

Abstract
Purpose
Patients with cancer who graciously consent for autopsy represent an invaluable resource for the study of cancer biology. To advance the study of tumor evolution, metastases, and resistance to treatment, we developed a next-generation rapid autopsy program integrated within a broader precision medicine clinical trial that interrogates pre- and postmortem tissue samples for patients of all ages and cancer types.
Materials and Methods
One hundred twenty-three (22%) of 554 patients who consented to the clinical trial also consented for rapid autopsy. This report comprises the first 15 autopsies, including patients with metastatic carcinoma (n = 10), melanoma (n = 1), and glioma (n = 4). Whole-exome sequencing (WES) was performed on frozen autopsy tumor samples from multiple anatomic sites and on non-neoplastic tissue. RNA sequencing (RNA-Seq) was performed on a subset of frozen samples. Tissue was also used for the development of preclinical models, including tumor organoids and patient-derived xenografts.
Results
Three hundred forty-six frozen samples were procured in total. WES was performed on 113 samples and RNA-Seq on 72 samples. Successful cell strain, tumor organoid, and/or patient-derived xenograft development was achieved in four samples, including an inoperable pediatric glioma. WES data were used to assess clonal evolution and molecular heterogeneity of tumors in individual patients. Mutational profiles of primary tumors and metastases yielded candidate mediators of metastatic spread and organotropism including CUL9 and PIGM in metastatic ependymoma and ANKRD52 in metastatic melanoma to the lung. RNA-Seq data identified novel gene fusion candidates.
Conclusion
A next-generation sequencing–based autopsy program in conjunction with a premortem precision medicine pipeline for diverse tumors affords a valuable window into clonal evolution, metastasis, and alterations underlying treatment. Moreover, such an autopsy program yields robust preclinical models of disease.
INTRODUCTION
The ability to comprehensively interrogate an individual patient’s cancer at molecular resolution is enabled by rapidly advancing sequencing technologies. The goal of the Caryl and Israel Englander Institute for Precision Medicine (IPM; New York, NY) is to harness as much information as possible from an individual patient’s tumor tissue not only in an effort to benefit the patient during life,1 but also to develop preclinical platforms2 and coclinical trials with the potential to benefit larger populations. An important arm of our precision medicine program permits postmortem tissue donation linked to robust downstream applications through a dedicated autopsy program.
Institutional rapid autopsy programs have been well described1,3-12 and have previously led to significant advances in cancer research in the areas of prostate,1,3,4,6,7,11 pancreas,10 breast,8 and pediatric brain tumors.5,9,12 This program builds on prior autopsy programs by enrolling patients of all ages and cancer types, capturing samples throughout a patient’s disease course, and leveraging multiple parallel platforms to generate high-throughput sequencing data and robust preclinical tumor models. We highlight our initial experience with 15 patients who underwent rapid autopsy and emphasize the following three themes: capturing the molecular status of a diverse set of neoplasms at the end point in disease progression relative to samples acquired earlier in the course of disease; the comparison of primary tumors to a multiplicity of metastatic sites at the molecular level to study processes underlying metastatic spread and organ-specific tumor tropism; and the development of tumor organoid models2 and patient-derived xenografts (PDXs), with the potential for high-throughput drug screening and functional validation studies.
MATERIALS AND METHODS
Basic Autopsy Protocol
Consent.
At the time that patients consent to enroll in the Precision Medicine Program (Weill Cornell Medicine [WCM] Institution Review Board No. 1305013903), they may also consent for the Rapid Autopsy Program (Data Supplement). The discussion of consent for autopsy may also be held at a later time. At our institution, the generic hospital autopsy consent form must also be signed by the next of kin. This institutional review board–approved autopsy tissue donor program is based in part on protocols developed at the University of Michigan by Drs Mark A. Rubin and Kenneth Pienta.3,4
Patient notification.
Once the treating physician notifies a contact member of a patient’s death by pager, a robotic calling system is activated to alert the autopsy team. Attending and resident autopsy pathologists, an autopsy technical assistant, and two to four members of the IPM laboratory are available at all times. The patients’ treating oncologists and surgeons may also be present. The postmortem interval goal for in-house patients is less than 1 hour.
Tissue collection.
Autopsy is performed using the en bloc Letulle dissection method. Tissues are snap frozen using Tissue-Tek O.C.T. embedding medium and Tissue-Tek cryomolds (Sakura Finetek, Torrance, CA). These are placed in a dry ice and methylbutane bath and stored at –80°C.1,6 In addition, fresh tissue is immediately transported to the laboratory for xenograft and tumor organoid development.2 See Data Supplement for sample autopsy findings.
Nucleic Acid Extraction
DNA extraction and whole-exome and whole-genome sequencing
As previously described,1 tumor and germline DNA is extracted using the Promega Maxwell 16 MDx DNA Purification Kits (Promega, Madison, WI). Sequencing is performed using Illumina HiSEquation 2500 Rapid Run Mode (2×101bp; Illumina, San Diego, CA). Short reads are aligned to GRC37/hg19 reference using Burrows-Wheeler aligner and processed accordingly to IPM-Exome-pipeline v0.9 as previously published.1,13 Three DNA samples from patient WCM0 corresponding to two liver metastases (LMs; LM1 and LM2) and germline DNA from blood underwent whole-genome sequencing (WGS) at the New York Genome Center following the sequencing protocols and analyses described in this section.14,15
RNA extraction and RNA sequencing.
Total RNA was prepared for RNA sequencing (RNA-Seq) in accordance with the standard Illumina mRNA sample preparation protocol (Illumina). Paired-end RNA-Seq at read lengths of 50 or 51 base pairs was performed with the HiSEquation 2000 (Illumina). Approximately 268 million paired-end reads were generated, corresponding to approximately 27 billion bases.16
DNA Sequencing and Data Analysis
Sequence data processing pipeline.
All exome data of the study samples were processed through the computational analysis pipeline of IPM, as previously described (IPM-Exome-pipeline v0.9).1,13 We implemented an approach to improve on single nucleotide variant calls in the presence of multisample patient data.17,18 WGS samples were analyzed at the New York Genome Center.15,19,20
Phylogenetic analysis.
Tumor samples are represented in a phylogenetic tree as nodes with no children, whereas the internal nodes model inferred tumor cell populations on the basis of observed single nucleotide variants using the parsimony ratchet method.21,22 See Data Supplement for gene clonality assessment.
RNA-Seq Data Analysis
All reads were independently aligned with STAR_2.4.0f123 for sequence alignment against the human genome build hg19, downloaded via the University of California Santa Cruz Genome Browser. For fusion analysis, we used STAR-fusion (STAR-Fusion_v0.5.1),24,25 FusionCatcher (v0.99.3e),26 and FusionSeq.27-31
Patient-Derived, Tumor Organoid, and Xenograft Development Platform
As previously described,2 fresh tumor tissue is dissociated before seeding cells into growth factor–reduced Matrigel (Corning, Corning, NY). For the establishment of PDX (Institutional Animal Care and Use Committee No. 2013-0116), we inject 1 million tumor organoid cells in a 1:1 mix with Matrigel in the flanks of NU/NU nude mice (Charles Rivers Laboratories, Wilmington, MA).
RESULTS
Consent
One hundred twenty-three (22%) of 554 patients who consented to the clinical trial have also consented for rapid autopsy. Of those patients for whom we have documented death in addition to premortem consent, we have performed autopsy on approximately 54% (see Materials and Methods for details on consent).
