well as data on previously reported 8TQ metal complexes, we predicted that 8TQ would bind the catalytic Zn2+ ion of Rpn11 in a bidentate fashion through the endocyclic nitrogen and exocyclic sulfur donor atoms.40-42 To validate this hypothesis, a model was sought to allow for structural characterization of the mode of binding. Tris(pyrazolyl)borate (Tp) complexes have been shown to serve as useful metalloenzyme active site mimics, giving some insight into bond lengths and angles for MBPs coordinated to metalloenzyme active site metal ions.43-48 A Zn2+ complex with the ligand hydrotris(5,3-methylphenylpyrazolyl)borate (TpMe,Ph)43 was combined with 8TQ to obtain the complex [(TpMe,Ph)Zn(8TQ)]. The metal complex was readily crystallized and revealed a five-coordinate Zn2+ center with a trigonal bipyramidal coordination geometry (Figure 3). 8TQ was bound in the expected bidentate manner, with the sulfur donor atom positioned in the equatorial plane (2.29 Å, Zn-S distance) and the endocyclic nitrogen atom serving as an axial donor (2.17 Å, Zn-N distance). The structure of this metalloenzyme model complex supports the hypothesis that 8TQ inhibited Rpn11 by metal coordination of the active site Zn2+ ion.
Figure 3.
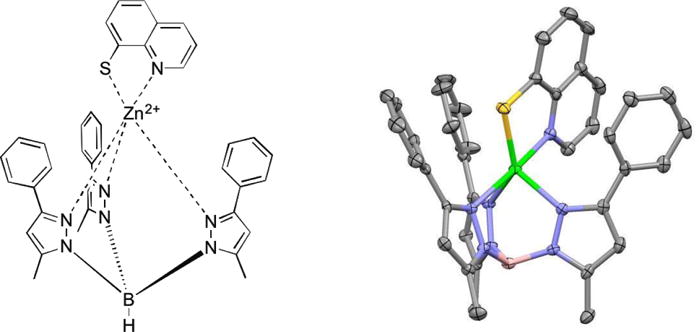
Chemical illustration (left) and image of the X-ray structure (right) of [(TpMe,Ph)Zn(8TQ)]. Thermal ellipsoids are shown at 50% probability. Hydrogen atoms are omitted for clarity. Color scheme: boron (pink), carbon (gray), nitrogen (blue), sulfur (yellow), and zinc (green).
Additional evidence for the mode of inhibition was obtained from SAR studies using 8TQ derivatives. Derivatives of 8TQ were prepared including fragments where the metal-coordinating atoms were removed, moved, or otherwise modified (Table 1). For example, compound 2 replaced the endocyclic nitrogen with a C-H group, giving a naphthyl derivative, which is incapable of the bidentate mode of binding exhibited by 8TQ (Figure 3). Similarly, in compounds 3, 4, and 5 the thiol moiety was replaced by a methyl, hydroxyl, or amine group, respectively, giving a series of isosteric compounds that lack the requisite thiol donor atom. In compound 6 the thiol moiety was alkylated with a methyl group (Scheme 1), which prevents formation of the anionic thiolate donor atom for binding Zn2+ (Figure 3). Finally, compound 7c places the coordinating nitrogen atom on the opposite side of the quinoline ring from the thiol moiety (Scheme 2), which produces an isosteric compound, but does not allow for bidentate binding of the ligand to the metal ion. As summarized in Table 1, compounds 2-6 and 7c all exhibited a complete loss of activity (IC50 >100 μM) against Rpn11, further validating the importance of the bidentate binding of 8TQ through the nitrogen and sulfur pair of donor atoms. Further confirmation of this hypothesis was demonstrated by the activity of compound 8e, which has an additional nitrogen atom at the 5-position of the ring (Scheme 3), but otherwise can maintain the 8TQ binding motif. Compound 8e inhibits Rpn11 with an IC50 value of 15±3.4 μM. The ~6-fold weaker activity of 8e when compared to 8TQ is attributed to the ability of 8e to tautomerize to the 1,5-Naphthyridine-4(1H)-thione form.
Table 1.
8TQ fragment derivatives used to examine the role of metal binding in Rpn11 inhibition.
| Cmpd | Structure | Rpn11 IC50 (μM) | HCT 116 Cytotoxicity (μM) | Cmpd | Structure | Rpn11 IC50 (μM) | HCT 116 Cytotoxicity (μM) |
|---|---|---|---|---|---|---|---|
| 8TQ |
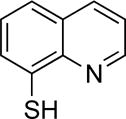
|
2.8±0.36 | 1.3 | 5 |
|
>100 | — |
| 2 |
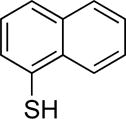
|
>100 | >100 | 6 |
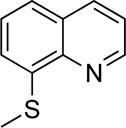
|
>100 | >100 |
| 3 |
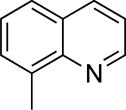
|
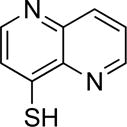
>100 | >100 | 7c |
|
>100 | >100 |
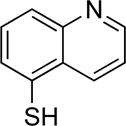
| 4 |
|
>100 | 6 | 8e |
|
15±3.4 | — |
Scheme 1. Synthesis of compound 6.
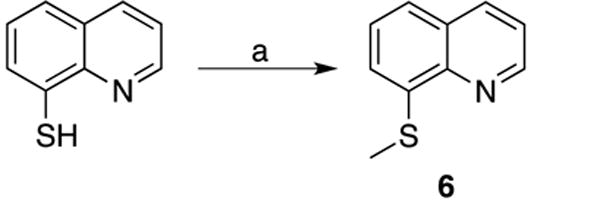
(a) CH3I, EtOH, H2O, 2M NaOH, 25 °C.
Scheme 2. Synthesis of compound 7c.
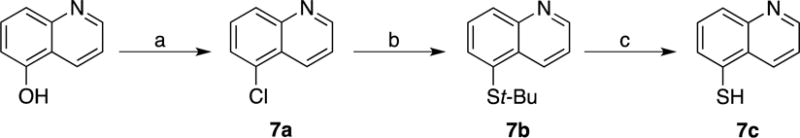
(a) POCl3, 100 °C; (b) t-BuSH, NaH, DMF, 140 °C; (c) 12M HCl, 100 °C.
Scheme 3.
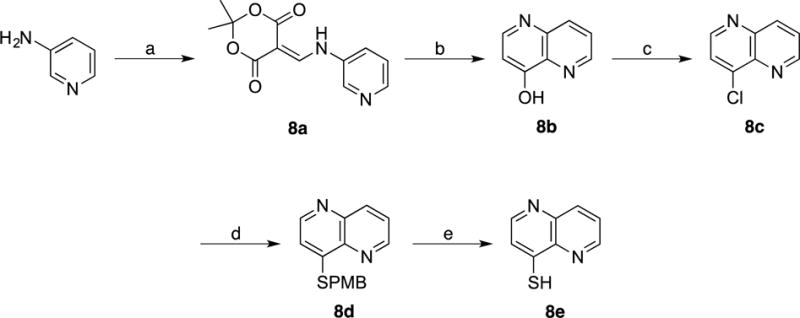
(a) 2,2,6-Trimethyl-4H-1,3-dioxin-4-one (Meldrum’s acid), Triethyl orthoformate, 105 °C; (b) Dowtherm A, 250 °C; (c) POCl3, Toluene, 110 °C; (d) 4-Methoxyphenyl)methanethiol (p-MBSH), NaH, DMF, 25 °C; (e) m-Cresol, TFA, reflux.
Synthesis of Methyl Derivatives and Cross Inhibition Studies
Having established a rudimentary structure-activity relationship (SAR) for the requisite metal-binding features of the 8TQ scaffold, a sublibrary of 8TQ derivatives with simple modifications to the scaffold was prepared in an effort to probe for possible hydrophobic (methyl groups) contacts within the active site, as well as to determine the best positions on the 8TQ ring to add substituents for subsequent rounds of derivatization. Functionalization of the 8TQ fragment was achieved largely via the Skraup and Doubner-Von Miller reactions using aniline derivatives as starting materials. Compounds 9a and 10a were synthesized starting with 2-fluoroaniline with the quinoline ring forming upon addition of a methyl-α,β-unsaturated aldehyde in the presence of aqueous HCl (Scheme 4). The 4-methyl quinoline analog (11a) was synthesized in similar fashion, by combining 2-fluoroaniline with an α,β-unsaturated ketone (Scheme 4).
Scheme 4. Synthesis of 2-, 3-, and 4-methyl-8-thioquinoline (apolar) derivatives of 8TQ.
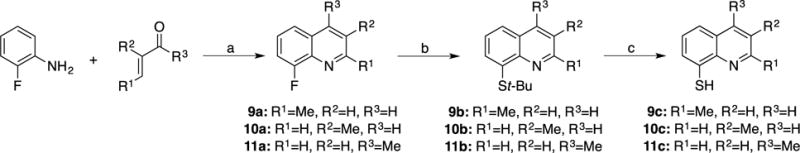
(a) Toluene, 6M HCl, 110 °C; (b) t-BuSH, NaH, DMF, 140 °C; (c) 12M HCl, 100 °C.
Compounds 12a and 13a were obtained by starting with methyl functionalized 2-chloro or 2-fluoroaniline in the presence of glycerol utilizing nitrobenzene as the solvent and oxidant (Scheme 5). Substitution of the resulting methyl-8-fluoro or methyl-8-chloroquinolines (9a, 10a, 11a, 12a, and 13a) to obtain the thiol functionality was obtained through a nucleophilic aromatic substitution reaction utilizing tert-Butyl thiol (t-BuSH). This was followed by a deprotection reaction under refluxing concentrated HCl to yield the free thiol (9c, 10c, 11c, 12c, and 13c).
Scheme 5. Synthesis of 5- and 6-methyl-8-thioquinoline.
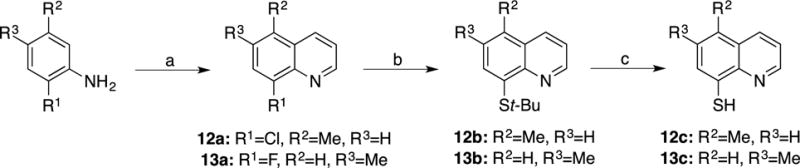
(a) Glycerol, Nitrobenzene, 150 °C; (b) t-BuSH, NaH, DMF, 140 °C; (c) 12M HCl, 10 0°C.
In addition to evaluation against Rpn11, the selectivity of these compounds against off-target metalloenzymes was also examined by performing inhibition assays against a host of other metalloenzymes (Table 2). These off-target metalloenzymes were selected because they possess a diverse set of structures and functions, utilize a metal ion in a catalytic role, are clinically relevant targets, and have readily available assays. The metalloenzymes examined included the Zn-dependent JAMM domain enzyme (Csn5), two histone deaceylases (HDAC-1, HDAC-6), a matrix metalloproteinase (MMP-2), carbonic anhydrase (hCAII), and a non-heme, Fe-dependent lipoxygenase (5-LO). In addition, to assess the effects of these compounds in a cellular model, a human colon carcinoma cell line (HCT 116) was utilized to measure the anti-proliferative activity of the fragments.
Table 2.
Enzyme inhibition data for 8TQ and 8TQ derivatives. Data listed includes inhibitory values against off-target metalloenzymes and cytotoxicity against the HCT 116 cell line. All IC50 values listed are in μM. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
| Cmpd | Structure | Rpn11 | Csn5 | HDAC1 | HDAC6 | MMP2 | 5-LO | hCAII | HCT 116 Cytotoxicity |
|---|---|---|---|---|---|---|---|---|---|
| 8TQ | H | 2.8±0.4 | 10.3±2.3 | >200 | >200 | >200 | >200 | >200 | 1.6±0.7 |
| 9c | 2-Me | >100 | >100 | N.D | N.D | >200 | N.D | N.D | >50 |
| 10c | 3-Me | 1.6±0.6 | 7.1±1.6 | >50 | >200 | >200 | >200 | >40 | 2.6±0.5 |
| 11c | 4-Me | 5.7±2.0 | 25.1±6.1 | >200 | >200 | >200 | >200 | >200 | >10 |
| 12c | 5-Me | 2.5±1.3 | 2.9±1.1 | >200 | >200 | >200 | >200 | >200 | 3.4±1.1 |
| 13c | 6-Me | 0.9±0.3 | 1.6±0.6 | >50 | >50 | >200 | >200 | >100 | 2.1±1.2 |
The results from these experiments are summarized in Table 2. The data demonstrate that the 8TQ scaffold was highly specific for the JAMM metalloproteins (Rpn11 and Csn5) over other metalloenzymes. Some discrimination between Rpn11 and Csn5 was observed, even with the relatively simple methyl substitutions, which suggests that specificity could be developed using this scaffold. Inhibition data also suggests that the Rpn11 active site was quite plastic and tolerated substitution at multiple positions on the 8TQ ring. Compound 9c did not inhibit the JAMM domain proteins, which correlates with loss of cytotoxicity toward the HCT 116 cell line (Table 2). Introduction of even a small methyl group resulted in complete loss of activity, which suggests that functionalization at the 2-position is not tolerated.
Synthesis of an 8TQ Sublibrary
Given the high affinity of 8TQ toward JAMM domain proteins, derivatives were sought that could improve potency while also adding selectivity for Rpn11. Compounds containing functional groups at the 3- and 4-positions were primarily pursued due to the synthetic accessibility of these derivatives over other active compounds (e.g. 5- and 6-position derivatives, Table 2). In addition, derivatives of the 2-position were prepared to confirm the negative SAR obtained with the methyl derivatives. Despite the good activity observed with 5- (12c) and 6- (13c) position derivatives, these were not further explored in this synthetic campaign.
Compounds 14b and 16b were synthesized starting from commercially available 2- or 3- carboxyl-8-fluoroquinoline as detailed in Scheme 6. Compound 18d was obtained via a Pfitzinger ring expansion reaction of 7-fluoroisatin and pyruvate under basic conditions to yield 18a. This was decarboxylated under aqueous conditions to afford 18b, which then yielded 18d over two steps (Scheme 7). The corresponding methyl ester derivatives were obtained via Fisher esterification (15, 17 and 19). Lastly, the 2-, 3-, and 4-carboxylate-8-thioquinoline compounds were coupled to amines mainly via the assistance of carbodiimidazole (CDI) or 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) coupling reagents. Alkyl amines were coupled mainly through the use of CDI at room temperature; however, less nucleophilic amines (aromatic) were coupled using HATU with heating.
Scheme 6. Synthesis of 2- and 3-carboxyl-8-thioquinoline (polar) derivatives.
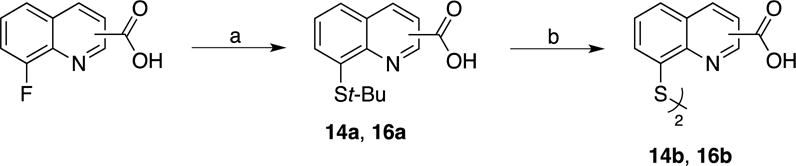
(c) t-BuSH, NaH, DMF, 140 °C; (d) 12M HCl, 110 °C.
Scheme 7. Synthesis of 4-carboxyl-8-thioquinoline.
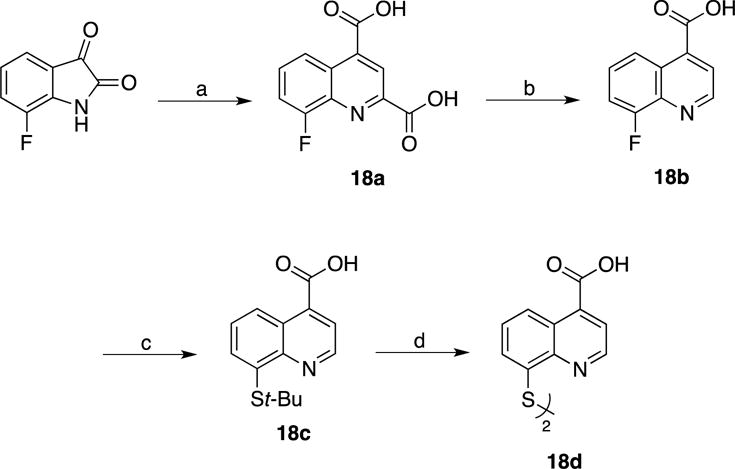
(a) Sodium Pyruvate, 5M NaOH, 110 °C; (b) H2O, 170 °C; (c) t-BuSH, NaH, DMF, 140 °C; (d) 12M HCl, 110 °C.
It should be noted that all of the aforementioned compounds were isolated as disulfide dimers, as evidenced by mass spectrometry. Under the Rpn11 assay conditions, which contained 1 mM of dithiothreitol (DTT) as a reductant, the disulfides were reduced to the monomeric active species. Screening of the aforementioned derivatives (14b, 15, 20, 21) demonstrated that functionalization at the 2-position was not well tolerated, consistent with the findings on the simple methyl derivative (9c). All the compounds functionalized at the 2-position were consistently less active than 8TQ (Table 3). In addition, activity generally decreased with increasing functional group size at the 2-position, possibly due to a clash with the protein active site.