Diversity of Tumor Types and Metastatic Sites
The first 15 autopsies performed comprise a diverse set of neoplasms and metastatic sites (Fig 1). Primary tumors were available in 12 (80%) of 15 patients from prior surgical resections and/or from recurrent or residual primary tumors obtained at autopsy. In patients with known metastatic disease before death, matched primary and metastatic tumor from individual patients was available in 10 (77%) of 13 patients, and metastatic disease was collected at autopsy in 100% (n = 13) of these patients. Patient ages ranged from less than 10 to greater than 89 years at death, and patients were variably treated with prior surgery, radiation, and/or chemotherapeutic agents (Data Supplement).
Fig 1.
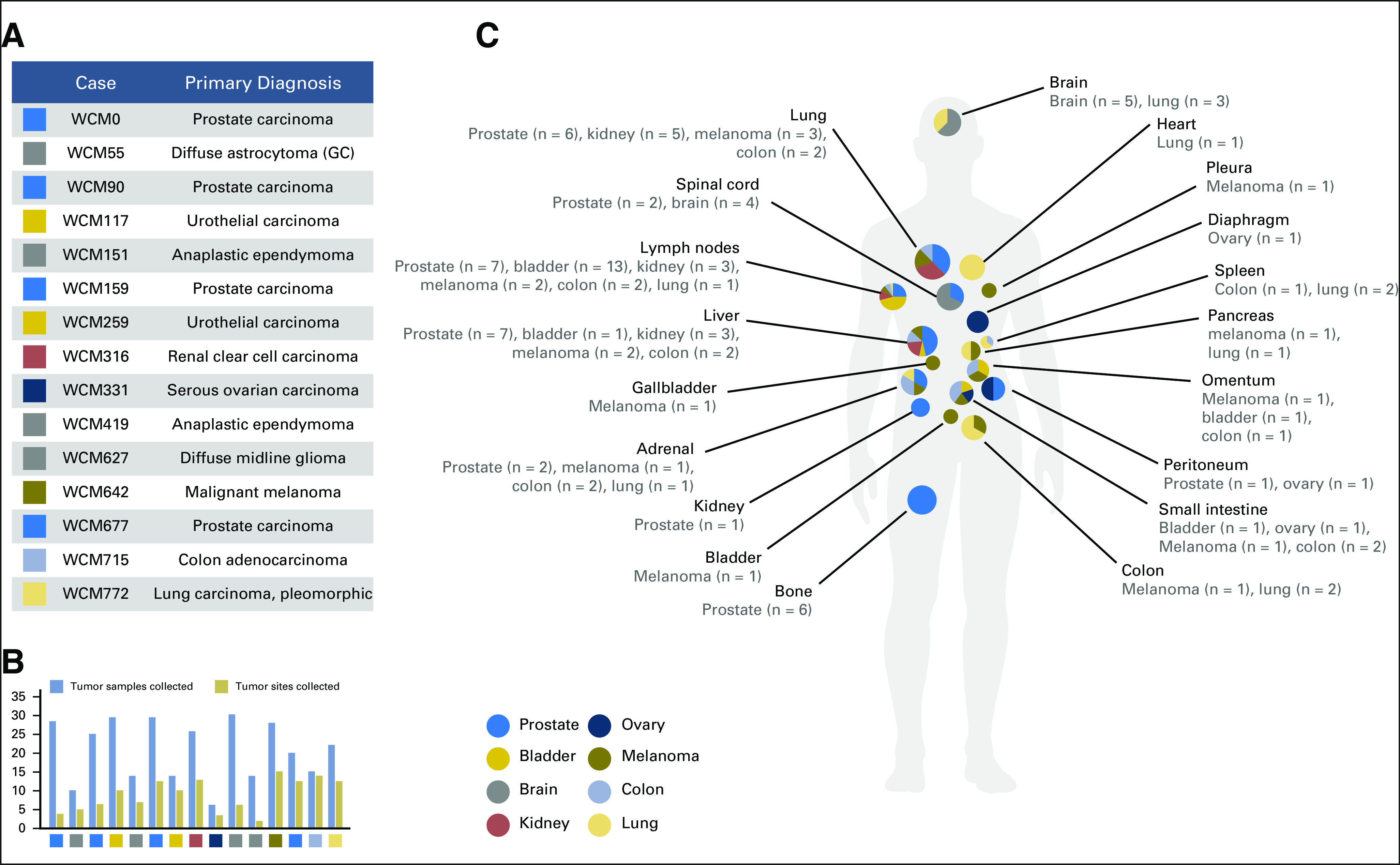
Rapid autopsies of Weill Cornell Medicine (WCM) study cohort. (A) The list of 15 autopsies shows the research identifier (WCM number) and primary diagnosis. (B) Bar chart showing the total number of frozen tumor samples collected and the number of distinctly annotated anatomic sites from which tumor was obtained for each autopsy. (C) Schematic illustration of anatomic sites of metastatic samples that were collected at autopsy. The pie charts represent the breakdown with respect to primary tumor origin, which correspond to the color key on the left side. GC, gliomatosis cerebri pattern of glioma infiltration.
Tissue Collection and DNA/RNA Extraction Results
Overall postmortem time interval (PMI) ranged from 0.5 to 18 hours. PMI for in-house patients averaged 3.1 hours (range, 0.5 to 6.5 hours), and PMI for outside patients averaged 8.42 hours (range, 2 to 18 hours). Delays were primarily attributable to transportation, administrative wait times, and/or accommodations of requests by the patient’s family. Transport was successfully managed from up to 50 miles from our Manhattan-based institution.
This initial cohort yielded a total of 346 frozen aliquots over 131 distinctly annotated anatomic sites, of which 310 contained tumor upon histologic review of frozen sections (Fig 1B). One hundred thirteen distinct samples underwent whole-exome sequencing (WES), including 82 frozen tumor and 10 frozen non-neoplastic control tissue samples obtained at autopsy, six samples derived from peripheral blood, and 15 formalin-fixed paraffin-embedded or frozen samples from prior surgical procedures. DNA yield and quality are provided in the Data Supplement. In addition to WES, RNA-Seq was performed on a subset of samples (n = 72). Data regarding RNA concentration, RNA integrity number values, 260/280 values, and 260/230 values over all RNA samples are provided in the Data Supplement. Finally, three samples from patient WCM0 (two LMs and one blood normal) underwent WGS through the Illumina Genome Network.
Average Coverage and Capture Efficiency
Overall 99.5% (> 170 million) and 95.8% (> 70 million) of paired-end reads generated could be mapped. The average coverage and capture efficiency for each patient are provided in the Data Supplement. WGS was done at greater than 90× coverage for both tumor samples (LM1 approximately 105×, LM2 approximately 90×) and greater than 40× for the blood normal (approximately 45×), with mapping rates between 94.5% and 95.6%.
Collection of Rare Disease Entities
A unique aspect of tumor acquisition at autopsy is the ability to procure material in inoperable patients. A salient example is WCM627, a pediatric patient with an inoperable infiltrative astrocytoma centered within the pons. In this patient, 14 frozen tissue aliquots were procured from the primary tumor mass within the brainstem at autopsy. Despite transporting the patient from an outside institution, our PMI for this patient was only 2 hours, and the effort led to viable cell cultures.
Tumor Clonal Architecture and Evolution
Autopsy material in combination with premortem surgical material is crucial to the study of the spatiotemporal evolution of cancer. In our cohort, prior surgical material was available in 13 (87%) of 15 patients. In one illustrative patient (WCM419), a young girl with recurrent anaplastic ependymoma, material was available from eight distinct surgical resection procedures spanning more than 5 years in addition to material obtained at autopsy (Fig 2). Multiple sites of disease were available for study including the posterior fossa (the primary site), metastatic lesions within the spinal cord, and a metastatic deposit involving the lateral ventricle obtained at autopsy (Figs 2A to 2C). Histologic examination of all tumor material showed prominent perivascular pseudorosette formation (Fig 2D). WES was performed on formalin-fixed paraffin-embedded material derived from prior surgical specimens as well as frozen autopsy tissue from two distinct sites (Figs 2E and 2F). Reconstruction of the evolutionary tree of this tumor revealed a complex branching pattern where several mutations (ERBB3, DNMT3A, BRCA1, NOTCH1, and RUNX1T1) in the primary tumor samples were not shared at different time points (0 to 64 months) or with descendent clones (Fig 2G). Moreover, we identified two genes, CUL9 and PIGM, as harboring alterations specific to three or greater metastatic sites of disease and absent from all tissues resected from the primary site of disease.
Fig 2.
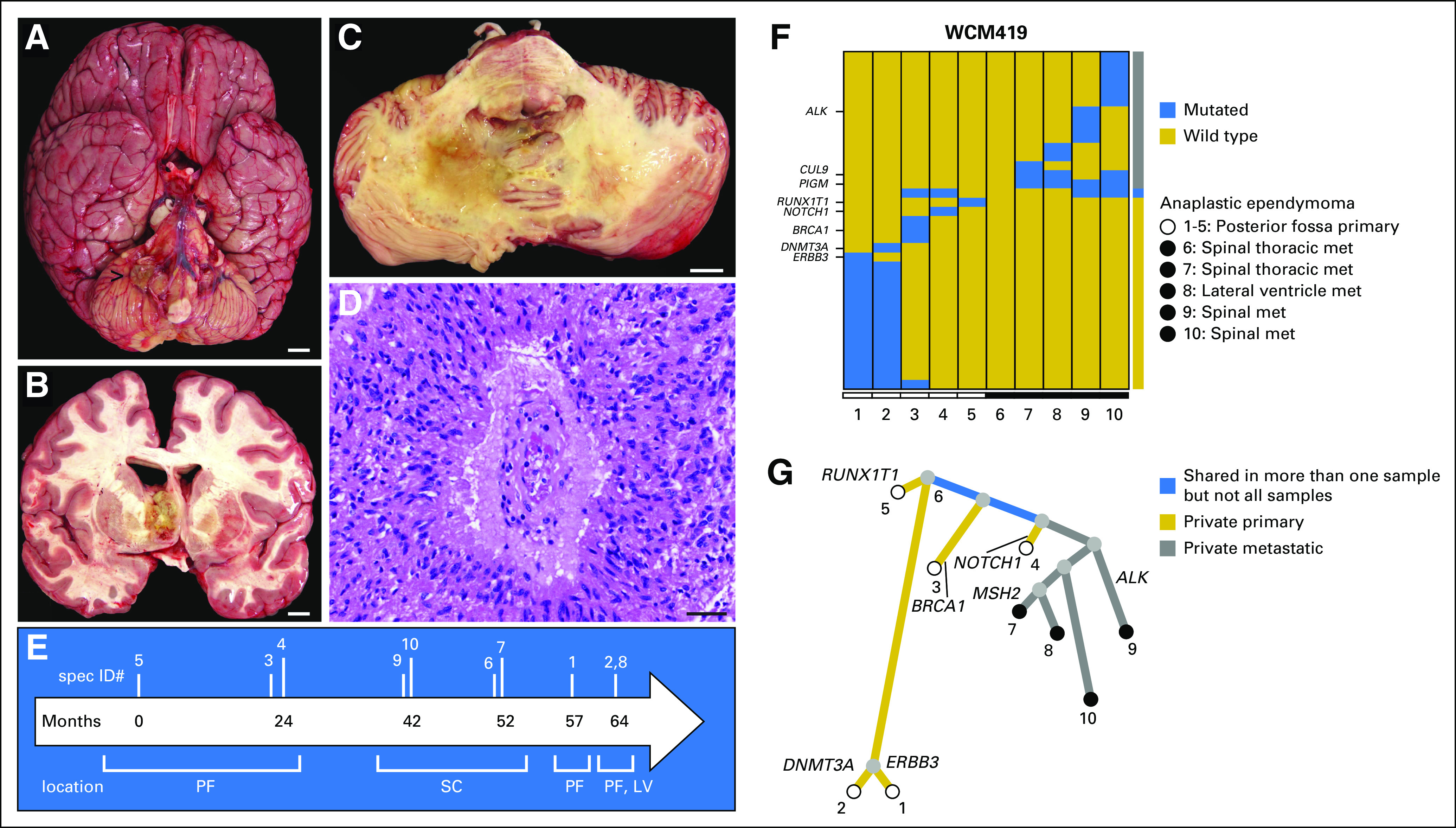
Clonal archeology of a patient with anaplastic ependymoma (rapid autopsy WCM419). Multiple sites of disease were available for study, including (A and B) the posterior fossa (primary site; [A] ventral surface of the whole brain with exophytic tumor component indicated by the arrow; [B] metastatic deposits within the supratentorial compartment involving the lateral ventricle and subventricular brain parenchyma, and lesions within the spinal cord (not shown), (C) axial cross-section through the pons and cerebellum). (D) Characteristic tumor histomorphology demonstrates spindled glial cells with fibrillar processes and perivascular pseudorosette formation (hematoxylin and eosin stain). (E) Timeline from diagnosis (month 0) to autopsy (month 64). Vertical lines indicate the relative timing of procedures from which samples were obtained during the course of disease. (F) Whole-exome sequencing was performed on prior surgical resection formalin-fixed paraffin-embedded specimens including primary tumor in posterior fossa (samples 1, 3, 4, and 5) and metastases to spinal cord (samples 6, 7, 9, and 10), as well as on frozen autopsy tissue from two distinct sites, including recurrent primary tumor in the posterior fossa (sample 2) and metastasis to the lateral ventricle (sample 8). Both the color-coded bar on the right and the phylogenetic tree below indicate whether mutations are private to primary or private to metastases or shared in more than one sample. There is no significant mutational overlap between the 10 samples. (G) Reconstruction of evolutionary tree. The length of the branches represents the distance between two tumors on the basis of the number of shared mutations. A complex branching pattern is present in this anaplastic ependymoma. Several mutations (ERBB3, DNMT3A, BRCA1, NOTCH1, and RUNX1T1) in the primary tumor samples are not shared either between them at different time points (0 to 64 months) or with descendent clones. None of the mutations detected was shared by all tumor samples. Scale bars = (A-C), 1 cm; (D), 40 μm. LV, lateral ventricle; met, metastasis; PF, posterior fossa; SC, spinal cord.