Table 3.
Rpn11 inhibitory activity of 8TQ derivatives functionalized at the 2-, 3-, and 4-positions of the ring system.
| |||||||
|---|---|---|---|---|---|---|---|
| Structure | Cmpd | Position | Rpn11 IC50 (μM) | Structure | Cmpd | Position | Rpn11 IC50 (μM) |
| Me | 9c | 2 | >100 |
|
— | 2 | — |
| 10c | 3 | 1.6±0.6 | 30 | 3 | 0.9±0.1 | ||
| 11c | 4 | 5.7±2.0 | 23 | 4 | 2.2±0.6 | ||
|
14b | 2 | >20 |
|
20 | 2 | >100 |
| 16b | 3 | 1.1±0.1 | 31 | 3 | 0.8±0.3 | ||
| 18d | 4 | 1.3±0.2 | 22 | 4 | 1.0±0.2 | ||
|
15 | 2 | >50 |
|
21 | 2 | >100 |
| 17 | 3 | 0.9±0.1 | 39 | 3 | 1.1±0.2 | ||
| 19 | 4 | 1.0±0.1 | — | 4 | — | ||
Derivatization at the 3- and 4-positions was consistently well tolerated. In order to confine the scope of these initial synthetic efforts, additional exploration of derivatives was restricted to the 3-position. To accomplish this, compound 16b was coupled to a series of amines with the assistance of CDI or HATU coupling reagents (Scheme 8). All of the compounds prepared via Scheme 8 were also isolated as disulfide dimers. A diverse set of amines, predominantly derivatives with substituted aryl groups or heterocycles with varying linker lengths (Table 4), was explored. In order to identify a potent yet selective Rpn11 inhibitor, all compounds were screened in cell-free assays against Rpn11, Csn5, and AMSH. The Rpn11 assay was carried out in the same manner in which the initial 96-fragment screen was performed. To measure Csn5 activity, a fluorescent substrate termed SCFSkp2-Nedd8OG was engineered. To produce this substrate, Nedd8 containing a unique N-terminal cysteine was labeled with Oregon Green 488, and then conjugated to SCFSkp2 as previously described.49 For AMSH the substrate termed DiUBK63TAMRA was purchased from commercial sources. DiUBK63TAMRA is labeled with a FRET pair (TAMRA/QXL) that upon cleavage by AMSH produces a fluorescent signal. The SAR obtained was used to increase activity against Rpn11 while discriminating against the other JAMM domain proteins Csn5 and AMSH. In addition, a cell-based assay was utilized to measure inhibition of the proteasome in cells. For this, we utilized a HeLa cell line that stably expresses UbG76V-GFP (Green Fluorescent Protein), which serves as a fluorescent signal for proteasome activity.50 These cells were treated with β5 inhibitor MG132 to accumulate UbG76V-GFP. The MG132 was then washed out and either DMSO or one of our compounds was added, and the decay of GFP fluorescence was monitored. Under normal conditions, the accumulated UbG76V-GFP is rapidly degraded by proteasome. However, if the proteasome function is blocked, the degradation rate of the reporter protein is reduced. The IC50 values reported in Table 4 represent the concentration of test agent at which the degradation rate was reduced by half.
Scheme 8. Synthesis of 3-carboxamide derivatives.
(a) CDI, DMF, 25 °C; (b) HATU, Et3N, DMF, 60 °C.
Table 4.
Inhibitory activity against Rpn11, Csn5, and AMSH for 3-position substituted 8TQ derivatives. Cellular levels of proteasome inhibition are also listed. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
| |||||
|---|---|---|---|---|---|
| Cmpd | Structure | Rpn11 IC50 (μM) | Csn5 IC50(μM) | AMSH IC50 (μM) | UbG76VGFP Hela Cell IC50 (μM) |
| 24 | H | 1.0±0.2 | 15.3±4.2 | 1.7±0.4 | 1.2±0.2 |
| 25 | Me | 1.6±0.3 | 68±14 | 4.1±1.3 | 2.6±0.5 |
| 26 |
|
>2 | >100 | 2.2±0.3 | >0.3 |
| 27 |
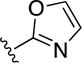
|
2.6±0.4 | 15±1 | 5.0±1.0 | >10 |
| 28 |
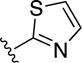
|
0.4±0.1 | 20±3 | 4.7±1.1 | 1.2±0.3 |
| 29 |
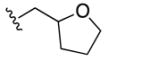
|
4.6±1.4 | 41±5 | 6.8±0.5 | 1.0±0.2 |
| 30 |
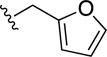
|
0.9±0.1 | <0.5 | 0.9±0.1 | 1.0±0.2 |
| 31 |
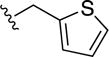
|
0.8±0.1 | 16±6 | 1.3±0.3 | 5.0±0.8 |
| 32 |
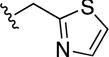
|
0.5±0.1 | 24±7 | 7.8±2.2 | 2.9±0.8 |
| 33 |
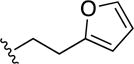
|
1.2±0.7 | 6.9±1.8 | 1.7±0.3 | 1.4±0.3 |
| 34 |
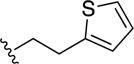
|
0.3±0.1 | 4.0±2.9 | 1.3±0.2 | 5.0±0.4 |
| 35 |
|
0.4±0.1 | 30±3 | 4.5±0.5 | 0.6±0.1 |
| 36 |
|
4.0±0.2 | >50 | 17±3 | >10 |
| 37 |
|
3.2±0.2 | >100 | 3.5±0.5 | >10 |
| 38 |
|
0.2±0.1 | 7±2 | 0.5±0.1 | >10 |
| 39 |
|
1.1±0.1 | 11±4 | >10 | >10 |
| 40 |
|
<0.2 | 18±6 | 0.9±0.1 | >10 |
| 41 |
|
0.8±0.2 | >100 | 3.5±0.7 | >10 |
| 42 |
|
<0.2 | 0.5±0.1 | 0.6±0.2 | >10 |
| 43 |
|
0.9±0.2 | 7±1 | <0.2 | >10 |
| 44 |

|
0.8±0.3 | 0.3±0.2 | 0.9±0.3 | >10 |
| 45 |
|
6.4±1.2 | >100 | 26±9 | >10 |
| 46 |
|
>5 | >30 | 3.5±0.6 | >10 |
Evaluation of the series of compounds in Table 4 identified 28 and 35 as two promising leads. Both compounds showed sub-micromolar IC50 values against Rpn11 in the biochemical assay and selectivity over Csn5 and AMSH. These compounds were then screened for cytotoxicity against 293T and A549 cells (Figure 4). Compound 28 had an IC50 of 6.4 μM and 5.8 μM against 293T and A549 cells, respectively. Meanwhile compound 35 demonstrated slightly lower IC50 values than 28 against both cell lines, at 2.1 μM and 3.8 μM, respectively. Ultimately, compound 35 was selected as the lead compound due to its better selectivity for Rpn11 over other JAMM proteins, efficacy in the cell-based assays, and more active cytotoxic profile.
Figure 4.
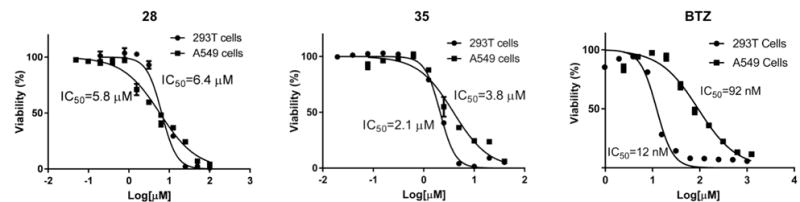
Cytotoxicity assay against 293T and A549 cancer cell lines with compounds 28 and 35, Bortezomib (BTZ) was also included as an internal control.
Synthesis and Evaluation of Control Compounds
A small series of control compounds (analogs of 35) were synthesized (Schemes 9-12) to re-evaluate the role of the MBP in this lead compound. An 8-hydroxyquinoline analog (47) was prepared in order to determine the importance of the softer Lewis base thiol (versus the harder Lewis base oxygen donor in 8-hydroxyquinoline). Compounds 48 and 49f prevent metal coordination, as they are elaborated analogs of inactive MBP fragments 6 and 7c (Table 1). Similarly, compound 50f was prepared as an elaborated analog of the less active MBP fragment 8e. Evaluation of these compounds yielded essentially no inhibition against Rpn11, Csn5, or AMSH (Table 5), recapitulating the SAR obtained with the original MBP fragments (Table 1).
Scheme 9. Synthesis of an 8-Hydroxyquinoline MBP based analog.
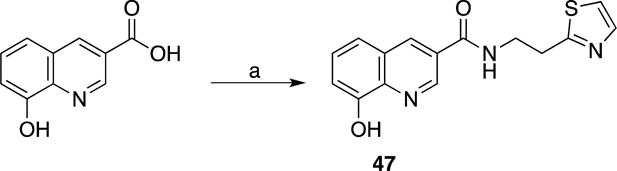
(a) 2-(Thiazol-2-yl)ethan-1-amine, HATU, Et3N, DMF, 25 °C.
Scheme 12. Synthesis of compound 50f, an MBP analog of lead compound 35.
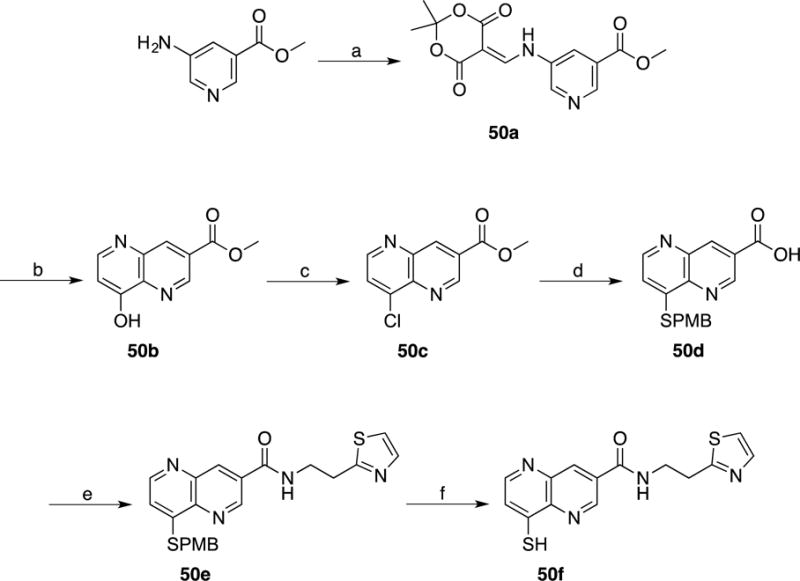
(a) Meldrum’s acid, Triethylformate, 100 °C; (b) Dowtherm A, 250 °C; (c) POCl3, Toluene, 110 °C; (d) p-MBSH, NaH, DMF, 25 °C; (e) 2-(Thiazol-2-yl)ethan-1-amine, pyridine, EDC, HOBT, DMF, 25 °C; (f) m-Cresol, TFA, reflux.
Table 5.
Inhibitory activity against Rpn11, Csn5, and AMSH for control compounds. Cellular levels of proteasome inhibition are also listed.
| Cmpd | Rpn11 IC50 (μM) | Csn5 IC50(μM) | AMSH IC50 (μM) | UbG76VGFP Hela Cell IC50 (μM) |
|---|---|---|---|---|
| 47 | >40 | >100 | >100 | >100 |
| 48 | >40 | >100 | >100 | >100 |
| 49f | >100 | 35±9 | >100 | >100 |
| 50f | 82±14 | >100 | >100 | >100 |
As a final experiment to demonstrate the importance of metal coordination in this class of inhibitors, an inhibition assay was carried out utilizing compound 35 in the presence of a soluble, small molecule coordination compound Zn(cyclen)2+ (cyclen = 1,4,7,10-tetraazacyclododecane). In this experiment, if metal coordination is critical for the activity of compound 35 then Zn(cyclen)2+ can act as a ‘decoy’ of a Zn-metalloprotein active site, thereby titrating 35 away from Rpn11 and reducing the apparent activity of the inhibitor. When inhibition of 35 against Rpn11 was measured in the presence of Zn(cyclen)2+ (100 μM, Figure 5), a significant loss in activity against Rpn11 was observed (IC50 = 77.4 μM vs. 0.39 μM, Figure 5). The observed IC50 value shift is attributed to the ability of Zn(cyclen)2+ to compete/titrate 35 away from Rpn11.
Figure 5.
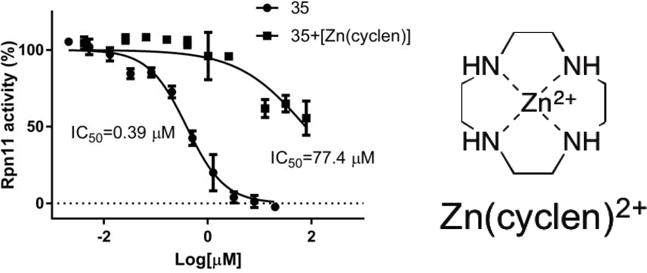
Inhibition of 35 against Rpn11 in the presence of Zn(cyclen)2+. Inhibition of 35 was also measured in the absence of Zn(cyclen)2+.
DISCUSSION
In order to discover a fragment that inhibits Rpn11, a FBDD approach using a 96-component library of MBPs led to the identification of the highly efficient 8TQ fragment for inhibiting Rpn11. Upon identifying 8TQ as an anchoring scaffold, derivatives were prepared to evaluate the hypothesis that metal binding was the source of 8TQ activity. Indeed, the SAR obtained from compounds 2-6 (Table 1) indicated that bidentate metal binding was essential for the observed inhibitory activity by 8TQ. Only derivative 8e, which possesses the same N,S donor atom set maintains some activity against Rpn11. In addition, the bioinorganic model complex [(TpMe,Ph)Zn(8TQ)] clearly supports the ability of 8TQ to form a ternary complex with a Zn2+ ion bound in a protein-like coordination environment (TpMe,Ph) (Figure 3). All of these results point to metal coordination as the mechanism of action for 8TQ against Rpn11.
Upon validating the mode of inhibition by 8TQ, efforts were made to develop a rudimentary SAR around the 8TQ scaffold to increase activity against Rpn11 while diminishing activity towards the most prominent off-targets, namely, the other JAMM family members Csn5 and AMSH. The initial strategy involved probing for hydrophobic and hydrophilic contacts near the active site, while also examining steric limitations. The quinoline ring was appended with methyl groups on the 2-, 3-, 4-, 5-, and 6-positions of the ring (Scheme 4 and 5, 9c, 10c, 11c, 12c, and 13c) or carboxylic acids on the 2-, 3-, and 4-positions of the ring (Scheme 6 and 7, 14b, 16b, and 18d). Derivatives with 3- and 4-subsitutents, including methyl (10c and 11c), carboxylate (16b and 18b). and methyl ester (17 and 19) substituents were all well tolerated, providing a consistent SAR, while substitution at the 2-position was not as well tolerated. The behavior of carboxylate derivatives with aromatic substituents was also consistent with the observed SAR, with substituents at the 3- and 4- being well tolerated. Among these, comparing the same substitutents at the 3- and 4-positions (16b to 18d, 17 to 19, and 30 to 23) suggested that 3-position derivatives might possess marginally better activity (Table 3), and hence derivatives of the 3-position became the focus of this study.
In addition to Rpn11, active JAMM domains are found in six other human proteins: the Csn5 subunit of the COP9-signalosome, the Brcc36 subunit of the BRCC and BRISC complexes, the closely-related AMSH and AMSH-LP proteins, MYSM1, and MPND. Of these, suitable biochemical assays are available for all but MYSM1 and MPND. AMSH is highly homologous to AMSH-LP, so we excluded this target from further consideration and focused our attention on Csn5 and AMSH as the major off-target concerns.51 However, 8TQ did show significant inhibition of one other JAMM domain protein (BRISC, Figure S1).