Molecular Features Among Multiple Metastatic Sites
Even in the absence of a definitive primary site, it is possible to gain insights into the evolutionary dynamics of tumor growth in distinct anatomic compartments. A case in point is WCM642, a 50-year-old male patient who presented with disseminated disease from a primary tumor of unknown origin (Fig 3). Prior biopsy demonstrated an epithelioid neoplasm without a definitive diagnosis. At autopsy, tumor was frozen from 15 distinctly annotated anatomic locations including adrenal gland and three distinct lobes of the lungs (Figs 3A and 3B), parietal pleura, omentum, lymph nodes (three distinct sites), small intestine, large intestine, liver (two distinct lobes), gallbladder, and pancreas. Diagnosis of this tumor was only ascertained at autopsy. At autopsy, cells demonstrated both epithelioid and sarcomatoid morphology (Figs 3C and 3D) and focal positive staining for S100 (not shown), which had been negative in the biopsy. Sequencing revealed mutations in NRAS and IDH1 (Fig 3E) and contributed to the final diagnosis of malignant melanoma.
Fig 3.
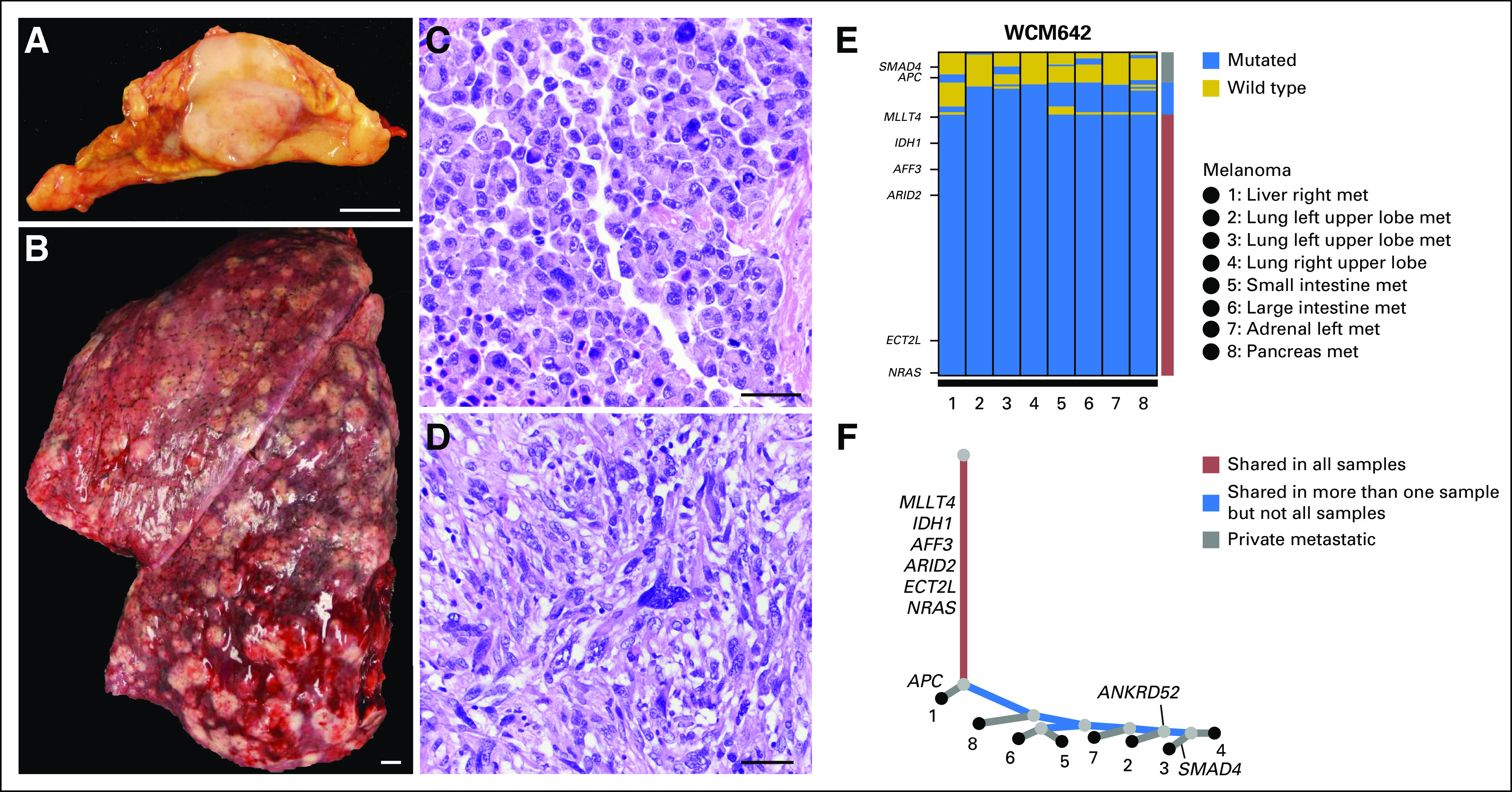
Molecular heterogeneity in a case of metastatic malignant melanoma (rapid autopsy WCM642). A 50-year-old male patient presented with disseminated disease from primary tumor of unknown origin. A previous biopsy (not shown) showed an unclassified malignant and epithelioid neoplasm. At autopsy, tissue samples from 17 distinct anatomic locations were snap frozen, including (A) adrenal gland and (B) lung. The tumor demonstrated heterogeneous histology with both (C) epithelioid and (D) spindled areas (hematoxylin and eosin stain). Tumor cells demonstrated focal staining for S100 (not shown), which had been negative in the biopsy. (E) Whole-exome sequencing results from eight distinct anatomic sites were obtained. Both the color-coded bar on the right and the phylogenetic tree below indicate whether mutations are private to any metastatic site or are shared in more than one sample or in all samples. Significant mutational overlap is present between all metastatic samples. (F) Reconstruction of the clonal architecture demonstrates that mutations in MLLT4, IDH1, AFF3, ARID2, ECT2L, and NRAS are shared in all samples. Few mutations are shared in more than one sample but not all samples (eg, APC in right liver metastasis (met); SMAD4 in left upper lobe of lung). The primary tumor was not identified and unavailable for sequencing analysis. Scale bars = (A and B), 1 cm; (C and D), 50 μm.
Reconstruction of the clonal architecture of eight metastases from this patient demonstrated several mutations that were shared in all samples (MLLT4, IDH1, AFF3, ARID2, ECT2L, and NRAS), as well as mutations restricted to particular metastatic sites (eg, APC in an LM and SMAD4 in lung; Fig 3F). Remarkably, the branch points of the phylogenetic tree reflect the anatomic distribution of metastases, with those encompassing the digestive system grouped separately from those to the adrenal gland and lungs. Moreover, metastases to the small and large intestines are more closely related with each other than with those to the liver and pancreas. These findings also correlate with site-specific alterations such as ANKRD52, which was mutated in three of three distinct lung metastases but in zero of five other metastatic sites tested.
In other patients, such as WCM0 (small-cell carcinoma of prostate) and WCM772 (pleomorphic carcinoma of lung), RNA-Seq data were used to further enhance comparisons across metastatic sites. In particular, for both of these patients, genes were identified that showed consistent overexpression across metastatic tumors for each patient at the transcriptional level relative to the expression of these genes over a large set of RNA-Seq samples. The latter analysis derived from diverse tumors over the accrued transcriptome profiling from the precision medicine clinical trial cohort (n = 300 samples). In the case of WCM0, CDKN2C was identified as an outlier across multiple sites, and in WCM772, MET was consistently overexpressed (Fig 4A). For WCM772, these data correlated with evidence of amplification of the MET gene derived from WES, whereas for WCM0, no such correlation was observed (Data Supplement). Additional RNA-Seq analysis of WCM0 samples identified novel gene fusion candidates such as ZNF526-MEGF8 (Figs 4B to 4D). In addition, WGS on two LMs from this patient yielded additional candidates, including the discovery of a gene fusion involving STYK1, a putative serine/threonine and tyrosine receptor protein kinase of potential clinical significance in castrate-resistant prostate cancer32 (Fig 5).