Compounds with substituents at the 3-position (24, 25), including carboxamide substituents, 30, 31, and 39, demonstrated a small increase in activity over 8TQ. With this preliminary SAR, a variety of substituents were explored via an amide linkage as illustrated in Table 3. The introduction of 5- or 6-membered heterocyclic, aromatic rings, such as thiophene, thiazole, furan, oxazole, and pyridine (27-37) improved activity. In addition, compounds 27-37 all demonstrated better solubility in aqueous solution (data not shown). A trend was observed wherein the heterocycles containing a thiophene ring (31 and 34) demonstrated better activity than furan-based analogs (30 and 33). Introduction of thiazole heterocycles demonstrated similar inhibition to thiophene containing compounds; however, thiazole-containing compounds (28, 32, and 35) all demonstrated improved selectivity for Rpn11 over Csn5 and AMSH. The introduction of a phenyl (39) or funtionalized phenyl (40-44) aromatic groups also improved the activity of the compounds; however, solubility in aqueous solution was poor (data not shown). Compounds 40 and 42 showed the best activity against Rpn11 (IC50 value <200 nM, Table 3); however, the phenyl substituted compounds demonstrated poor selectivity over AMSH and also failed to show any cell-based activity. A pair of saturated ring derivatives was also explored (29 and 45), but these compounds consistently demonstrated poor activity. From this series of aryl substituted compounds (24-45), compound 35 showed the best overall characteristics and performance. Compound 35 showed submicromolar activity in the Rpn11 biochemical assay (IC50 value 0.39±0.04 μM) and ~100-fold selectivity over Csn5 and ~10-fold selectivity over AMSH. Compound 35 also demonstrated cytotoxicity towards 293T and A549 cells with an IC50 of 2.1 and 3.8 μM respectively. Lastly, a series of 35 MBP analogs further validated the SAR and mode of inhibition. Compound 47 utilizes a harder Lewis base 8-hydroxyquinoline MBP that displays poor activity against Rpn11. This suggests that the soft Lewis base character of the 8-thioquinoline allows for better affinity for the active site Zn2+ ion. Evaluation of 48 and 49f revalidate the necessity of placing the coordinating atoms at the 1- and 8- positions. Finally, compound 50f uses fragment 8e (Scheme 3) instead of 8TQ as the MBP. Compound 50f showed activity against Rpn11 (~82 μM), but was significantly less active when compared to 35 (0.39 μM). This is consistent with the activity of the core scaffolds, fragments 8TQ and 8e, where 8TQ shows better activity against Rpn11 then 8e, again wholly consistent with metal coordination and formation of a 35-Rpn11 ternary complex as the mechanism of action of these inhibitors.
CONCLUSIONS
Proteasome inhibitors represent an expanding area with a broad therapeutic potential; however, limitations with current FDA approved inhibitors have generated interest in developing novel compounds. By utilizing a FBDD approach, a first-in-class, Rpn11-selective inhibitor with sub-micromolar IC50 values that is cytotoxic towards cancer cell lines has been obtained. By utilizing a modest library of <100 fragments we identified a fragment with low micromolar IC50 values for Rpn11. The power of this approach was underscored by a subsequent high-throughput screen of >300,000 compounds, which yielded a thioester derivative of 8TQ as the only hit that satisfied all criteria.25 A series of compounds helped establish rudimentary SAR, and from this an inhibitor that blocks proliferation of cancer cells was obtained. Further biological characterization of the lead compound, 35, are described elsewhere.25 Through inhibition of Rpn11 the ubiquitin tagged to the protein destined for degradation cannot be removed, thereby causing the proteasome to become inhibited. This represents a completely new mode of action for a proteasome inhibitor and thus has potential for novel applications in the chemotherapy of cancer.
EXPERIMENTAL SECTION
General Experimental Details
All reagents and solvents were obtained from commercial sources and used without further purification. Microwave reactions were performed in 10 mL or 35 mL microwave vials using a CEM Discover S reactor. Column chromatography was performed using a Teledyne ISCO CombiFlash Rf system with prepacked silica cartridges or High Performance Gold C18 columns. 1H/13C NMR spectra were recorded at ambient temperature on a 400 or 500 Varian FT-NMR instrument located in the Department of Chemistry and Biochemistry at the U.C. San Diego. Mass spectra were obtained at the Molecular Mass Spectrometry Facility (MMSF) in the Department of Chemistry and Biochemistry at the University of California, San Diego. Further details on synthesis may be found in the Supporting Information. The purity of all compounds used in assays was determined to be ≥95% by 1H NMR spectroscopy and confirmed by high-resolution mass spectrometry (HRMS) and liquid chromatography-mass spectrometry (LC-MS) analysis using an Agilent 6230 Accurate-Mass LC-TOFMS at the MMSF (U.C. San Diego).
Synthetic Procedures and Compound Characterization
8-(Methylthio)quinoline (6)
To a solution of 8-thioquinoline (0.07 g, 0.35 mmol) in a mixture of EtOH, H2O, and 2M NaOH (4 mL, 2:1:1 ratio) was added CH3I (120 mL, 1.8 mmol). The solution was stirred at room temperature for 24 h and then evaporated to dryness. The reaction mixture was then dissolved in CH2Cl2 and washed with H2O (3×50mL). The combined organic layers were dried and concentrated in vacuo. The crude was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.006 g (10%). 1H NMR (500 MHz, CDCl3): δ 8.95 (dd, J = 4.2, 1.8 Hz, 1H), 8.14 (dd, J = 8.3, 1.7 Hz, 1H), 7.57 (dd, J = 8.2, 1.3 Hz, 1H), 7.50 (dd, J = 8.1, 7.3 Hz, 1H), 7.45 (dd, J = 8.2, 4.2 Hz, 1H), 7.41 (dd, J = 7.4, 1.3 Hz, 1H), 2.59 (s, 3H). ESI-MS (+): m/z 176.11 [M+H]+.
5-Chloroquinoline (7a)
A solution of 5-Hydroxyquinoline (0.1 g, 0.68 mmol) in POCl3 (5 mL) was stirred at 100 °C for 2 h. H2O was added slowly to the reaction mixture to neutralize POCl3 and the resulting solution was evaporated to dryness. To the resulting crude was added MeOH, which caused the formation of a white precipitate. The solid was isolated by filtration to afford product. Yield = 0.08 g (71%). 1H NMR (400 MHz, DMSO-d6): δ 8.93 – 8.89 (m, 1H), 8.47 (t, J = 6.8 Hz, 1H), 7.78 (d, J = 5.5 Hz, 1H), 7.74 – 7.67 (m, 1H), 7.60 – 7.54 (m, 1H), 7.46 (d, J = 6.6 Hz, 1H). ESI-MS (+): m/z 164.05 [M+H]+.
5-(tert-Butylthio)quinolone (7b)
To a solution of 7a (0.07 g, 0.43 mmol) in DMF (7 mL) was added NaH (0.035 g, 1.45 mmol) and tert-butylthiol (t-BuSH, 0.97 mL, 0.86 mmol) under nitrogen atmosphere. The reaction was stirred at 140 °C for 18 h. The resulting solution was then concentrated in vacuo and the crude material was purified by via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.04 g (43%). 1H NMR (400 MHz, CD3OD): δ 8.89 (dd, J = 4.3, 1.8 Hz, 1H), 8.58 – 8.53 (m, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.55 – 7.46 (m, 1H), 7.38 (dd, J = 8.4, 4.3 Hz, 1H), 6.85 (d, J = 7.7 Hz, 1H), 1.53 (s, 9H). ESI-MS (+): m/z 218.15 [M+H]+.
Quinoline-5-thiol (7c)
A solution of 7b (0.04 g, 0.17 mmol) in conc. HCl (11 mL) was stirred at 90 °C for 19 h. The resulting solution was neutralized to pH 9-10 with NaOH and extracted twice with CHCl3 (3×10mL). The combined organic layers were dried and the solution was concentrated in vacuo. The crude material was then recrystallized from EtOAc. Yield = 0.01 g (48%). 1H NMR (500 MHz, CDCl3): δ 8.95 – 8.90 (m, 1H), 8.66 – 8.56 (m, 1H), 7.75 – 7.70 (m, 1H), 7.60 – 7.50 (m, 1H), 7.45 – 7.36 (m, 1H), 6.93 – 6.85 (m, 1H). ESI-MS (+): m/z 162.10 [M+H]+.
2,2-Dimethyl-5-((pyridin-3-ylamino)methylene)-1,3-dioxane-4,6-dione (8a)
To a preheated (~100 °C) mixture of 3-Aminopyridine (0.37 g, 4.0 mmol) and 2,2-Dimethyl-[1,3]dioxane-4,6-dione (Meldrum’s acid, 0.69 g, 4.8 mmol) was added Triethyl Orthoformate (4.0 mL, 24.0 mmol). The solution was stirred at 100 °C for 2 h. The reaction proceeded by changing color from yellow to wine red accompanying the formation of a yellow precipitate. After cooling to room temperature, the excess liquid of Triethyl Orthoformate was removed via vacuum distillation. The resulting solid was purified via silica gel chromatography eluting a gradient of 70 to 100% EtOAc in Hexanes. Yield = 0.72 g (72%). 1H NMR (400 MHz, CDCl3): δ 11.25 (d, J = 10.4 Hz, 1H), 8.63 (d, J = 14.00 Hz, 1H), 8.61 (d, J = 3.2 Hz, 1H), 8.55 (dd, J = 4.8 Hz, J = 1.6 Hz, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.41 (dd, J = 8.20, 4.2 Hz, 1H), 1.77 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.2, 163.0, 152.9, 147.6, 140.6, 134.6, 124.9, 124.1, 105.2, 88.5, 26.9. ESI-MS (+): m/z 248.90 [M+H]+.
1,5-Naphthyridin-4-ol (8b)
To a flask containing 8a (2.6 g, 10.4 mmol) under nitrogen atmosphere was added Dowtherm A (150 mL) and placed in a pre-heated oil bath at 250 °C. The reaction mixture was stirred at reflux for 1 h. A color change from orange yellow to dark brown was observed. The resulting solution was cooled to room temperature and filtered to isolate solid product. The solid was rinsed with Diphenyl Ether and Acetone to give the desired product as a dark solid. Yield = 1.14 g (75%). 1H NMR (400 MHz, CD3OD + one drop TFA): δ 9.07 (d, J = 4.8 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.61 (d, J = 7.2 Hz, 1H), 8.22 (dd, J = 8.8 Hz, J = 4.8 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H). 13C NMR (125 MHz, CD3OD + one drop TFA): δ 172.3, 147.5, 145.5, 138.6, 134.8, 134.4, 130.1, 112.2. ESI-MS (+): m/z 147.29 [M+H]+.
4-Chloro-1,5-naphthyridine (8c)
To a solution of 8b (0.8 g, 5.47 mmol) in Toluene (20 mL) was added POCl3 (1.02 mL, 10.95 mmol) at room temperature. The solution was stirred at 110 °C for 2 h, then allowed to cool to room temperature, resulting in the formation of a precipitate. The solution and dark solid was quenched with sat. NaHCO3 and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified via silica gel chromatography eluting a gradient of 20 to 40% EtOAc in CH2Cl2. Yield = 0.37 g (41%). 1H NMR (400 MHz, CDCl3): δ 8.92 (dd, J = 4.4, 1.2 Hz, 1H), 8.69 (d, J = 4.8 Hz, 1H), 8.26 (dd, J = 8.8, 1.6 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.56 (dd, J = 8.4, 4.0 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 151.4, 151.3, 150.5, 144.7, 143.9, 140.7, 137.8, 125.2, 124.3. ESI-MS (+): m/z 165.28 [M+H]+.
4-((4-Methoxybenzyl)thio)-1,5-naphthyridine (8d)
To a solution of 8c (900 mg, 5.47 mmol) in DMF (30 mL) was added (4-Methoxyphenyl)methanethiol (p-MBSH, 1.1 mL, 8.20 mmol) at room temperature. The solution was stirred for 2 h, then quenched with MeOH and concentrated in vacuo. The resulting residue was diluted with H2O and neutralized with 1N HCl to pH ~ 8. The aqueous solution was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude material was purified via silica gel chromatography eluting a gradient of 25 to 70% EtOAc in Hexanes. Yield = 1.07 g (70%). 1H NMR (400 MHz, CDCl3): δ 8.90 (dd, J = 4.4, 1.6 Hz, 1H), 8.69 (d, J = 4.8 Hz, 1H), 8.33 (dd, J = 8.4, 1.6 Hz, 1H), 7.63 (dd, J = 8.4, 4.8 Hz, 1H), 7.39 (d, J = 9.2 Hz, 2H), 7.35 (d, J = 4.8 Hz, 1H), 6.86 (dd, J = 8.8 Hz, 2H), 4.24 (s, 2H), 3.77 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.2, 151.4, 150.1, 149.5, 142.6, 141.7, 137.7, 130.1, 130.1, 126.9, 125.0, 118.1, 114.3, 55.4, 34.8. ESI-MS (+): m/z 283.05 [M+H]+.
1,5-Naphthyridine-4-thiol (8e)
To a solution of 8d (0.7 g, 2.48 mmol) in TFA (20 mL) was added m-Cresol (1.3 mL, 12.41 mmol) at room temperature. The solution was then stirred at reflux for 16 h and then allowed to cool. The resulting reaction mixture was concentrated and diluted with the EtOAc. The solution was neutralized with sat. NaHCO3, which resulted in the formation of an orange red precipitate. The precipitate was collected via vacuum filtration and washed with H2O and Acetone to yield the desired product. Yield = 0.38 g, (94%). 1H NMR (400 MHz, CDCl3): δ 8.70-8.63 (m, 1H), 8.03 (dd, J = 7.8, 2.2 Hz, 1H), 7.95 (d, J = 4.8 Hz, 1H), 7.46 (dd, J = 8.2, 4.2 Hz, 1H), 7.41 (d, J = 5.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 174.2, 148.4, 147.2, 146.7, 143.4, 137.42, 128.2, 122.8. ESI-MS (+): m/z 163.19 [M+H]+.
8-Fluoro-2-methylquinoline (9a)
To a solution of 2-Fluoroaniline (1 g, 9 mmol) in Toluene (40 mL) was added 6M HCl (12 mL) and Crotonaldehyde (1.47 mL, 1.8 mmol). The heterogeneous mixture was stirred at 110 °C for 2 h. The aqueous layer was separated, neutralized to pH 9, and extracted with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.71 g (49%). 1H NMR (400 MHz, DMSO-d6): δ 8.29 (d, J = 9.5 Hz, 1H), 7.71 (d, J = 7.0 Hz, 1H), 7.56 – 7.40 (m, 3H), 2.66 (s, 3H). ESI-MS (+): m/z 162.2 [M+H]+.
8-(tert-Butylthio)-2-methylquinoline (9b)
To a solution of 9a (0.195 g, 1.21 mmol) in DMF (20 mL) was added NaH (0.097 g, 4.04 mmol) and t-BuSH (0.272 mL, 2.42 mmol) under nitrogen atmosphere. The solution was stirred at 140 °C for 18 h. The reaction mixture was evaporated to dryness and the crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.22 g (77%). 1H NMR (400 MHz, CDCl3): δ 8.74 (d, J = 8.3 Hz, 1H), 8.27 (dd, J = 7.2, 1.3 Hz, 1H), 8.17 – 8.04 (m, 1H), 7.81 (t, J = 7.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 3.59 (s, 3H), 1.43 (s, 9H). ESI-MS(+): m/z 231.91 [M+H]+.
2-Methylquinoline-8-thiol (9c)
A solution of 9b (0.04 g, 0.17 mmol) in conc. HCl (11 mL) was stirred at 90 °C for 19 h. The solution was neutralized to pH 9-10 and extracted EtOAc (3×10mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was recrystallized from EtOH. Yield = 0.02 g (66%). 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.41 – 7.30 (m, 2H), 2.85 (s, 3H). APCI-MS(-): m/z 174.10 [M-H]-.
8-Fluoro-3-methylquinoline (10a)
To a solution of 2-Fluoroaniline (1.0 g, 9 mmol) in Toluene (40 mL) was added 6M HCl (12 mL) and Methacrolein (1.5 mL, 1.8 mmol). The heterogeneous mixture was stirred at 110 °C for 2.5 h. The aqueous layer was separated, neutralized to pH 9 and extracted with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.65 g (45%). 1H NMR (500 MHz, CDCl3): δ 8.53 (d, J = 2.3 Hz, 1H), 7.56 (s, 1H), 7.22 – 7.17 (m, 1H), 7.17 – 7.10 (m, 1H), 7.08 – 6.98 (m, 1H), 2.22 – 2.21 (s, 3H). ESI-MS(+): m/z 162.19 [M+H]+.
8-(tert-Butylthio)-3-methylquinoline (10b)
To a solution of 10a (0.5 g, 3.1 mmol) in DMF (50 mL) was added NaH (0.25 g, 10.3 mmol) and t-BuSH (0.698 mL, 6.2 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 140 °C for 18 h. The resulting solution was evaporated to dryness and the crude material purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.56 g (78%). 1H NMR (500 MHz, CDCl3): δ 8.90 (d, J = 2.3 Hz, 1H), 7.96 (dd, J = 7.2, 1.5 Hz, 1H), 7.92 (d, J = 1.1 Hz, 1H), 7.74 (dd, J = 8.2, 1.5 Hz, 1H), 7.47 (dd, J = 8.1, 7.2 Hz, 1H), 2.53 (s, 3H), 1.37 (s, 9H). ESI-MS(+): m/z 231.92 [M+H]+.
3-Methylquinoline-8-thiol (10c)
A solution of 10b (0.08 g, 0.35 mmol) in conc. HCl (25 mL) was stirred at 90 °C for 19 h. The reaction mixture was neutralized to pH 9 with NaOH and extracted with EtOAc (3×10mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.02 g (33%). 1H NMR (500 MHz, CDCl3): δ 8.73 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.60 (d, J = 8.0 Hz, 1H), 7.47 – 7.45 (m, 1H), 7.35 – 7.31 (m, 1H), 5.58 (s, 1H), 2.48 (s, 3H). ESI-MS(+): m/z 176.16 [M+H]+.
8-Fluoro-4-methylquinoline (11a)
To a solution of 2-Fluoroaniline (1.0 g, 9 mmol) in Toluene (40 mL) was added 6M HCl (12 mL) and Methyl Vinyl ketone (1.5 mL, 1.8 mmol). The heterogeneous mixture was stirred at 110 °C for 16 h. The aqueous layer was separated, neutralized to pH 9 with 6M NaOH and extracted with CH2Cl2 (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.4 g (28%). 1H NMR (500 MHz, DMSO-d6): δ 8.74 (d, J = 4.4 Hz, 1H), 7.83 – 7.77 (m, 1H), 7.57 – 7.46 (m, 2H), 7.39 (dd, J = 4.3, 1.0 Hz, 1H), 2.61 (s, 3H). ESI-MS(+): m/z 162.23 [M+H]+.
8-(tert-Butylthio)-4-methylquinoline (11b)
To a solution of 11a (255 mg, 1.58 mmol) in DMF (25 mL) was added NaH (0.13 g, 5.29 mmol) and t-BuSH (0.356 mL, 3.16 mmol) under nitrogen atmosphere. The solution was stirred at 140 °C for 18 h. The reaction mixture was evaporated to dryness and the crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.2 g (55%). 1H NMR (500 MHz, CDCl3): δ 8.88 (d, J = 4.3 Hz, 1H), 8.05 – 7.93 (m, 2H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.22 (dd, J = 4.3, 1.0 Hz, 1H), 2.69 (s, 3H), 1.36 (s, 9H). ESI-MS(+): m/z 231.90 [M+H]+.
4-Methylquinoline-8-thiol (11c)
A solution of 11b (0.08 g, 0.35 mmol) in conc. HCl (25 mL) was stirred at 90 °C for 19 h. The reaction mixture was neutralized to pH 9 with NaOH and extracted with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.02 g (30%). 1H NMR (500 MHz, CDCl3): δ 8.76 (d, J = 4.4 Hz, 1H), 7.76 – 7.66 (m, 2H), 7.39 (dd, J = 8.4, 7.3 Hz, 1H), 7.25 (dd, J = 4.4, 1.0 Hz, 1H), 2.69 (s, 3H). ESI-MS(+): m/z 176.17 [M+H]+.
8-Chloro-5-methylquinoline (12a)
To a solution of 2-Chloro-5-methylaniline (1 g, 14.1 mmol) in 75% Sulfuric acid (8 mL) was added Nitrobenzene (1.44 mL, 14.1 mmol) and Glycerol (2.06 mL, 28.2 mmol). The heterogeneous mixture was stirred at 150 °C for 2 h. This was allowed to cool, then H2O was added to the mixture and extracted with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 1.0 g (40%). 1H NMR (500 MHz, CDCl3): δ 9.04 (dd, J = 4.2, 1.7 Hz, 1H), 8.32 (dd, J = 8.5, 1.7 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.49 (dd, J = 8.5, 4.2 Hz, 1H), 7.28 (dd, J = 7.8, 0.9 Hz, 1H), 2.65 (d, J = 1.0 Hz, 3H). ESI-MS(+): m/z 178.21 [M+H]+.
8-(tert-Butylthio)-5-methylquinoline (12b)
To a solution of 12a (1 g, 5.62 mmol) in DMF (100 mL) was added NaH (0.45 g, 18.8 mmol) and t-BuSH (1.26 mL, 3.16 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 140 °C for 18 h. The resulting solution was evaporated to dryness and the crude material purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.19 g (14%). 1H NMR (500 MHz, CDCl3): δ 9.05 (dd, J = 4.2, 1.7 Hz, 1H), 8.34 (dd, J = 8.5, 1.7 Hz, 1H), 7.72 (d, J = 7.6 Hz, 1H), 7.50 (dd, J = 8.5, 4.2 Hz, 1H), 7.29 (dd, J = 7.6, 1.0 Hz, 1H), 2.66 (d, J = 1.0 Hz, 3H), 1.34 (s, 9H). ESI-MS(+): m/z 231.91 [M+H]+.
5-Methylquinoline-8-thiol (12c)
A solution of 12b (0.08 g, 0.35 mmol) in conc. HCl (25 mL) was stirred at 100 °C for 19 h. The resulting solution was neutralized to pH 9 with NaOH and extracted with EtOAc (3×50mL). The combined organic layers were dried and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.05 g (81%). 1H NMR (500 MHz, CDCl3): δ 8.94 (dd, J = 4.3, 1.8 Hz, 1H), 8.09 (dd, J = 8.3, 1.8 Hz, 1H), 7.79 (d, J = 1.8 Hz, 1H), 7.45 (dd, J = 8.3, 4.3 Hz, 1H), 7.38 (s, 1H), 2.39 (s, 3H). ESI-MS(+): m/z 176.00 [M+H]+.
8-Fluoro-6-methylquinoline (13a)
To a solution of 2-Fluoro-6-methylaniline (0.5 g, 4.0 mmol) in 75% Sulfuric acid (4 mL) was added Nitrobenzene (0.409 mL, 4.0 mmol) and Glycerol (588 mL, 8.0 mmol). The heterogeneous mixture was stirred at 150 °C for 3 h, then allowed to cool to room temperature. H2O was added to the reaction mixture and with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.17 g (27%). 1H NMR (500 MHz, CDCl3): δ 8.79 (dd, J = 4.2, 1.6 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.92 (s, 1H), 7.34 (dd, J = 8.4, 4.2 Hz, 1H), 7.15 (dd, J = 11.5, 1.8 Hz, 1H), 2.43 (d, J = 1.0 Hz, 3H). ESI-MS(+): m/z 162.19 [M+H]+.
8-(tert-Butylthio)-6-methylquinoline (13b)
To a solution of 13a (0.14 g, 0.87 mmol) in DMF (14 mL) was added NaH (0.07 g, 2.91 mmol) and t-BuSH (0.196 mL, 1.74 mmol) under nitrogen atmosphere. The solution was stirred at 140 °C for 18 h. The resulting solution was evaporated to dryness and the crude material purified via silica gel column chromatography using a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.16 g (78% yield). 1H NMR (500 MHz, CDCl3): δ 8.98 (dd, J = 4.2, 1.8 Hz, 1H), 8.06 (dd, J = 8.2, 1.8 Hz, 1H), 7.89 (d, J = 2.0 Hz, 1H), 7.58 (d, J = 1.0 Hz, 1H), 7.37 (dd, J = 8.2, 4.2 Hz, 1H), 2.54 (s, 3H), 1.37 (s, 9H). ESI-MS(+): m/z 231.91 [M+H]+.
6-Methylquinoline-8-thiol (13c)
A solution of 13b (0.08 g, 0.35 mmol) in conc. HCl (25 mL) was stirred at 100 °C for 19 h. The crude material was neutralized to pH 9 with NaOH and extracted with EtOAc (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.03 g (46%). 1H NMR (400 MHz, CDCl3): δ 8.84 (dd, J = 4.3, 1.6 Hz, 1H), 8.03 (dd, J = 8.2, 1.6 Hz, 1H), 7.55 (d, J = 1.8 Hz, 1H), 7.38 (dd, J = 8.2, 4.2 Hz, 1H), 7.31 (d, J = 0.9 Hz, 1H), 5.61 (s, 1H), 2.46 (s, 3H). ESI-MS(+): m/z 176.17 [M+H]+.
8-(tert-Butylthio)quinoline-2-carboxylic acid (14a)
To a solution of 8-Fluoroquinoline-2-carboxylic acid (0.42 g, 2.19 mmol) in DMF (40 mL) was added NaH (0.18 g, 7.29 mmol) and t-BuSH (0.495 mL, 4.4 mmol) under nitrogen atmosphere. The solution was stirred at 140 °C for 18 h. The reaction mixture was evaporated to dryness and the crude material was taken in H2O and acidified with 1M HCl until a precipitate was formed (pH 2). The precipitate was filtered and dried under vacuum. Yield = 0.45 g (78%). 1H NMR (400 MHz, CDCl3): δ 8.42 (dd, J = 8.5, 2.4 Hz, 1H), 8.32 (dd, J = 6.6, 3.7 Hz, 1H), 8.12 (d, J = 6.8 Hz, 1H), 7.94 (d, J = 7.2 Hz, 1H), 7.70 – 7.62 (m, 1H), 1.35 – 1.30 (m, 9H). ESI-MS(+): m/z 261.96 [M+H]+.
8,8′-Disulfanediylbis(quinoline-2-carboxylic acid) (14b)
A solution of 14a (0.24 g, 0.92 mmol) in conc. HCl (40 mL) was stirred at 110 °C for 12 h. The solution was neutralized to pH 9 and washed with EtOAc (3×50mL). The aqueous layer was then acidified to pH 2-3 and the precipitate was collected via vacuum filtration. The product was isolated as a disulfide dimer as evidenced by mass spectrometry. Yield = 0.15 g (80%). 1H NMR (400 MHz, DMSO-d6): δ 8.61 (d, J = 8.5 Hz, 1H), 8.21 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H). ESI-MS (-): m/z 407.05 [M-H] -.
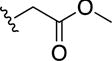
Dimethyl 8,8′-disulfanediylbis(quinoline-2-carboxylate) (15)
In a 10 mL microwave tube was placed 14b (0.02 g, 0.97 mmol) and MeOH (2 mL), followed by 15 drops of conc. H2SO4. The solution was placed in a microwave reactor and heated to 90 °C with stirring for 24 min. The solution was evaporated to dryness and the crude material was taken up in CHCl3 and washed with a sat. NaHCO3 (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Yield = 0.02 g (84%). 1H NMR (400 MHz, CDCl3): δ 8.33 (dd, J = 8.7, 1.4 Hz, 1H), 8.26 (dd, J = 8.5, 1.4 Hz, 1H), 7.95 (d, J = 7.5 Hz, 1H), 7.68 (d, J = 8.2 Hz, 1H), 7.47 (td, J = 7.8, 1.5 Hz, 1H), 4.10 (s, 3H). ESI-MS(+): m/z 437.16 [M+H]+.
8-(tert-Butylthio)quinoline-3-carboxylic acid (16a)
To a solution of 8-Fluoroquinoline-3-carboxylic acid (1 g, 5.2 mmol) in DMF (40 mL) was added NaH (0.5 g, 20.8 mmol) and t-BuSH (2.35 mL, 20.8 mmol) under nitrogen atmosphere. The reaction mixture was stirred at 140 °C for 18 h. The solution was evaporated to dryness and the crude material was taken up in H2O and acidified with 6M HCl until a precipitate was formed (pH 2). The precipitate was filtered and dried under vacuum. Yield = 1.47 g (100%). 1H NMR (400 MHz, DMSO-d6): δ 9.35 (d, J = 2.0 Hz, 1H), 8.96 (d, J = 1.9 Hz, 1H), 8.20 – 8.15 (m, 1H), 8.10 (d, J = 7.2 Hz, 1H), 7.67 (dd, J = 8.2, 7.2 Hz, 1H), 1.30 (s, 9H). ESI-MS(+): m/z 261.97 [M+H]+.
8,8′-Disulfanediylbis(quinoline-3-carboxylic acid) (16b)
A solution of 16a (0.6 g, 2.3 mmol) in conc. HCl (50 mL) was stirred at 110 °C for 12 h. The reaction mixture was neutralized to pH 9 and washed with EtOAc (3×50mL). The aqueous layer was then acidified to pH 2-3 and the observed precipitate collected via vacuum filtration. The crude material was recrystallized from EtOH. The product was isolated as a disulfide dimer as evidenced by mass spectrometry. Yield = 0.18 g (38%). 1H NMR (400 MHz, DMSO-d6): δ 9.38 (d, J = 1.6 Hz, 1H), 9.02 (d, J = 1.6 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H). ESI-MS(+): m/z 409.01 [M+H]+.
Dimethyl 8,8′-disulfanediylbis(quinoline-3-carboxylate) (17)
In a 10 mL microwave tube was placed 16b (0.020 g, 0.97 mmol) and MeOH (2 mL) followed by 15 drops of conc. H2SO4. The solution was placed in a microwave reactor and heated to 90 °C with stirring for 20 min. The solution was evaporated to dryness and the crude material was taken up in CHCl3 and washed with a sat. solution of NaHCO3 (3×50mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was recrystallized from EtOH. Yield = 0.004 g (19%). 1H NMR (400 MHz, DMSO-d6): δ 9.41 (d, J = 2.0 Hz, 1H), 9.10 (d, J = 2.0 Hz, 1H), 8.08 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.69 – 7.59 (m, 1H), 3.98 (s, 3H). ESI-MS(+): m/z 437.11 [M+H]+.
8-Fluoroquinoline-4-carboxylic acid (18b)
To a solution of 7-Fluoroisatin (0.5 g, 3.03 mmol) in H2O (10 mL) in a 35 mL microwave tube was added 5M NaOH (2.52 mL, 15.1 mmol) and Sodium Pyruvate (0.4 g, 3.66 mmol). The mixture was placed in a microwave reactor and heated to 110 °C with stirring for 10 min. After cooling to room temperature, the suspension containing the dicarboxylic acid derivative was acidified to pH 2 and the dark solid was filtered off to afford 18a. A portion of 18a (0.17 g) was then placed in a 10 mL microwave tube and H2O (2 mL) was added. The resulting suspension was placed in a microwave reactor and heated to 170 °C (or 280 psi) with stirring for 5 min. The brown solid was collected via vacuum filtration. Yield = 60% over 2 steps. 1H NMR (400 MHz, DMSO-d6): δ 9.07 (d, J = 4.1 Hz, 1H), 8.48 (d, J = 7.4 Hz, 1H), 8.00 (d, J = 4.1 Hz, 1H), 7.73 – 7.62 (m, 2H). ESI-MS(+): m/z 192.27 [M+H]+.
8-(tert-Butylthio)quinoline-4-carboxylic acid (18c)
To a solution of 18b (0.3 g, 1.57 mmol) in DMF (30 mL) was added NaH (0.15 g, 6.3 mmol) and t-BuSH (0.707 mL, 36.3 mmol) under nitrogen atmosphere. The mixture was stirred at 140 °C for 18 h. The solution was evaporated to dryness and the crude material was taken up in H2O and acidified with HCl until a precipitate formed (pH 2). The precipitate was collected via vacuum filtration and discarded. The filtrate was extracted with EtOAc (3×50mL), dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. H2O was then added to the crude material resulting in a yellow precipitate. The precipitate was collected by vacuum filtration. Yield = 0.2 g (49% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.07 (d, J = 4.1 Hz, 1H), 8.60 (d, J = 8.6 Hz, 1H), 8.05 (d, J = 7.1 Hz, 1H), 7.90 (d, J = 4.3 Hz, 1H), 7.68 (t, J = 7.9 Hz, 1H), 1.30 (s, 9H). ESI-MS(+): m/z 261.93 [M+H]+.
8,8′-Disulfanediylbis(quinoline-4-carboxylic acid) (18d)
A solution of 18c (0.2 g, 0.76 mmol) in conc. HCl (18 mL) was stirred at 110 °C for 12 h. The crude material was neutralized to pH 9 and washed EtOAc (3×10mL). The aqueous layer was then acidified to pH 2-3 with HCl and the resulting precipitate was collected via vacuum filtration. The product was isolated as a disulfide dimer as evidenced by mass spectrometry. Yield = 0.09 g (58% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.13 (d, J = 4.5 Hz, 1H), 8.49 (d, J = 8.5 Hz, 1H), 8.05 (d, J = 4.4 Hz, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H). ESI-MS(-): m/z 406.96 [M-H] -.
Dimethyl 8,8′-disulfanediylbis(quinoline-4-carboxylate) (19)
In a 10 mL microwave tube was placed 18d (0.02 g, 0.97 mmol) and MeOH (2 mL), followed by 15 drops of conc. H2SO4. The reaction mixture was placed in a microwave reactor and heated to 90 °C with stirring for 20 min. The solution was evaporated to dryness and the crude material was taken up in CHCl3 and washed with a sat. solution of NaHCO3 (3×50mL). The collected organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was recrystallized from EtOH. Yield = 0.02 g (100% yield). 1H NMR (400 MHz, CDCl3): δ 8.99 (dd, J = 4.2, 1.7, 0.8 Hz, 1H), 8.12 (dd, J = 8.3, 1.7, 0.8 Hz, 1H), 7.89 (d, J = 8.8 Hz, 1H), 7.53 (dd, J = 8.3, 4.2 Hz, 1H), 7.27 (d, J = 1.8 Hz, 1H), 4.04 (d, J = 0.8 Hz, 3H). ESI-MS(+): m/z 437.02 [M+H]+.