Fig 4.
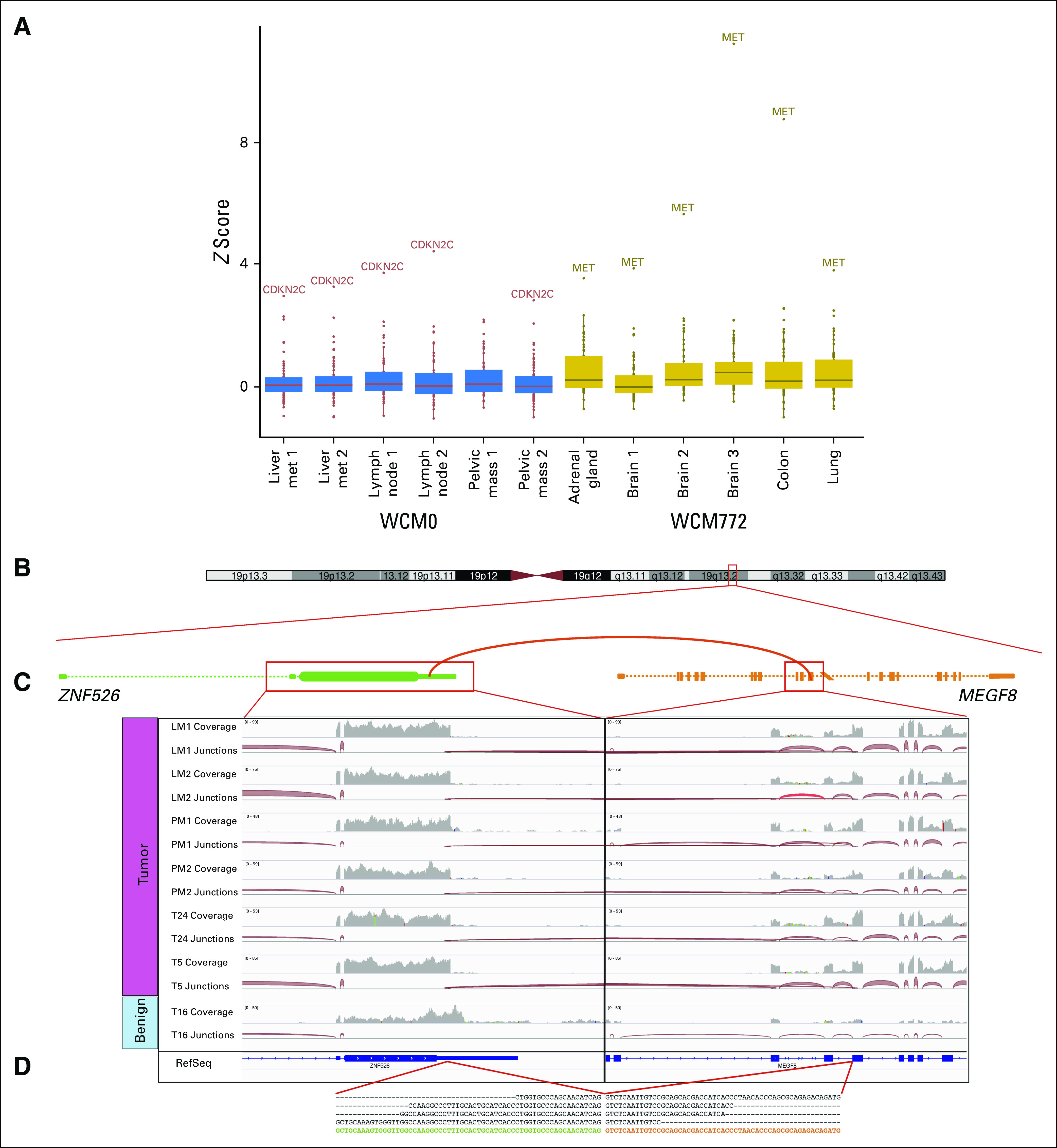
Examples of recurrent outliers and identification of novel gene fusion through RNA sequencing analysis. (A) Gene expression outlier analysis across 300 RNA sequencing samples derived from a diversity of tumors within the Institute for Precision Medicine cohort highlights WCM0 (small-cell carcinoma of prostate) and WCM772 (pleomorphic carcinoma of lung), two patients who show CDKN2C and MET as recurrent outliers in distinct anatomic samples, respectively. z scores were calculated for 70 druggable cancer genes, and outliers were selected at a cutoff of z score greater than 2.5 and fragments per kilobase million greater than 50. (B) RNA sequencing analysis by FusionCatcher and FusionSeq allows for the identification of novel gene fusion candidates. Schematic of the fusion between ZNF526 and MEGF8 in WCM0 showing the connected exons. (C) Integrative Genomics Viewer snapshots illustrating the data from six tumor specimens and one benign tissue specimen from the same patient. The coverage track summarizes the number of reads per nucleotide, whereas the junction tracks show how reads are connected by splicing. Note that the tumor samples all have connections between the two genes, whereas the benign specimen has none. (D) The sequence of the junction with some representative sequenced reads. met, metastasis.
Fig 5.