8,8′-Disulfanediylbis(N-(thiophen-2-ylmethyl)quinoline-2-carboxamide) (20)
To a solution of 14b (0.05 g, 0.24 mmol) in dry CH2Cl2 (2 mL) was added Oxalyl Chloride (0.320 mL, 2.90 mmol) and 12 drops of dry DMF under nitrogen atmosphere. The solution was stirred at room temperature for 2 h. The solution was then evaporated to dryness to remove the excess of Oxalyl Chloride. The resulting acyl chloride solution was then added to a solution of 2-Thiophenemethylamine (0.300 mL, 2.9 mmol) in dry CH2Cl2 (6 mL) under nitrogen atmosphere and stirred at room temperature for 18 h. The solution was then washed with 1M HCl to remove excess of amine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.06 g (79%). 1H NMR (400 MHz, CDCl3): δ 8.59 (t, J = 6.2 Hz, 1H, NH), 8.45 – 8.40 (m, 1H), 8.39 – 8.32 (m, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.72 – 7.66 (m, 1H), 7.51 – 7.41 (m, 1H), 7.25 – 7.23 (m, 1H), 7.15 – 7.11 (m, 1H), 7.03 – 6.95 (m, 1H), 4.95 (d, J = 5.8 Hz, 2H). ESI-MS(+): m/z 598.93 [M+H]+, 621.03 [M+Na]+.
8,8′-Disulfanediylbis(N-benzylquinoline-2-carboxamide) (21)
To a solution of 14b (0.05 g, 0.24 mmol) in dry CH2Cl2 (2 mL) was added Oxalyl Chloride (0.42 mL, 0.48 mmol) and 5 drops of dry DMF under nitrogen atmosphere. The solution was stirred at room temperature for 2 h. The solution was evaporated to dryness to remove the excess of Oxalyl Chloride. The resulting acyl chloride solution was then added to a solution of Benzylamine (0.319 mL, 2.9 mmol) in dry CH2Cl2 (5 mL) under nitrogen atmosphere and the mixture was stirred at room temperature for 18 h. The solution was then washed with 1M HCl to remove excess of Benzylamine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.05 g (74%). 1H NMR (400 MHz, CDCl3): δ 8.60 (t, J = 6.2 Hz, 1H, NH), 8.43 (d, J = 8.5 Hz, 1H), 8.36 (d, J = 8.6 Hz, 1H), 7.86 (dd, J = 7.5, 1.2 Hz, 1H), 7.69 (dd, J = 8.2, 1.2 Hz, 1H), 7.49 – 7.43 (m, 3H), 7.40 – 7.34 (m, 2H), 7.30 (d, J = 7.2 Hz, 1H), 4.80 (d, J = 6.2 Hz, 2H). ESI-MS(+): m/z 587.08 [M+H]+, 609.11 [M+Na]+.
8,8′-Disulfanediylbis(N-(thiophen-2-ylmethyl)quinoline-4-carboxamide) (22)
To a solution of 18d (0.05 g, 0.24 mmol) in dry CH2Cl2 (2 mL) was added Oxalyl Chloride (0.640 mL, 5.76 mmol) and 15 drops of dry DMF under nitrogen atmosphere. The solution was stirred at room temperature for 2 h. The solution was evaporated to dryness to remove the excess of Oxalyl Chloride and dry CH2Cl2 was added to the crude material (2 mL). The resulting acyl chloride solution was added to a solution of 2-Thiophenemethylamine (0.600 mL, 5.76 mmol) in dry CH2Cl2 (10 mL) under nitrogen atmosphere and stirred at room temperature for 2 days. The solution was then washed with 1M HCl to remove excess of amine, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude material was purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.03 g (41% yield). 1H NMR (400 MHz, DMSO-d6): δ 9.47 (d, J = 5.9 Hz, 1H), 9.08 (d, J = 3.9 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 4.2 Hz, 1H), 7.59 – 7.51 (m, 1H), 7.43 (d, J = 5.1 Hz, 1H), 7.07 (d, J = 3.3 Hz, 1H), 6.98 (s, 1H), 4.70 (d, J = 5.8 Hz, 2H). ESI-MS(+): m/z 599.05 [M+H]+.
8,8′-Disulfanediylbis(N-(furan-2-ylmethyl)quinoline-4-carboxamide) (23)
To a solution of 18d (0.2 g, 0.98 mmol) in DMF (10 mL) was added Carbonyldimidazole (CDI, 0.24 g, 1.46 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this reaction mixture was added Furan-2-ylmethanamine (0.146 mmol) and stirred for an additional 12 h. The resulting solution was concentrated under reduced pressure, then purified via silica gel column chromatography eluting a gradient of 0 to 100% EtOAc in Hexanes. Yield = 0.15 g (54%). 1H NMR (400 MHz, DMSO-d6): δ 9.33 (t, J = 5.6 Hz, 1H), 9.08 (d, J = 4.4 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.76 (d, J = 7.6 Hz, 1H), 7.69-7.61 (m, 2H), 7.57 (t, J = 8.0 Hz, 1H), 6.43-6.36 (m, 2H), 4.55 (d, J = 5.6 Hz, 2H). ESI-MS(+): m/z 566.17 [M+H]+.
8,8′-Disulfanediylbis(quinoline-3-carboxamide) (24)
To a solution of 16b (0.05g, 0.24 mmol) in DMF (5 mL) was added CDI (0.06g, 0.37 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this was added NH4OH (0.305g, 2.44 mmol) and allowed to stir for 1 h. The resulting solution was concentrated under reduced pressure and purified via reverse-phase chromatography eluting a gradient of 0 to 100% Acetonitrile in H2O. Yield = 0.016 g (32%). 1H NMR (400 MHz, DMSO-d6): δ 9.37 (d, J = 2.0 Hz, 1H), 8.91 (d, J = 2.0 Hz, 1H), 8.38 (b, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.82 (m, 2H), 7.61 (t, J = 8.0 Hz, 1H). HR-ESI-MS calcd for [C20H15N4O2S2]+: 407.0631; Found: 407.0637.
8,8′-Disulfanediylbis(N-methylquinoline-3-carboxamide) (25)
To a solution of 16b (0.05 g, 0.24 mmol) in DMF (5 mL) was added Hydroxybenzotriazole (HOBT, 0.06g, 0.37 mmol) and EDC (Ethyl-3-(3-dimethylaminopropyl)carbodiimide, 0.07g, 0.37 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this reaction mixture was added Methylamine (1M THF solution, 0.49 mmol) and allowed to stir for 1 h. The resulting solution was concentrated under reduced pressure and purified via reverse-phase chromatography eluting a gradient of 0 to 100% Acetonitrile in H2O. Yield = 0.018 g (34%). 1H NMR (400 MHz, DMSO-d6): δ 9.35 (s, 1H), 8.89 (m, 2H), 7.95 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 2.84 (s, 3H). HR-ESI-MS calcd for [C22H19N4O2S2]+: 435.0944; Found: 435.0947.
Dimethyl 2,2′-((8,8′-disulfanediylbis(quinoline-8,3-diyl-3-carbonyl))bis(azanediyl))diacetate (26)
To a solution of 16b (0.04 g, 0.17 mmol) in DMF (4 mL) was added HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxidhexafluorophosphate, 0.08 g, 0.21 mmol), HOBT (0.03 g, 0.21 mmol), Et3N (0.073 mL, 0.52 mmol) and Methyl 2-aminoacetate (24 mg, 0.19 mmol) and allowed to stir at room temperature for 1 h. The resulting solution was concentrated under reduced pressure, then the crude material was then dissolved in CH2Cl2 and washed with 1M HCl solution. The product, which precipitated out of the organic layer, was recrystallized from MeOH. Yield = 0.005 g (11%). 1H NMR (400 MHz, DMSO-d6): δ 9.42 (t, J = 5.9 Hz, 1H, NH), 9.37 (s, 1H), 8.93 (s, 1H), 7.97 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.62 (t, J = 7.7 Hz, 1H), 4.13 (d, J = 5.7 Hz, 2H), 3.68 (s, 3H). HR-ESI-MS calcd for [C26H23N4O6S2]+: 551.1054; Found: 551.1052.
Procedure for the amide coupling (Method A)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added CDI (0.24 g, 1.46 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. The resulting solution was concentrated under reduced pressure, then purified via reverse-phase chromatography eluting a gradient of 0 to 100% Acetonitrile in H2O.
Procedure for the amide coupling (Method B)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added HATU (0.56 g, 1.46 mmol) and Et3N (0.204 mL, 1.46 mmol) and the mixture was stirred at 60° C for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. The resulting solution was concentrated under reduced pressure, then purified via reverse-phase chromatography eluting a gradient of 0 to 100% Acetonitrile in H2O.
8,8′-Disulfanediylbis(N-(oxazol-2-yl)quinoline-3-carboxamide) (27)
Product afforded via Method B. Yield = 0.13 g (48%). 1H NMR (400 MHz, DMSO-d6): δ 9.47 (s, 1H), 9.06 (d, J = 1.6 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 7.93 (s, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.25 (s, 1H). HR-ESI-MS calcd for [C26H17N6O4S2]+ : 541.0747; Found: 541.0749.
8,8′-Disulfanediylbis(N-(thiazol-2-yl)quinoline-3-carboxamide) (28)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added HATU (0.56 g, 1.46 mmol) and Et3N (0.204 mL, 1.46 mmol) and the mixture was stirred at 60° C for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. To the resulting solution was added H2O, which resulted in the formation of a precipitate. The precipitate was isolated through vacuum filtration to afford desired product. Yield = 0.12 g (43%). 1H NMR (400 MHz, DMSO-d6): δ 9.51 (s, 1H), 9.15 (s, 1H), 7.98 (d, J = 8.0 Hz,1H), 7.88 (d, J = 8.0 Hz, 1H), 7.64-7.59 (m, 2H), 7.32 (d, J = 2.4 Hz, 1H). 13C NMR (100 MHz, DMSO- d6): δ 164.5, 159.6, 149.2, 146.6, 138.2, 137.7, 135.0, 128.7, 127.8, 127.4, 127.0, 126.8, 114.7. HR-ESI-MS calcd for [C26H16N6O2S4Na]+: 595.0110; Found: 595.0103.
8,8′-Disulfanediylbis(N-((tetrahydrofuran-2-yl)methyl)quinoline-3-carboxamide) (29)
Product afforded via Method A. Yield = 0.14 g (51%). 1H NMR (400 MHz, DMSO-d6): δ 9.43 (d, J = 2 Hz, 1H), 8.88 (d, J = 2 Hz, 1H), 7.94-7.90 (m, 3H), 7.60 (t, J = 8 Hz, 1H), 4.13-3.47 (m, 5H), 2.09-1.67 (m, 4H). HR-ESI-MS calcd for [C30H31N4O4S2]+ : 575.1781; Found: 575.1780.
8,8′-Disulfanediylbis(N-(furan-2-ylmethyl)quinoline-3-carboxamide) (30)
Product afforded via Method A. Yield = 0.084 g (30%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (d, J = 2.4 Hz, 1H), 8.99 (d, J = 2.4 Hz, 1H), 8.50 (br, 1H), 8.35 (dd, J = 7.2, 1.2 Hz, 1H), 8.28 (dd, J = 7.2, 1.2 Hz, 1H), 7.84 (t, J = 7.6 Hz, 1H), 7.50 (dd, J = 1.6, 0.8 Hz, 1H), 6.40 (m, 2H), 4.67 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C30H22N4O4S2Na]+: 589.0975; Found: 589.0972.
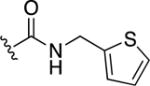
8,8′-Disulfanediylbis(N-(thiophen-2-ylmethyl)quinoline-3-carboxamide) (31)
Product afforded via Method A. Yield = 0.13 g (44%). 1H NMR (400 MHz, DMSO-d6): δ 9.58 (t, J = 5.6 Hz, 1H), 9.37 (d, J = 2.4 Hz, 1H), 8.91 (d, J = 2.4 Hz, 1H), 7.96 (d, J = 8 Hz, 1H), 7.84 (d, J = 8 Hz, 1H), 7.62 (t, J = 8 Hz, 1H), 7.42 (d, J = 6 Hz, 1H), 7.09-6.97 (m, 2H), 4.73 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C30H23N4O2S4]+: 599.0698; Found: 599.0701.
8,8′-Disulfanediylbis(N-(thiazol-2-ylmethyl)quinoline-3-carboxamide) (32)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added CDI (0.24 g, 1.46 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. To the reaction mixture was added H2O, which resulted in the formation of a precipitate. The precipitate was isolated through vacuum filtration to afford final product. Yield = 0.035 g (12%). 1H NMR (400 MHz, DMSO-d6): δ 9.83 (t, J = 5.6 Hz, 1H), 9.40 (d, J = 2 Hz, 1H), 8.95 (d, J = 2 Hz, 1H), 7.97 (d, J = 8 Hz, 1H), 7.86 (d, J = 8 Hz, 1H), 7.76 (d, J = 3.2 Hz, 1H), 7.66 (d, J = 3.2 Hz, 1H), 7.63 (t, J = 8 Hz, 1H), 4.86 (d, J = 6 Hz, 2H). HR-ESI-MS calcd for [C28H21N6O2S4]+: 601.0603; Found: 601.0600.
8,8′-Disulfanediylbis(N-(2-(furan-2-yl)ethyl)quinoline-3-carboxamide) (33)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added CDI (0.24 g, 1.46 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. The reaction solution was concentrated and diluted with Diethyl Ether, which resulted in the formation of a precipitate. The precipitate was isolated through vacuum filtration and purified via reverse-phase chromatography eluting a gradient of 0 to 100% Acetonitrile in H2O. Yield = 0.065 g (22%). 1H NMR (400 MHz, DMSO-d6): δ 9.32 (d, J = 2 Hz, 1H), 9.04 (t, J = 5.2 Hz, 1H), 8.84 (d, J = 2 Hz, 1H), 7.94 (d, J = 8 Hz, 1H), 7.83 (d, J = 8 Hz, 1H), 7.61-7.54 (m, 2H), 6.36 (t, J = 2.8 Hz, 1H), 6.21 (d, J = 2.8 Hz, 1H), 3.62 (q, J = 6 Hz, 2H), 2.96 (q, J = 7.2 Hz, 2H). HR-ESI-MS calcd for [C32H26N4O4S2Na]+: 617.1288; Found: 617.1283.
8,8′-Disulfanediylbis(N-(2-(thiophen-2-yl)ethyl)quinoline-3-carboxamide) (34)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added CDI (0.24 g, 1.46 mmol) and stirred at room temperature for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. To the resulting solution was added H2O, which resulted in the formation of a precipitate. The precipitate was isolated through vacuum filtration and further purified via reverse-phase chromatography using a gradient of 0 to 100% Acetonitrile in H2O. Yield = 0.075 g (24%). 1H NMR (400 MHz, DMSO-d6): δ 9.35 (s, 1H), 9.10 (t, J = 5.2 Hz, 1H), 8.87 (s, 1H), 7.95 (d, J = 6.4 Hz, 1H), 7.84 (d, J = 6.4 Hz, 1H), 7.62 (t, J = 5.6 Hz, 1H), 7.34-6.96 (m, 3H), 3.60 (t, J = 6 Hz, 2H), 3.14 (t, J = 6 Hz, 2H). HR-ESI-MS calcd for [C32H27N4O2S4]+ : 627.1011; Found: 627.1013.
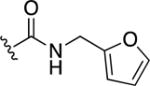
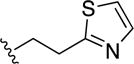
8,8′-Disulfanediylbis(N-(2-(thiazol-2-yl)ethyl)quinoline-3-carboxamide) (35)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added CDI (0.24 g, 1.46 mmol) and the mixture was stirred at room temperature for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.15 mmol) and the solution was stirred for an additional ~12 h. To the resulting solution was added H2O, which resulted in the formation of a precipitate. The precipitate was isolated through vacuum to yield the final product. Yield = 0.18 g (57%). 1H NMR (400 MHz, DMSO-d6): δ 9.33 (s, 1H), 9.11 (t, J = 5.2 Hz, 1H), 8.86 (s, 1H), 7.95 (d, J = 8 Hz, 1H), 7.84 (d, J = 8 Hz, 1H), 7.74 (d, J = 3.2 Hz, 1H), 7.62-7.58 (m, 2H), 3.74 (q, 2H), 3.34 (t, 2H). 13C NMR (100 MHz, DMSO- d6): δ 167.7, 163.3, 148.9, 146.3, 143.0, 136.6, 134.9, 128.6, 128.6, 127.6, 127.5, 126.3, 120.4, 40.1, 32.9. HR-ESI-MS calcd for [C30H25N6O2S4]+: 629.0916; Found: 629.0913.
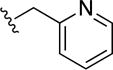
8,8′-Disulfanediylbis(N-(pyridin-2-ylmethyl)quinoline-3-carboxamide) (36)
Product afforded via Method B. Yield = 0.06 g (21%). 1H NMR (400 MHz, DMSO-d6): δ 9.52 (d, J = 2.4 Hz, 1H), 9.07 (d, J = 2.4 Hz, 1H), 8.77-8.58 (m, 2H), 8.36 (dd, J = 7.2, 1.2 Hz, 1H), 8.32 (dd, J = 7.2, 1.2 Hz, 1H), 7.86-7.82 (m, 2H), 7.55 (d, J = 8 Hz, 1H), 7.36 (t, J = 6 Hz, 3H), 4.82 (d, J = 6 Hz, 2H). HR-ESI-MS calcd for [C32H25N6O2S2]+: 589.1475; Found: 589.1478.
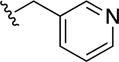
8,8′-Disulfanediylbis(N-(pyridin-3-ylmethyl)quinoline-3-carboxamide) (37)
Product afforded via Method B. Yield = 0.11 g (38%). 1H NMR (400 MHz, DMSO-d6): δ 9.46 (d, J = 2.4 Hz, 1H), 8.98 (d, J = 2.4 Hz, 1H), 8.70-8.52 (m, 2H), 8.34 (dd, J = 7.2, 1.2 Hz, 1H), 8.27 (dd, J = 7.2, 1.2 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.83 (t, J = 7.6 Hz, 1H), 7.43 (dd, J = 7.6, 4.8 Hz, 1H), 4.73 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C32H25N6O2S2]+: 589.1475; Found: 589.1477.
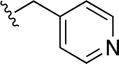
8,8′-Disulfanediylbis(N-(pyridin-4-ylmethyl)quinoline-3-carboxamide) (38)
Product afforded via Method B. Yield = 0.06 g (21%). 1H NMR (400 MHz, DMSO-d6): δ 9.88 (t, J = 5.6 Hz, 1H), 9.46 (d, J = 2.4 Hz, 1H), 9.03 (d, J = 2.4 Hz, 1H), 8.84-8.74 (m, 2H), 7.99 (d, J = 8 Hz, 1H), 7.92-7.91 (m, 2H), 7.87 (d, J = 8 Hz, 1H), 7.65 (t, J = 8 Hz, 1H), 4.80 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C32H25N6O2S2]+: 589.1475; Found: 589.1477.
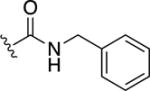
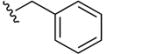
8,8′-Disulfanediylbis(N-benzylquinoline-3-carboxamide) (39)
To a solution of 16b (0.2 g, 0.98 mmol) in DMF (10 mL) was added HATU (0.56 g, 1.46 mmol) and Et3N (0.204 mL, 1.46 mmol) and the mixture was stirred at 60° C for ~15 min under nitrogen atmosphere. To this solution was added the corresponding amine (0.146 mmol) and the solution was stirred for an additional 12 h. The reaction mixture was concentrated and the crude material was dissolved in CH2Cl2. The product, which precipitated out of the organic layer, was collected via vacuum filtration. Yield = 0.03 g (42%). 1H NMR (400 MHz, DMSO-d6): δ 9.49 (t, J = 5.9 Hz, 1H,), 9.41 (d, J = 2.1 Hz, 1H), 8.95 (d, J = 2.2 Hz, 1H), 7.94 (s, 1H), 7.86-7.81 (m, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.41-7.32 (m, 4H), 7.26 (t, J = 7.1 Hz, 1H), 4.58 (d, J = 5.8 Hz, 2H). HR-ESI-MS calcd for [C34H27N4O2S2]+: 587.1570; Found: 587.1571.
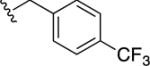
8,8′-Disulfanediylbis(N-(4-fluorobenzyl)quinoline-3-carboxamide) (40)
Product afforded via Method A. Yield = 0.04 g (14%). 1H NMR (400 MHz, DMSO-d6): δ 9.48 (t, J = 5.6 Hz, 1H), 9.39 (d, J = 1.6 Hz, 1H), 8.92 (d, J = 1.6 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 7.44-7.15 (m, 4H), 4.56 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C34H25F2N4O2S2]+: 623.1382; Found: 623.1384.
8,8′-Disulfanediylbis(N-(4-(trifluoromethyl)benzyl)quinoline-3-carboxamide) (41)
Product afforded via Method A. Yield = 0.09 g (26%). 1H NMR (400 MHz, DMSO-d6): δ 9.58 (t, J = 5.6 Hz, 1H), 9.40 (s, 1H), 8.94 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 2H), 7.62-7.61 (m, 3H), 4.67 (d, J = 5.2 Hz, 2H). ESI-MS(+): m/z 723.25 [M+H]+. HR-ESI-MS calcd for [C36H24F6N4O2S2Na]+: 745.1137; Found: 745.1141.
8,8′-Disulfanediylbis(N-(4-methoxybenzyl)quinoline-3-carboxamide) (42)
Product afforded via Method A. Yield = 0.15 g (47%). 1H NMR (400 MHz, DMSO-d6): δ 9.40-9.38 (m, 2H), 8.91 (d, J = 1.6 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 6.92 (d, J = 8.0 Hz, 2H), 4.50 (d, J = 5.6 Hz, 2H) 3.72 (s, 3H). HR-ESI-MS calcd for [C36H30N4O4S2Na]+: 669.1601; Found: 669.1603.
8,8′-Disulfanediylbis(N-(benzo[d][1,3]dioxol-5-ylmethyl)quinoline-3-carboxamide) (43)
Product afforded via Method A. Yield = 0.12 g (37%). 1H NMR (400 MHz, Acetone-d6): δ 9.38 (s, 2H), 9.01 (s, 1H), 8.27-8.26 (m, 2H), 7.81 (d, J = 7.6 Hz, 1H), 6.95 (s, 1H), 6.87-6.85 (m, 2H), 5.98 (s, 2H), 4.46 (d, J = 5.6 Hz, 2H). HR-ESI-MS calcd for [C36H27N4O6S2]+: 675.1372; Found: 675.1374.
8,8′-Disulfanediylbis(N-(4-morpholinobenzyl)quinoline-3-carboxamide) (44)
Product afforded via Method A. Yield = 0.18 g (50%). 1H NMR (400 MHz, DMSO-d6): δ 9.40-9.33 (m, 2H), 8.91 (s, 1H), 7.94 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 8.0 Hz, 2H), 4.47 (d, J = 5.6 Hz, 2H), 3.72 (t, J = 4.8 Hz, 4H), 3.05 (t, J = 4.8 Hz, 4H). HR-ESI-MS calcd for [C42H40N6O4S2 Na]+: 779.2445; Found: 779.2443.
8,8′-Disulfanediylbis(N-(2-morpholinoethyl)quinoline-3-carboxamide) (45)
Product afforded via Method A. Yield = 0.07 g (24%). 1H NMR (400 MHz, Acetone-d6): δ 9.45 (s, 1H), 8.96 (s, 1H), 8.36 (d, J = 8 Hz, 1H), 8.30 (d, J = 8 Hz, 1H), 7.86 (t, J = 8 Hz, 1H), 3.79-3.75 (m, 6H), 3.06(t, J = 6 Hz, 2H). HR-ESI-MS calcd for [C32H37N6O4S2]+: 633.2312; Found: 633.2316.
Dibenzyl2,2′-((2,2′-((8,8′-disulfanediylbis(quinoline-8,3-diyl-3-carbonyl))bis(azanediyl))bis(acetyl))bis(azanediyl))diacetate (46)
To a solution of 16b (0.05 g, 0.24 mmol) in dry DMF (5 mL) was added HATU (0.11 g, 0.29 mmol), HOBT (0.04 g, 0.29 mmol), Et3N (0.068 mL, 0.48 mmol) and H2N-Gly-Gly-Bz (0.11 g, 0.26 mmol). The reaction was stirred at room temperature for 4 h. The resulting mixture was evaporated to dryness and the crude material was dissolved in CH2Cl2 and washed with 1M HCl, H2O and then Brine. The product, which precipitated out of the organic layer, was filtered off and dried under vacuum. Yield = 0.07 g (69%). 1H NMR (400 MHz, DMSO-d6): δ 9.40 (d, J = 2.1 Hz, 1H), 9.29 (t, J = 5.9 Hz, 1H), 8.94 (d, J = 2.2 Hz, 1H), 8.51 (t, J = 6.0 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.88 – 7.82 (m, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.37 – 7.28 (m, 5H), 5.14 (s, 2H), 4.03 (d, J = 5.9 Hz, 2H), 3.95 (d, J = 5.9 Hz, 2H). HR-ESI-MS calcd for [C42H37N6O8S2]+: 817.2109; Found: 817.2106.
8-Hydroxy-N-(2-(thiazol-2-yl)ethyl)quinoline-3-carboxamide (47)
To a solution of 8-Hydroxyquinoline-3-carboxylic acid (0.1 g, 0.529 mmol) in DMF (3 mL) was added Et3N (0.088 mL, 0.634 mmol) and HATU (0.24 g, 0.634 mmol) then allowed to stir at room temperature for ~15 min. To this was then added 2-(Thiazol-2-yl)ethan-1-amine (0.08 g, 0.634 mmol) and allowed to stir for 18 h. The resulting solution was concentrated in vacuo and purified via silica gel chromatography eluting a gradient of 0-20% MeOH in CH2Cl2. Yield = 0.04 g (30%). 1H NMR (400 MHz, DMSO-d6): δ 9.35 (t, J = 3.6 Hz, 1H), 9.26 (s, 1H), 9.17 (s, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.66-7.63 (m, 3H), 7.40 (t, J = 3.6 Hz, 1H), 3.74 (q, J = 5.6 Hz, 2H), 3.36 (t, J = 6 Hz, 2H). HR-ESI-MS calcd for [C15H12N3O2S]-: 298.0656; Found: 298.0658.
8-(Methylthio)-N-(2-(thiazol-2-yl)ethyl)quinoline-3-carboxamide (48)
A solution of 35 (0.1 g, 0.159 mmol) in MeOH (10 mL) was cooled down to 0 °C and placed under nitrogen atmosphere. To this was added NaBH4 (0.06 g, 1.59 mmol) and allowed to stir for ~ 20 min. The solution was then allowed to heat up to room temperature and stirred for an additional 1 h. The resulting solution was then concentrated in vacuo, dissolved in THF (10 mL) and then added CH3I (0.23 g, 1.59 mmol) and allowed to stir at room temperature for 1 h. This solution was then refluxed for 18 h, concentrated and purified via silica gel chromatography eluting a gradient of 0-90% EtOAc in Hexanes. Yield = 0.23 g (22%). 1H NMR (400 MHz, CDCl3): δ 9.27 (s, 1H), 8.65 (s, 1H), 7.92 (br, 1H), 7.76 (d, J = 1.6 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 7.6 Hz, 1H), 7.27 (d, J = 1.6 Hz, 1H), 4.00 (q, J = 5.6 Hz, 2H), 3.39 (t, J = 6 Hz, 2H), 2.57 (s, 3H). HR-ESI-MS calcd for [C16H15N3OS2Na]+: 352.0549; Found: 352.0547.
8-Bromo-5-fluoro-2-methylquinoline (49a)
A solution of 2-Bromo-5-fluoroaniline (10 g, 52.6 mmol) in 6M HCl (50 mL) and Toluene (50 mL) was refluxed for ~30 min. To this was then added Crotonaldehyde (6.54 mL, 79 mmol) and stirred at reflux for 18 h. The resulting solution was partitioned in a separatory funnel and the organic layer was discarded. The remaining aqueous solution was made basic with 6M NaOH, then extracted with EtOAc. The organic layer was then isolated and dried with MgSO4, then filtered and purified via silica gel chromatography eluting a gradient 0-10% EtOAc in Hexanes to afford product as an off-white solid. Yield = 5.096 g (40%). 1H NMR (400 MHz, CDCl3): δ 8.31 (d, J = 8.4 Hz, 1H), 7.94 (q, J = 5.6 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.07 (t, J = 8.8 Hz, 1H), 2.83 (s, 3H). ESI-MS(+): m/z 242.28 [M+H]+.
8-Bromo-5-fluoroquinoline-2-carboxylic acid (49b)
To a solution of 49a (2g, 8.33 mmol) in Pyridine (30 mL) was added selenium dioxide (2.77 g, 24.99 mmol) and heated to reflux for 16 h. The resulting solution was then concentrated in vacuo. The crude was taken up in H2O and heated to 70 °C for 30 min. The solution was hot filtered in order to remove excess SeO2 byproduct and afford product as a light tan solid. Yield = 2.25 g (95%). 1H NMR (400 MHz, CDCl3): δ 8.80 (d, J = 8.4 Hz, 1H), 8.38 (d, J = 8.4 Hz, 1H), 8.25 (q, J = 5.2 Hz, 1H), 7.52 (t, J = 8.8 Hz, 1H). ESI-MS(-): m/z 268.12 [M-H]-.
5-Fluoroquinoline-2-carboxylic acid (49c)
To a solution of 49b (2.1 g, 7.90 mmol) in MeOH (30 mL) was added 1M NaOH (17.8 mL, 17.8 mmol) followed by Pd(OH)2/C (0.277 g, 0.395 mmol,) and allowed to stir at room temperature under a hydrogen atmosphere (balloon) for 2.5 h. The resulting solution was filtered over celite and rinsed with H2O and MeOH. The filtrate was recovered and concentrated in vacuo to remove organic solvent. The resulting aqueous solution was then acidified with 1M HCl to make solution slightly acidic. The acidic solution was then extracted with EtOAc. The combined organic layers were dried with MgSO4 and filtered to remove solids. The filtrate was concentrated to afford product as a tan solid. Yield = 0.389 g (26%). 1H NMR (400 MHz, Acetone-d6): δ 8.74 (d, J = 8.8 Hz, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.03 (d, J = 8.8 Hz, 1H), 7.92 (q, J = 6 Hz, 1H), 7.54 (t, J = 8 Hz, 1H). ESI-MS(-): m/z 190.22 [M-H]-.
5-Fluoro-N-(2-(thiazol-2-yl)ethyl)quinoline-2-carboxamide (49d)
To a solution of 49c (0.15 g, 0.785 mmol) in DMF (5 mL) was added Et3N (0.131 mL, 0.942 mmol) followed by HATU (0.358 g, 0.942 mmol) and then 2-(Thiazol-2-yl)ethan-1-amine (0.121 g, 0.942 mmol) and allowed to stir at room temperature for 15 h. To the resulting solution was added sat. NaHCO3 (aqueous) and allowed to stir for ~10 min. This was then extracted with EtOAc and dried with MgSO4, filtered, then concentrated and purified via silica gel chromatography eluting a gradient of 0-50% EtOAc in Hexanes. Yield = 0.1 g (42%). 1H NMR (400 MHz, CDCl3): δ 8.73 (br, 1H), 8.59 (d, J = 8.8 Hz, 1H), 8.36 (d, J = 8.8 Hz, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.71-7.66 (m, 1H), 7.30 (d, J = 8.8 Hz, 1H), 7.25 (d, J = 3.2 Hz, 1H), 4.02 (q, J = 6.8 Hz, 1H), 3.43 (t, J = 6.8 Hz, 1H). ESI-MS(+): m/z 302.11 [M+H]+.
5-((4-Methoxybenzyl)thio)-N-(2-(thiazol-2-yl)ethyl)quinoline-2-carboxamide (49e)
To a solution of 49d (0.1 g, 0.332 mmol) in DMF (5 mL) was added p-MBSH (0.195 mL, 1.327 mmol) and NaH (0.05 g, 1.327 mmol) then allowed to stir at 140 °C for 22 h. The resulting solution was concentrated in vacuo and purified via silica gel chromatography eluting a gradient of 0-50% EtOAc in Hexanes. Yield = 0.07 g (47%). 1H NMR (400 MHz, CDCl3): δ 8.73 (br, 1H), 8.59 (d, J = 8.8 Hz, 1H), 8.36 (d, J = 8.8 Hz, 1H), 7.91 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 3.2 Hz, 1H), 7.71-7.66 (m, 1H), 7.30 (d, J = 8.8 Hz, 1H), 7.25 (d, J = 3.2 Hz, 1H), 4.02 (q, J = 6.8 Hz, 2H), 3.43 (t, J = 6.8 Hz, 2H). ESI-MS(+): m/z 436.13 [M+H]+.
5,5′-Disulfanediylbis(N-(2-(thiazol-2-yl)ethyl)quinoline-2-carboxamide) (49f)
To a solution of 49e (0.05 g, 0.115 mmol) in TFA (5 mL) was added m-Cresol (0.1 g, 0.956 mmol) and refluxed for 22 h. The resulting solution was concentrated in vacuo and purified via silica gel chromatography eluting a gradient of 0-50% EtOAc in Hexanes. Yield = 0.004 g (12%). 1H NMR (400 MHz, DMSO-d6): δ 9.21 (t, J = 6 Hz, 1H), 8.66 (d, J = 8.8 Hz, 1H), 8.15 (d, J = 8 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.82-7.76 (m, 2H), 7.73 (d, J = 3.6 Hz, 1H), 7.59 (d, J = 3.6 Hz, 1H), 3.76 (q, J = 6.8 Hz, 2H), 3.33 (t, J = 6.8 Hz, 2H). HR-ESI-MS calcd for [C30H24N6O2S4Na]+: 651.0736; Found: 651.0736.