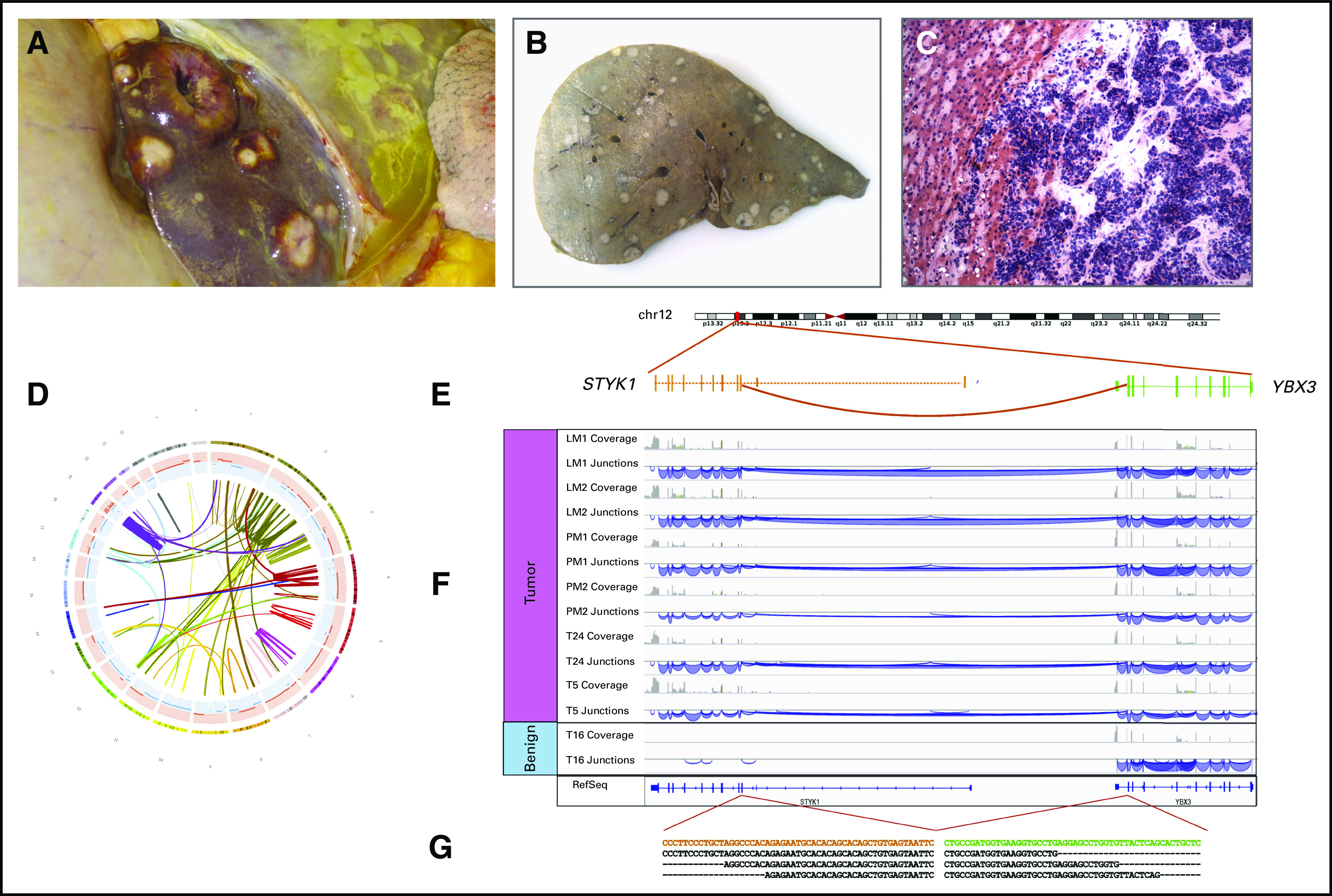
Whole-genome sequencing of small-cell carcinoma of prostate nominates a novel gene fusion with clinical relevance (rapid autopsy WCM0). A 55-year-old male patient status post prostatectomy presented with local recurrence, metastases in the pelvis and liver, and Cushing syndrome in the month before death. (A and B) Upon opening the abdominal cavity, numerous metastases were present in the liver. (A) Liver in situ with multiple lesions is shown. (B) Section of liver after fixation is shown. (C) On histologic examination, the tumor demonstrated small-cell neuroendocrine morphology (hematoxylin and eosin stain of frozen tissue, ×200 original magnification). (D) Circos plot of chained rearrangements in sample LM2 (liver metastasis). (E) Structural variants called with Clipping Reveals Structure algorithm led to identification of a novel intrachromosomal gene fusion. Schematic of the fusion between YBX3 and STYK1 showing the connected exons. (F) Integrative Genomics Viewer snapshots illustrating the data from six tumor specimens and one benign tissue specimen from the same patient. The coverage track summarizes the number of reads per nucleotide, whereas the junction tracks show how reads are connected by splicing. Note that the tumor samples all have connections between the two genes, whereas the benign specimen has none. (G) The sequence of the junction with some representative sequenced reads. Overexpression of STYK1 may serve as a potential molecular target in castrate-resistant prostate cancer.32
Tumor Organoid, PDX, and Cell Line Development
PDXs and cell lines are the most frequently used models in cancer research and anticancer drug screening. The tumor organoid model is a major in vitro breakthrough that has the potential to study in vivo biologic processes and may provide a new avenue for personalized cancer care.33,34
As demonstrated in patient WCM331, despite a delayed PMI of 6.5 hours as a result of transportation from an outside hospital, tumor organoid development of ovarian serous adenocarcinoma was achieved. Upon opening the peritoneal cavity, disseminated carcinomatosis was readily observed (Fig 6A), and tumor tissue was immediately placed into culture media. Viable organoids histologically comparable to metastatic autopsy tissue were produced and consistent with ovarian serous adenocarcinoma (Figs 6B to 6D). WES was performed on a metastatic omental lesion biopsied 2 years before autopsy, on several metastatic lesions obtained at autopsy, and on the tumor organoid material derived from a peritoneal nodule obtained at autopsy. The analysis demonstrates genetic alterations that are shared across these specimens (Figs 6E and 6F). Reconstruction of the clonal architecture of metastatic samples and tumor organoid demonstrates that PTPRB mutation is shared across all samples, whereas FOXO1 mutation is present in one metastasis (to small bowel serosa) and the tumor organoid only. At the same time, differences in certain alterations may reflect heterogeneity in disease across time and anatomic location. Alterations restricted to the tumor organoid material only (eg, PRDM16 and KMT2A) may reflect either additional tumor heterogeneity or alterations arising in vitro.
Fig 6.
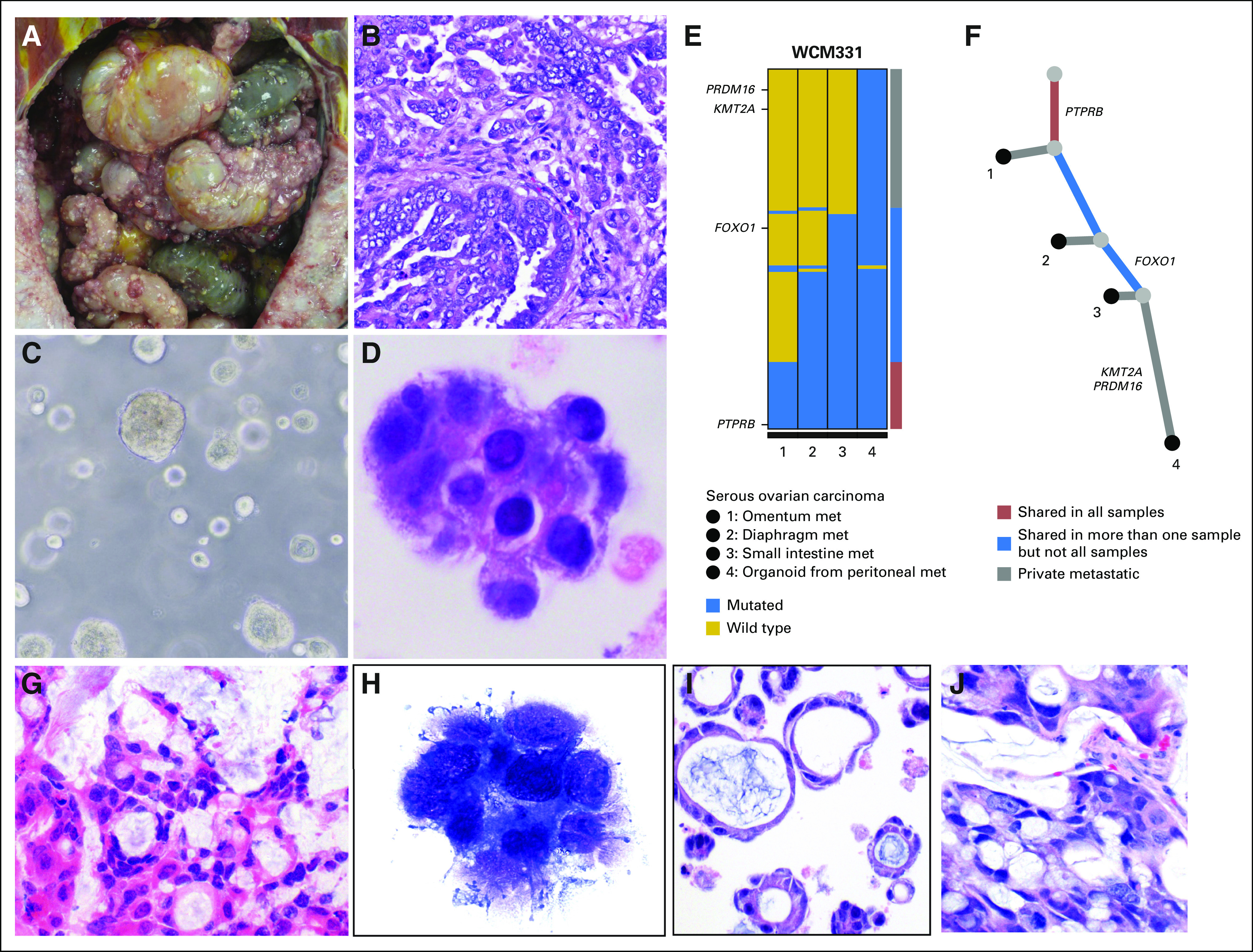
Tumor organoid and patient-derived xenograft development (rapid autopsies WCM331 and WCM715). (A) In patient WCM331, upon opening the peritoneal cavity, disseminated carcinomatosis was present. (B) Histologic examination confirmed high-grade serous adenocarcinoma of the ovary (hematoxylin and eosin [HE] stain, ×200 original magnification). (C) Viable tumor organoids were successfully produced. Image of patient-derived tumor organoid in a Matrigel scaffold (Corning, Corning, NY). (D) Cytologic comparison of these tumor organoids with representative sections from metastatic autopsy tissue revealed morphologically similar cells (Diff-Quik stain, ×400 original magnification). (E) Whole-exome sequencing was performed on formalin-fixed paraffin-embedded material derived from an omental metastasis biopsied 2 years before autopsy (sample 1), on frozen material from two metastases obtained at autopsy (samples 2 and 3), and on tumor organoid material derived from a peritoneal nodule obtained at autopsy (sample 4). Both the color-coded bar on the right and the phylogenetic tree below indicate whether mutations are private to the metastatic sites or are shared in more than one sample or in all samples. Multiple genetic alterations are shared across these specimens ranging from one (eg, FOXO1) to all samples (eg, PTPRB). This molecular evidence supports that the tumor organoid recapitulates the patient’s metastatic disease. (F) In the absence of material from the primary tumor, partial reconstruction of the clonal evolution of metastatic samples (including the tumor organoid as surrogate of one of the metastasis at autopsy) demonstrates that PTPRB mutation is shared in all samples and FOXO1 mutation is present in two metastases. Some alterations were seen in the tumor organoid only (eg, PRDM16 and KMT2A). (G) In case WCM715, an HE-stained section of a liver metastasis obtained at autopsy showed metastatic mucinous adenocarcinoma of the colon (×200 original magnification). (H) Smear preparation of tumor organoid culture (Diff-Quik stain) to confirm tumor growth (×400 original magnification). (I) HE-stained section of formalin-fixed paraffin-embedded organoid (×200 original magnification). (J) HE-stained section of patient-derived xenograft derived from organoid implantation in mouse demonstrates comparable histomorphology with original liver metastasis and derived tumor organoid (×200 original magnification). met, metastases.