Methyl 5-(((2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)methyl)amino)nicotinate (50a)
To a preheated (~100 °C) mixture of 3-Aminopyridine (0.61 g, 4.0 mmol) and Meldrum’s acid (0.69 g, 4.8 mmol) was added Triethyl Orthoformate (4.0 mL, 24.0 mmol). The mixture was stirred at 100 °C for 2 h. The reaction proceeded by changing color from yellow to wine red accompanying the formation of yellow precipitate. After cooling to room temperature, the excess liquid of Triethyl Orthoformate was removed via vacuum distillation. The resulting solid was purified via silica gel chromatography eluting a gradient of 30 to 70% EtOAc in CH2Cl2. Yield = 1.6 g (88%). 1H NMR (400 MHz, CDCl3): δ 11.32 (d, J = 13.6 Hz, 1H), 9.09 (s, 1H), 8.75 (s, 1H), 8.68 (d, J = 13.6 Hz, 1H), 8.22 (s, 1H), 3.98 (s, 3H), 1.75 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 165.6, 164.7, 163.1, 152.8, 148.5, 144.10, 134.9, 127.2, 125.3, 105.9, 89.7, 53.1, 27.4. ESI-MS(+): m/z 306.97 [M+H]+.
Methyl 8-hydroxy-1,5-naphthyridine-3-carboxylate (50b)
To a solution of 50a (0.5 g, 1.63 mmol) was added Dowtherm A (150 mL) under nitrogen atmosphere and heated to 250 °C for 1 h. During the reaction, the color of the solution changed from orange yellow to dark brown. After cooling down to room temperature, the reaction solution was filtered to afford product. The solid was rinsed with Diphenyl Ether and Acetone. Yield = 0.15 g (45%). ESI-MS(+): m/z 205.29 [M+H]+.
Methyl 8-chloro-1,5-naphthyridine-3-carboxylate (50c)
To a solution of 50b (0.1g, 0.49 mmol) in Toluene (10 mL) was added POCl3 (0.14 mL, 1.47 mmol) at room temperature. The solution was stirred at 110 °C for 2 h, then allowed to cool to room temperature, which caused formation of a precipitate. The solution and dark solid was quenched with sat. NaHCO3 and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, then filtered and concentrated under reduce pressure. The residue was purified via silica gel chromatography eluting a gradient of 20 to 50% EtOAc in CH2Cl2. Yield = 0.078 g (71%). 1H NMR (400 MHz, CDCl3): δ 9.49 (t, J = 2.0 Hz, 1H), 8.95 (d, J = 2.0 Hz, 1H), 8.86 (d, J = 4.8 Hz, 1H), 7.77 (d, J = 4.8 Hz, 1H), 3.99 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 165.0, 151.9, 151.2, 144.3, 144.0, 143.0, 140.2, 127.3, 126.2, 53.1. ESI-MS(+): m/z 223.26 [M+H]+
8-((4-Methoxybenzyl)thio)-1,5-naphthyridine-3-carboxylic acid (50d)
To a solution of 50c (0.18 g, 0.81 mmol) in DMF (10 mL) was added p-MBSH (0.23 mL, 1.62 mmol) at room temperature. The solution was stirred for 2 h, then acidified with 1N HCl to pH ~ 4, then concentrated under reduced pressure. The resulting residue was diluted with H2O and the solid was collected to give the desired product. Yield = 0.26 g (99%). 1H NMR (400 MHz, DMSO-d6): δ 9.30 (s, 1H), 8.84 (d, J = 3.2 Hz, 1H), 8.75 (s, 1H), 7.75 (d, J = 3.2 Hz, 1H), 7.45 (d, J = 6.4 Hz, 2H), 6.92 (d, J = 6.4 Hz, 2H), 4.37 (s, 2H), 3.74 (s, 3H). 13C NMR (125 MHz, DMSO-d6): δ 166.2, 159.0, 151.9, 150.9, 149.4, 143.1, 141.4, 139.2, 130.7, 128.1, 127.9, 120.5, 114.5, 55.5, 33.8. ESI-MS(+): m/z 325.07 [M+H]+.
8-((4-Methoxybenzyl)thio)-N-(2-(thiazol-2-yl)ethyl)-1,5-naphthyridine-3-carboxamide (50e)
To a solution of 50d (0.25 g, 0.77 mmol) in DMF (5 mL) was added 2-(Thiazol-2-yl)ethan-1-amine (0.11 g, 0.84 mmol), Pyridine (0.25 mL, 3.06 mmol), HOBT (0.24 g, 1.54 mmol) and EDC (0.3 g, 1.54 mmol) at room temperature. The solution was stirred for 2 h, then concentrated under reduced pressure. The resulting residue was diluted with H2O and extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The resulting residue was purified via silica gel chromatography eluting a gradient of 30 to 70% EtOAc in CH2Cl2. Yield = 0.17 g (51%). 1H-NMR (400 MHz, DMSO-d6) δ 9.24 (s, 1H), 9.18 (t, J = 4.4 Hz, 1H), 8.83 (d, J = 4.0 Hz, 1H), 7.74 (d, J = 2.8 Hz, 1H), 7.72 (d, J = 4.0 Hz, 1H), 7.60 (d, J = 2.8 Hz, 1H), 7.45 (d, J = 6.8 Hz, 2H), 6.92 (d, J = 6.8 Hz, 2H), 4.36 (s, 2H), 3.74 (s, 3H), 3.72-3.66 (m, 2H), 3.34-3.29 (m, 2H). 13C NMR (125 MHz, DMSO-d6): δ 167.5, 164.7, 159.0, 151.7, 150.8, 148.6, 142.8, 142.3, 141.5, 136.3, 131.0, 130.7, 127.9, 120.2, 120.1, 114.5, 55.5, 33.7, 32.7. ESI-MS(+): m/z 437.07 [M+H]+.
8-Mercapto-N-(2-(thiazol-2-yl)ethyl)-1,5-naphthyridine-3-carboxamide (50f)
To a solution of 50d (0.14 g, 0.32 mmol) in TFA (20 mL) was added m-Cresol (0.17 mL, 1.60 mmol) at room temperature. The reaction was then heated at reflux for 16 h, then concentrated under reduced pressure and diluted with the EtOAc. The solution was neutralized with sat. NaHCO3, which caused an orange red solid to precipitate. The solid was collected via filtration and washed with H2O and Acetone to afford product. Yield = 0.087 g (86%). 1H NMR (400 MHz, DMSO-d6): δ 12.97 (br, 1H), 9.19 (t, J = 4.4 Hz, 1H), 9.09 (s, 1H), 8.44 (s, 1H), 7.92 (d, J = 5.2 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.50 (d, J = 5.2 Hz, 1H), 3.72-3.66 (m, 2H), 3.34-3.29 (m, 2H). 13C NMR (125 MHz, DMSO-d6): δ 167.4, 164.3, 147.0, 146.6, 142.8, 134.5, 133.0, 132.1, 129.1, 128.6, 120.3, 32.6. HR-ESI-MS calcd for [C14H12N4OS2Na]+: 339.0345; Found: 339.0348.
Synthesis of [(TpMe,Ph)Zn(8TQ)]
[(TpMe,Ph)ZnOH] was synthesized as previously reported.43 [(TpMe,Ph)ZnOH] (0.1 g, 0.177 mmol) was dissolved in CH2Cl2 (15 mL) to yield a colorless solution. To this was added a solution of MeOH (10 mL) containing 8-Mercaptoquinoline-HCl (0.035 g, 0.177 mmol) and Et3N (25 μL, 0.177 mmol). The colorless solution obtained a bright yellow color. The reaction mixture was stirred for 20 h at room temperature under a nitrogen atmosphere. The reaction mixture was then concentrated in vacuo and yielded a bright yellow solid. The solid was dissolved in minimal amount of benzene (~5 mL). Blocks were grown out of a solution of the complex in Benzene diffused with Pentane.
Rpn11 Activity Assay
To measure Rpn11 activity, a synthetic peptide substrate, termed Ub4-pepOG, was engineered. This substrate consists of four linear ubiquitins connected to a short peptide sequence containing a unique cysteine to which is conjugated a single Oregon Green 488 fluorophore molecule. The peptide bond between the fourth ubiquitin and the downstream peptide is cleaved by 26S proteasome in vitro, which can be observed by SDS-PAGE and fluorescent polarization measurement. The fluorescent peptide released upon cleavage of Ub4-pepOG consists of only 30 amino acids; therefore the decrease of polarization observed in fluorescence polarization assays arose mainly from deubiquitination of the peptide and could be observed even when the proteolytic activity of the 20S CP was inhibited. The fluorescence polarization assay was performed as previously described37 at 30 °C in a low-volume 384 well solid black plate. Briefly, components were added to each well in the following sequence: 1) 5 μL inhibitor compound in buffer containing 3% DMSO or 3% DMSO in buffer as a control; and 2) 5 μL of 26S proteasome (Enzo life sciences) in buffer (20 nM proteasome was pre-incubated with epoxomicin at room temperature for 1 hour, then dilute 10-fold in 1× Assay Buffer). Substrate (5 μL, 3 nM Ub4-pepOG) in buffer was then added to initiate the reaction. To evaluate the effects of Zn(cyclen)2+ on Rpn11 activity, the assay was carried out in the same manner as described with the addition of 100 μM Zn(cyclen)2+ in the titration reaction. Fluorescence polarization was measured using a plate reader with excitation at 480 nm and emission at 520 nm. To calculate the IC50 of Rpn11 inhibitors, eight to twelve-point titration was performed for each compound, up to a concentration of 100 μM. Rpn11 activity was normalized to the DMSO control and fitted using a dose-response curve. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
CSN5 Activity Assay
A fluorescent substrate termed SCFskp2-Nedd8OG was engineered to measure Csn5 activity in vitro.38 To produce this substrate, Nedd8 containing a unique N-terminal cysteine was labeled with Oregon Green 488, and then conjugated to SCFSkp2 as previously described.49 This assay measures the decrease in fluorescence polarization due to the decrease in apparent molecular weight of the Oregon Green fluorophore (from the ~175 kDa substrate to ~9kDa Nedd8OG) as a result of Csn5-dependent cleavage of the isopeptide bond which links Nedd8OG to SCFSkp2. Assays were performed in a low-volume 384 well solid black plate comprising equal volumes of compound, substrate (SCFskp2-Need8OG) and enzyme (Csn5). Fluorescence polarization was recorded using the same protocol as for the Rpn11 activity assay at 30 °C. IC50 was calculated as described above. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
AMSH Activity Assay
AMSH is known to selectively cleave diubiquitin linked via K63. A substrate termed DiUbK63TAMRA was purchased from Boston Biochem to assay AMSH activity in vitro. DiUbK63TAMRA was labeled with the FRET pair TAMRA/QXL. Upon AMSH cleavage, TAMRA was separated from the quencher QXL which resulted in an increase of TAMRA fluorescence intensity, which was monitored using a fluroescence plate reader (excitation at 540 nm and emission at 590 nm). The assay was performed in a low-volume 384 well solid black plate at 30 °C and analyzed as described above. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
BRISC activity assay
Purified BRISC complex (consisting of KIAA0157, BRCC-45, BRCC-36 and MERIT-40) was kindly provided by Dr. Elton Zeqiraj (University of Leeds, U.K.). DiUbK63TAMRA (Boston Biochem, MA) was used to measure the activity of BRISC complex. Upon cleavage, TAMRA was separated from the quencher QXL which resulted in an increase of TAMRA fluorescence intensity monitored using excitation at 540 nm and emission at 590 nm. The assay was performed in a low-volume 384 well solid black plate at 30 °C and analyzed as described above. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
HDAC1 and 6 Activity Assay
HDAC1 and 6 were purchased from BPS Bioscience (BPS Bioscience catalog #50051 and 50006) and the assay was carried out as instructed by manufacturer. The enzyme was diluted with 25 mM Tris-Cl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.1 mg/mL BSA, pH 8.0 buffer and its activity was measured by utilizing Substrate 3 (BPS Bioscience catalog #50037). The assays were carried out in black, low binding NUNC 96-well plates. Each well contained a volume of 50 μL including buffer, HDAC (3.8 ng/well of HDAC-1, 50 ng/well of HDAC-6), inhibitor, and substrate (20 μM). Prior to adding substrate, the plate was preincubated for 5 min. Upon addition of substrate, the plate was incubated at 37 °C for 30 min. At this point, HDAC assay developer (50 μL, BPS Bioscience catalog #50030) was added to each well and the plate was incubated for 15 min at room temperature. The fluorescence was recorded with a BioTek FLx 800 microplate reader. The measured fluorescence was compared for samples versus controls containing no inhibitor (0% inhibition). Reported IC50 value represents the average values obtained from at least three independent measurements, with the standard deviation reported as the error.
MMP Activity Assay
MMP-2 and OmniMMP fluorogenic subsbtrate (P-126) were purchased from Enzo Life Sciences (Farmingdale, NY). The assay was carried out in white NUNC 96-well plates as previously described.52 Each well contained a volume of 90 μL including buffer (50 mM HEPES, 10 mM CaCl2, 0.05% Brij-35, pH 7.5), human recombinant MMP (1.16 U of MMP-2), and the fragment solution. The enzyme and inhibitor were incubated for 30 min at 37 °C, the reaction was then initiated by the addition of 10 μL (100 μL total volume of wells) of the fluorogenic OmniMMP substrate (4 μM final concentration, Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2-AcOH). Fluorescence measurements were recorded using a Bio-Tek FL×800 fluorescence plate reader every minute for 20 min with excitation and emission wavelengths at 320 and 400 nm, respectively. The rate of fluorescence increase was compared for samples versus negative controls (no inhibitor, arbitrarily set as 100% activity). Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
5-Lipoxygenase Activity Assay
The assay was performed according to a literature procedure at room temperature.53 Each well contained a volume of 80 μL including buffer (50 mM Tris, 2 mM EDTA, 2 mM CaCl2, pH 7.5), human recombinant 5-LO (0.2 U, Cayman Chemicals), reporter dye (20, 70 -dichlorofluorescin diacetate; H2DCFDA, 10 μM, In- vitrogen), fragment solution, arachidonic acid (AA, 3 μM, Fischer Scientific), and adenosine triphosphate (ATP, 10 μM, Sigma-Aldrich). H2DCFDA and 5-LO were incubated for 5 min prior to the addition of the fragment solution. This was followed by a second incubation for 10 min. The reaction was initiated by the addition of a substrate solution containing AA and ATP. The reaction was monitored using a Bio-Tek FL×800 fluorescence plate reader. Fluorescence measurements were recorded every minute for 20 min with excitation and emission wavelengths at 485 and 528 nm, respectively. The rate of fluorescence increase was compared for samples versus negative controls (no inhibitor, arbitrarily set as 100% activity). Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
hCAII Activity Assay
hCAII was expressed and purified as previously reported.54 Assays were carried out in 50 mM HEPES (pH 8.0). A BioTek Precision XS microplate sample processor was utilized. The compounds were incubated with protein (final concentrations of 100 nM for hCAII) for 10 min at 25 °C. A substrate (p-nitrophenylacetate; final concentration of 500 μM) was added, and hCAII-catalyzed cleavage was monitored by the increase in absorbance at 405 nm corresponding to formation of the p-nitrophenolate anion. The initial linear reaction rate was compared to that of wells containing no inhibitor (0% inhibition) and no protein (100% inhibition). The rate of non-hCAII-catalyzed PNPA hydrolysis in the presence of inhibitor was subtracted from each trial before determination of the percent inhibition. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
UbG76V-GFP degradation assay
To determine the potency of Rpn11 inhibitor in cells, a reporter degradation assay was employed. Briefly, stably transfected Hela cells expressing UbG76V-GFP were treated with the reversible proteasome inhibitor MG132 at 37 °C incubator with 5% CO2 in air, which increased cellular levels of UbG76V-GFP to yield a detectable fluorescent signal. After 4 h, MG132 was removed and a cycloheximide (CHX) chase was initiated with or without varying concentrations of inhibitor compound at 37 °C with 5% CO2 in air. Reporter degradation, monitored by the decay of GFP fluorescence, was measured to quantify Rnp11 inhibition using high throughput microscopy. The rate of fluorescence decrease was normalized to a DMSO control and analyzed using dose-response equation. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
Cytotoxicity assay
293T, A549 and HCT 116 cell lines were cultured in DMEM with 10% FBS in white, clear-bottom tissue culture-treated 96-well plates. Cells were treated with different concentrations of inhibitor compounds in triplicates for 72 h at 37 °C with 5% CO2 in air. CellTiter-Glo (Promega, Madison, WI) reagent was added to the 96 well plates to measure cell viability. Luminescence values were measured in PHERAstar microplate reader (BMG labtech, Ortenberg, Germany). Collected data was normalized to DMSO control and fit to a dose-response equation to determine IC50 values. Reported IC50 values represents the average value obtained from at least three independent measurements, with the standard deviation reported as the error.
Supplementary Material
Scheme 10. Synthesis of a methylated analog of lead compound 35.
(a) NaBH4, MeOH, 25 °C; (b) CH3I, THF, reflux.
Scheme 11. Synthesis of compound 49f, a structural-isomer analog of lead compound 35.
(a) Crotonaldehyde, Toluene, 6M HCl, reflux; (b) SeO2, Pyridine, reflux; (c) Pd(OH)2/C, 1M NaOH, MeOH, H2, 25 °C; (d) 2-(Thiazol-2-yl)ethan-1-amine, HATU, Et3N, DMF, 25 °C; (e) p-MBSH, NaH, DMF, 140 °C; (f) m-Cresol, TFA, reflux.