Another example of patient-derived preclinical model development from rapid autopsy tissue is illustrated in a patient with colon adenocarcinoma (WCM715; Fig 6G) for whom both tumor organoid (Figs 6H and 6I) and downstream PDX models (Fig 6J) were established. In addition to successful PDX development using cells directly harvested from rapid autopsy material (eg, prostate cancer in patient WCM677), cell line development was also pursued (eg, diffuse midline glioma in patient WCM627, with viable passages to date; data not shown).
DISCUSSION
Tissue procurement from patients with cancer at autopsy and within the context of a pan-cancer precision medicine clinical trial captures biologic data at a state in disease progression that is rarely studied. In contrast to previously reported autopsy programs, we have incorporated data analysis and development of preclinical models using multiple platforms,2 clinical-grade assays,13 and computational algorithms,13,35,36 leveraging the infrastructure of IPM. Here, we illustrate a proof of principle in developing a powerful model for interrogating a diverse set of primary tumor types and metastatic sites, including newly generated tumor organoids, PDX models, and cell culture. Indeed, with just the first 15 autopsies, we have revealed novel genetic candidates as potential mediators of metastatic spread in both ependymoma and melanoma, as well as provided the foundation for additional work investigating treatment effect in metastatic urothelial carcinoma.
As a testament to our ability to track patients through their disease course, we analyzed primary tumor samples in 77% of patients for whom metastatic disease was procured at autopsy and in 80% of patients overall. Moreover, prior surgically obtained premortem tumor material (either primary or metastatic) was available for study in 87% of patients, enabling in the vast majority of patients the potential to study tumor evolution over time and through diverse therapeutic modalities.
We have demonstrated that analysis of WES data of rapid autopsy material in the context of a precision medicine clinical trial permits the identification of genes that become altered at biologically significant branch points in tumor evolution, including those that may contribute to metastatic potential or treatment resistance. For example, analysis of WCM419 revealed alterations in CUL9 and PIGM that were present in most or all spatiotemporally distinct metastatic sites of ependymoma but absent from multiple resections at the primary site. CUL9, which encodes a putative E3 ubiquitin ligase that binds to p53, has been described as a tumor suppressor,37 whereas PIGM encodes a mannosyltransferase involved in glycosylphosphatidylinositol-anchor biosynthesis.38 In addition, we have identified gene candidates for further study that may mediate organotropism of metastases in melanoma; for example, we found that ANKRD52 mutations were restricted to lung metastases in a patient with widely metastatic melanoma. Finally, additional work on two patients from within this cohort (WCM117 and WCM259) has already yielded further insights into chemotherapy-resistant urothelial carcinoma, demonstrating enrichment in clonal alterations including targetable mutations (eg, L1CAM and integrin signaling pathways).21
Comparison of WES between primary tumors, metastatic sites, and preclinical in vitro models of disease also enables an assessment of the extent to which critical molecular features of a tumor are reiterated, for example, in tumor organoids. The latter seem to recapitulate the patient’s metastatic disease at the exome level, representing an opportunity to develop coclinical trials and other clinically relevant applications (discussed in additional detail by Pauli et al39). In addition, investigation of WGS and RNA-Seq data including gene expression outlier analysis across our precision medicine cohort has allowed the identification of novel gene fusions and overexpression of certain transcripts across metastatic sites with potential clinical relevance.
We have set the foundation to investigate several critical questions over a diverse set of neoplasms, including the temporal evolution of tumors, the clonal evolution of tumors with respect to widely disseminated metastatic disease sites, and the production and examination of tumor organoid models derived from these specimens. Performing additional drug screening on preclinical platforms developed from autopsy tissue has the potential of benefiting future patients with molecularly similar tumors. Analyses of the data in each one of these extremely valuable patients is ongoing, with the goal of gleaning as much information as possible from every patient.