Acknowledgments
The authors acknowledge Dr. Yongxuan Su (U.C. San Diego Molecular Mass Spectrometry Facility) for aid with HR-MS and HPLC fragment purity analysis. We would like to acknowledge Dr. Dave P. Martin and Dr. David T. Puerta for their helpful discussions and assistance with the synthesis and characterization of the [(TpMe,Ph)Zn(8TQ)] metal complex and the U.C. San Diego X-ray diffraction facilities for assistance with experiments. We also thank Dr. Elton Zeqiraj for providing purified BRISC complex. S.M.C. is a co-founder, has an equity interest, and receives income as member of the Scientific Advisory Board for Cleave Biosciences and is a co-founder, has an equity interest, and a member of the Scientific Advisory Board for Forge Therapeutics.
Funding Sources
This work was initiated with an American Recovery and Reinvestment Act (ARRA) supplement to R.J.D (3-R01-GM065997-07S1), followed by a grant to S.M.C. and R.J.D. from the National Cancer Institute (R01-CA164803). J.L. was supported in part by a Caltech Grubstake Award and CBEA Award from Amgen. R.J.D. is an Investigator of the Howard Hughes Medical Institute and this work was supported in part by HHMI.
ABBREVIATIONS
- UPS
Ubiqutin-proteasome system
- FBDD
Fragment based drug discovery
- MBP
Metal-binding pharmacophore
- 8TQ
8-Thioquinoline
- MM
Multiple myeloma
- SAR
Structure activity relationship
- HTS
High-throughput screen
- Tp
Tris(pyrazolyl)borate
- t-BuSH
tert-Butylthiol
- p-MBSH
4-Methoxyphenyl)methanethiol
- HCT-116
Human colorectal carcinoma cells
- Ub-GFP
Ubiquitinated-green fluorescent protein
- Cyclen
1,4,7,10-Tetraazacyclododecane
- 293T
Human embryonic kidney cells
- A549
Adenocarcinomic human alveolar basal epithelial cells
Footnotes
The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
Author Contributions
The manuscript was written by C.P., S.M.C., and R.J.D. The compounds were designed by C.P., M.R., and S.M.C. and synthesized by C.P., M.R., and Y.M. Inhibition and cellular assays were performed by J.L, A.M., T.-F.C., and F.P.
SUPPORTING INFORMATION
Compound Identification and Molecular formula strings
Additional information as indicated in the text
References
- 1.Kyle RA, Rajkurnar SV. Treatment of Multiple Myeloma: A Comprehensive Review. Clin Lymphoma Myeloma. 2009;9(4):278–288. doi: 10.3816/CLM.2009.n.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Cancer Society. Survival Rates by Stage for Multiple Myeloma. http://www.cancer.org/cancer/multiplemyeloma/detailedguide/multiple-myeloma-survival-rates (accessed September 16, 2016).
- 3.Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome Inhibitors: An Expanding Army Attacking a Unique Target. Chem Biol. 2012;19(1):99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome Inhibitors in Multiple Myeloma: 10 Years Later. Blood. 2012;120(5):947–959. doi: 10.1182/blood-2012-04-403733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Linden WA, Willems LI, Shabaneh TB, Li N, Ruben M, Florea BI, van der Marel GA, Kaiser M, Kisselev AF, Overkleeft HS. Discovery of a Potent and Highly Beta 1 Specific Proteasome Inhibitor from a Focused Library of Urea-Containing Peptide Vinyl Sulfones and Peptide Epoxyketones. Org Biomol Chem. 2012;10(1):181–194. doi: 10.1039/c1ob06554h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desvergne A, Genin E, Marechal X, Gallastegui N, Dufau L, Richy N, Groll M, Vidal J, Reboud-Ravaux M. Dimerized Linear Mimics of a Natural Cyclopeptide (TMC-95A) Are Potent Noncovalent Inhibitors of the Eukaryotic 20S Proteasome. J Med Chem. 2013;56(8):3367–3378. doi: 10.1021/jm4002007. [DOI] [PubMed] [Google Scholar]
- 7.Geurink PP, van der Linden WA, Mirabella AC, Gallastegui N, de Bruin G, Blom AEM, Voges MJ, Mock ED, Florea BI, van der Marel GA, Driessen C, van der Stelt M, Groll M, Overkleeft HS, Kisselev AF. Incorporation of Non-natural Amino Acids Improves Cell Permeability and Potency of Specific Inhibitors of Proteasome Trypsin-like Sites. J Med Chem. 2013;56(3):1262–1275. doi: 10.1021/jm3016987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawamura S, Unno Y, List A, Mizuno A, Tanaka M, Sasaki T, Arisawa M, Asai A, Groll M, Shuto S. Potent Proteasome Inhibitors Derived from the Unnatural cis-Cyclopropane Isomer of Belactosin A: Synthesis, Biological Activity, and Mode of Action. J Med Chem. 2013;56(9):3689–3700. doi: 10.1021/jm4002296. [DOI] [PubMed] [Google Scholar]
- 9.Ozcan S, Kazi A, Marsilio F, Fang B, Guida WC, Koomen J, Lawrence HR, Sebti SM. Oxadiazole-isopropylamides as Potent and Noncovalent Proteasome Inhibitors. J Med Chem. 2013;56(10):3783–3805. doi: 10.1021/jm400221d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallastegui N, Groll M. The 26S proteasome: Assembly and Function of a Destructive Machine. Trends Biochem Sci. 2010;35(11):634–642. doi: 10.1016/j.tibs.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka K, Mizushima T, Saeki Y. The Proteasome: Molecular Machinery and Pathophysiological Roles. Biol Chem. 2012;393(4):217–234. doi: 10.1515/hsz-2011-0285. [DOI] [PubMed] [Google Scholar]
- 12.Reyes-Turcu FE, Wilkinson KD. Polyubiquitin Binding and Disassembly By Deubiquitinating Enzymes. Chem Rev. 2009;109(4):1495–1508. doi: 10.1021/cr800470j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koulich E, Li XH, DeMartino GN. Relative Structural and Functional Roles of Multiple Deubiquitylating Proteins Associated With Mammalian 26S Proteasome. Mol Biol Cell. 2008;19(3):1072–1082. doi: 10.1091/mbc.E07-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guterman A, Glickman MH. Complementary Roles for Rpn11 and Ubp6 in Deubiquitination and Proteolysis by the Proteasome. J Biol Chem. 2004;279(3):1729–1738. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- 15.Cenci S, Oliva L, Cerruti F, Milan E, Bianchi G, Raule M, Mezghrani A, Pasqualetto E, Sitia R, Cascio P. Pivotal Advance: Protein Synthesis Modulates Responsiveness of Differentiating and Malignant Plasma Cells to Proteasome Inhibitors. J Leukocyte Biol. 2012;92(5):921–931. doi: 10.1189/jlb.1011497. [DOI] [PubMed] [Google Scholar]
- 16.Deshaies RJ. Proteotoxic Crisis, the Ubiquitin-Proteasome System, and Cancer Therapy. BMC Biol. 2014;12:1–14. doi: 10.1186/s12915-014-0094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCullough J, Clague MJ, Urbe S. AMSH is an Endosome-Associated Ubiquitin Isopeptidase. J Cell Biol. 2004;166(4):487–492. doi: 10.1083/jcb.200401141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, Bernards R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell. 2005;123(5):773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Zhu P, Zhou WL, Wang JX, Puc J, Ohgi KA, Erdjument-Bromage H, Tempst P, Glass CK, Rosenfeld MG. A Histone H2A Deubiquitinase Complex Coordinating Histone Acetylation and H1 Dissociation in Transcriptional Regulation. Mol Cell. 2007;27(4):609–621. doi: 10.1016/j.molcel.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambroggio XI, Rees DC, Deshaies RJ. JAMM: A Metalloprotease-like Zinc Site in the Proteasome and Signalosome. PLOS Biol. 2004;2(1):113–119. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sobhian B, Shao GZ, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 Targets BRCA1 to Specific Ubiquitin Structures at DNA Damage Sites. Science. 2007;316(5828):1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cope GA, Suh GSB, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ. Role of Predicted Metalloprotease Motif of Jab1/Csn5 in Cleavage of Nedd8 From Cul1. Science. 2002;298(5593):608–611. doi: 10.1126/science.1075901. [DOI] [PubMed] [Google Scholar]
- 23.Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S. Structural Basis for Specific Cleavage of Lys 63-Linked Polyubiquitin Chains. Nature. 2008;455(7211):358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- 24.Shrestha RK, Ronau JA, Davies CW, Guenette RG, Strieter ER, Paul LN, Das C. Insights into the Mechanism of Deubiquitination by JAMM Deubiquitinases from Cocrystal Structures of the Enzyme with the Substrate and Product. Biochemistry. 2014;53(19):3199–3217. doi: 10.1021/bi5003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Yakushi T, Parlati F, Mackinnon AL, Perez C, Ma Y, Carter KP, Colayco S, Magnuson G, Brown B, Nguyen K, Vasile S, Suyama E, Smith LH, Sergienko E, Pinkerton AB, Chung TDY, Palmer A, Pass I, Hess S, Cohen SM, Deshaies RJ. Capzimin, A Potent and Specific Inhibitor of Proteasome Isopeptidase Rpn11. 2017 doi: 10.1038/nchembio.2326. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kisselev AF, Goldberg AL. Proteasome Inhibitors: From Research Tools to Drug Candidates. Chem Biol. 2001;8(8):739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 27.Adams J. The Development of Proteasome Inhibitors as Anticancer Drugs. Cancer Cell. 2004;5(5):417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 28.Crawford LJ, Walker B, Irvine AE. Proteasome Inhibitors in Cancer Therapy. J Cell Commun Signal. 2011;5(2):101–110. doi: 10.1007/s12079-011-0121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim HS, Archer CT, Kodadek T. Identification of a Peptoid Inhibitor of the Proteasome 19S Regulatory Particle. J Am Chem Soc. 2007;129(25):7750–7751. doi: 10.1021/ja072027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim HS, Cai D, Archer CT, Kodadek T. Periodate-Triggered Cross-Linking Reveals Sug2/Rpt4 as the Molecular Target of a Peptoid Inhibitor of the 19S Proteasome Regulatory Particle. J Am Chem Soc. 2007;129(43):12936–12937. doi: 10.1021/ja075469+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trader DJ, Simanski S, Kodadek T. A Reversible and Highly Selective Inhibitor of the Proteasomal Ubiquitin Receptor Rpn13 Is Toxic to Multiple Myeloma Cells. J Am Chem Soc. 2015;137(19):6312–6319. doi: 10.1021/jacs.5b02069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anchoori RK, Karanam B, Peng SW, Wang JW, Jiang R, Tanno T, Orlowski RZ, Matsui W, Zhao M, Rudek MA, Hung CF, Chen X, Walters KJ, Roden RBS. A bis-Benzylidine Piperidone Targeting Proteasome Ubiquitin Receptor RPN13/ADRM1 as a Therapy for Cancer. Cancer Cell. 2013;24(6):791–805. doi: 10.1016/j.ccr.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu YG, Carrasco RD, Richardson P, Linder S, Chauhan D, Anderson KC. A Novel Small Molecule Inhibitor of Deubiquitylating Enzyme USP14 and UCHL5 Induces Apoptosis in Multiple Myeloma and Overcomes Bortezomib Resistance. Blood. 2014;123(5):706–716. doi: 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobsen JA, Fullagar JL, Miller MT, Cohen SM. Identifying Chelators for Metalloprotein Inhibitors Using a Fragment-Based Approach. J Med Chem. 2011;54(2):591–602. doi: 10.1021/jm101266s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garner AL, Struss AK, Fullagar JL, Agrawal A, Moreno AY, Cohen SM, Janda KD. 3-Hydroxy-1-alkyl-2-methylpyridine-4(1H)-thiones: Inhibition of the Pseudomonas aeruginosa Virulence Factor LasB. ACS Med Chem Lett. 2012;3(8):668–672. doi: 10.1021/ml300128f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fullagar JL, Garner AL, Struss AK, Day JA, Martin DP, Yu J, Cai XQ, Janda KD, Cohen SM. Antagonism of a Zinc Metalloprotease Using a Unique Metal-Chelating Scaffold: Tropolones as Inhibitors of P. aeruginosa Elastase. Chem Commun. 2013;49(31):3197–3199. doi: 10.1039/c3cc41191e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.National Center for Biotechnology Information. PubChem BioAssay Database; AID=588493. https://pubchem.ncbi.nlm.nih.gov/bioassay/588493. (accessed September 16, 2016).
- 38.National Center for Biotechnology Information. PubChem BioAssay Database; AID=651999. https://pubchem.ncbi.nlm.nih.gov/bioassay/651999. (accessed September 16, 2016).
- 39.Hopkins AL, Groom CR, Alex A. Ligand Efficiency: A Useful Metric for Lead Selection. Drug Discovery Today. 2004;9(10):430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 40.Borgneczak K, Tjalve H. Effect of 8-Hydroxy-Quinoline, 8-Mercapto-Quinoline and 5-Chloro-7-Iodo-8-Hydroxy-Quinoline on the Uptake and Distribution of Nickel in Mice. Pharmacol Toxicol. 1994;74(3):185–192. doi: 10.1111/j.1600-0773.1994.tb01097.x. [DOI] [PubMed] [Google Scholar]
- 41.Prachayasittikul V, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. 8-Hydroxyquinolines: A Review of Their Metal Chelating Properties and Medicinal Applications. Drug Des Dev Ther. 2013;7:1157–1178. doi: 10.2147/DDDT.S49763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su CY, Liao S, Wanner M, Fiedler J, Zhang C, Kang BS, Kaim W. The Copper(I)/Copper(II) Transition in Complexes With 8-Alkylthioquinoline Based Multidentate Ligands. Dalton Trans. 2003;(2):189–202. [Google Scholar]
- 43.Puerta DT, Cohen SM. Elucidating Drug-Metalloprotein Interactions with Tris(pyrazolyl)borate Model Complexes. Inorg Chem. 2002;41(20):5075–5082. doi: 10.1021/ic0204272. [DOI] [PubMed] [Google Scholar]
- 44.Bridgewater BM, Parkin G. Lead Poisoning and the inactivation of 5-Aminolevulinate Dehydratase as Modeled by the Tris(2-mercapto-1-phenylimidazolyl)hydroborato Lead Complex, {[Tm-Ph]-Pb}[ClO4] J Am Chem Soc. 2000;122(29):7140–7141. [Google Scholar]
- 45.Hammes BS, Carrano CJ. Methylation of (2-Methylethanethiol-bis-3,5-dimethylpyrazolyl)methane Zinc Complexes and Coordination of the Resulting Thioether: Relevance to Zinc-Containing Alkyl Transfer Enzymes. Inorg Chem. 2001;40(5):919–927. doi: 10.1021/ic001178p. [DOI] [PubMed] [Google Scholar]
- 46.Trofimenko S. Recent Advances in Poly(Pyrazolyl)Borate (Scorpionate) Chemistry. Chem Rev. 1993;93(3):943–980. [Google Scholar]
- 47.Tesmer M, Shu MH, Vahrenkamp H. Sulfur-Rich Zinc Chemistry: New Tris(thioimidazolyl)hydroborate Ligands and Their Zinc Complex Chemistry Related to the Structure and Function of Alcohol Dehydrogenase. Inorg Chem. 2001;40(16):4022–4029. doi: 10.1021/ic0101275. [DOI] [PubMed] [Google Scholar]
- 48.Vahrenkamp H. Transitions, Transition Sstates, Transition State Analogues: Zinc Pyrazolylborate Chemistry Related to Zinc Enzymes. Acc Chem Res. 1999;32(7):589–596. [Google Scholar]
- 49.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural Insights Into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Cell. 2008;134(6):995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou TF, Deshaies RJ. Quantitative Cell-based Protein Degradation Assays to Identify and Classify Drugs That Target the Ubiquitin-Proteasome System. J Biol Chem. 2011;286(19):16546–16554. doi: 10.1074/jbc.M110.215319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies CW, Paul LN, Kim MI, Das C. Structural and Thermodynamic Comparison of the Catalytic Domain of AMSH and AMSH-LP: Nearly Identical Fold but Different Stability. J Mol Biol. 2011;413(2):416–429. doi: 10.1016/j.jmb.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puerta DT, Griffin MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohen SM. Heterocyclic Zinc-Binding Groups for Use in Next-Generation Matrix Metalloproteinase Inhibitors: Potency, Toxicity, and Reactivity. J Biol Inorg Chem. 2006;11(2):131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 53.Pufahl RA, Kasten TP, Hills R, Gierse JK, Reitz BA, Weinberg RA, Masferrer JL. Development of a Fluorescence-Based Enzyme Assay of Human 5-Lipoxygenase. Anal Biochem. 2007;364(2):204–212. doi: 10.1016/j.ab.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Martin DP, Hann ZS, Cohen SM. Metalloprotein-Inhibitor Binding: Human Carbonic Anhydrase II as a Model for Probing Metal-Ligand Interactions in a Metalloprotein Active Site. Inorg Chem. 2013;52(21):12207–12215. doi: 10.1021/ic400295f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.