Acknowledgment
We thank our patients and their families for participation in this study. We also acknowledge Douglas Scherr, MD, and Christopher Barbieri, MD, for contributing samples; to Yasmin Khakoo, MD, and our other referring clinicians; to our clinical pathology fellows for their assistance during rapid autopsies; to Terra McNary, Loredana Puca, Mirjam Blattner, Uday Bhanu Maachani, Nector Garcia, Anvioris Taveras, and Yelena Churakova for technical assistance; to funeral home transport personnel; to autopsy and laboratory personnel involved in this project; and to Rogério Paulo da Silva for art design.
Footnotes
Supported in part by the National Institutes of Health (NIH; Grant No. R01 CA116337 to H.B., F.D., and M.A.R.; Grant No. 5U01 CA111275-09 to J.M.M., M.A.R., and F.D.), the Conquer Cancer Foundation and the John and Elizabeth Leonard Family Foundation Young Investigator Award (B.M.F), NIH/National Center for Advancing Translational Sciences Grant No. KL2TR000458 (B.F), Damon Runyon-Gordon Family Clinical Investigator Award No. CI-67-13 (H.B.), Department of Defense Prostate Cancer Research Program Grant No. PC121341 (H.B.), Starr Cancer Consortium (H.B. and M.A.R), Nuovo Soldati Foundation (J.C.), Associazione Italiana per la Ricerca sul Cancro (Grant No. IG 13562 to F.D.), and the European Research Council (Consolidator Grant for Synthetic Lethal Phenotype Identification Through Cancer Evolution Analysis to F.D.). Also supported in part by the Translational Research Program at Weill Cornell Medicine Pathology and Laboratory Medicine.
AUTHOR CONTRIBUTIONS
Conception and design: David J. Pisapia, Steven Salvatore, Chantal Pauli, Brian D. Robinson, Robert Kim, Alexandros Sigaras, Bishoy Faltas, David M. Nanus, Prajwal Rajappa, Jeffrey Greenfield, Himisha Beltran, Mark A. Rubin, Juan Miguel Mosquera
Financial support: Bishoy Faltas, Jeffrey Greenfield, Francesca Demichelis, Himisha Beltran, Mark A. Rubin, Juan Miguel Mosquera
Administrative support: Steven Salvatore, Robert Kim, Juan Miguel Mosquera
Provision of study material or patients: Steven Salvatore, Scott T. Tagawa, Robert Kim, David M. Nanus, Olivier Elemento, Himisha Beltran, Juan Miguel Mosquera
Collection and assembly of data: David J. Pisapia, Steven Salvatore, Chantal Pauli, Erika Hissong, Ken Eng, Verena-Wilbeth Sailer, Brian D. Robinson, Kyung Park, Joanna Cyrta, Scott T. Tagawa, Myriam Kossai, Jacqueline Fontugne, Robert Kim, Alexandros Sigaras, Danielle Pancirer, Bishoy Faltas, Ana M. Molina, Prajwal Rajappa, Mark M. Souweidane, Himisha Beltran, Juan Miguel Mosquera
Data analysis and interpretation: David J. Pisapia, Steven Salvatore, Ken Eng, Davide Prandi, Verena-Wilbeth Sailer, Brian D. Robinson, Alexandros Sigaras, Rema Rao, Bishoy Faltas, Rohan Bareja, Prajwal Rajappa, Jeffrey Greenfield, Anne-Katrin Emde, Nicolas Robine, Olivier Elemento, Andrea Sboner, Francesca Demichelis, Himisha Beltran, Mark A. Rubin, Juan Miguel Mosquera
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Next-Generation Rapid Autopsies Enable Tumor Evolution Tracking and Generation of Preclinical Models
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
David J. Pisapia
No relationship to disclose
Steven Salvatore
No relationship to disclose
Chantal Pauli
No relationship to disclose
Erika Hissong
No relationship to disclose
Ken Eng
No relationship to disclose
Davide Prandi
No relationship to disclose
Verena-Wilbeth Sailer
No relationship to disclose
Brian D. Robinson
Stock and Other Ownership Interests: Metastat,
Consulting or Advisory Role: Progenics Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Methods for diagnosing and treating prostate cancer
Kyung Park
No relationship to disclose
Joanna Cyrta
No relationship to disclose
Scott T. Tagawa
Consulting or Advisory Role: Medivation, Astellas Pharma, Dendreon, Janssen, Bayer, Genentech, Sanofi, Endocyte, Immunomedics
Speakers' Bureau: Amgen
Research Funding: Eli Lilly (Inst), Sanofi (Inst), Janssen (Inst), Astellas Pharma (Inst), Progenics (Inst), Millennium (Inst), Amgen (Inst), Bristol-Myers Squibb (Inst), Dendreon (Inst), Rexahn Pharmaceuticals (Inst), Bayer (Inst), Genentech (Inst), Newlink Genetics (Inst), Inovio Pharmaceuticals (Inst), AstraZeneca (Inst), Immunomedics (Inst), Novartis (Inst), AVEO (Inst), Rexahn Pharmaceuticals (Inst), Boehringer Ingelheim (Inst), Merck (Inst), Stem CentRx (Inst)
Travel, Accommodations, Expenses: Sanofi
Myriam Kossai
No relationship to disclose
Jacqueline Fontugne
No relationship to disclose
Robert Kim
No relationship to disclose
Alexandros Sigaras
Employment: Weill Cornell Medical College
Rema Rao
No relationship to disclose
Danielle Pancirer
No relationship to disclose
Bishoy Faltas
No relationship to disclose
Rohan Bareja
No relationship to disclose
Ana M. Molina
Honoraria: ASCO
Consulting or Advisory Role: Eisai, Exelixis, Novartis
David M. Nanus
Consulting or Advisory Role: Genentech
Prajwal Rajappa
No relationship to disclose
Mark M. Souweidane
Consulting or Advisory Role: Aesculap
Travel, Accommodations, Expenses: Aesculap
Jeffrey Greenfield
No relationship to disclose
Anne-Katrin Emde
No relationship to disclose
Nicolas Robine
No relationship to disclose
Olivier Elemento
No relationship to disclose
Andrea Sboner
No relationship to disclose
Francesca Demichelis
Patents, Royalties, Other Intellectual Property: Co-inventor on a patent filed by the University of Michigan and the Brigham and Women’s Hospital covering the diagnostic and therapeutic fields for ETS fusions in prostate cancer. The diagnostic field has been licensed to Gen-Probe.
Himisha Beltran
Consulting or Advisory Role: Bayer, Janssen Oncology, Genzyme
Research Funding: Astellas Pharma (Inst), Eli Lilly (Inst), Janssen (Inst), Millennium (Inst), Stemcentryx Abbvie
Mark A. Rubin
Research Funding: Eli Lilly, Janssen
Juan Miguel Mosquera
No relationship to disclose
REFERENCES
- 1.Beltran H, Eng K, Mosquera JM, et al. : Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol 1:466-474, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pauli C, Puca L, Mosquera JM, et al. : An emerging role for cytopathology in precision oncology. Cancer Cytopathol 124:167-173, 2016 [DOI] [PubMed] [Google Scholar]
- 3. Rubin MA, Putzi M, Mucci N, et al: Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res 6:1038-1045, 2000. [PubMed]
- 4.Shah RB, Mehra R, Chinnaiyan AM, et al. : Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res 64:9209-9216, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Zarghooni M, Bartels U, Lee E, et al. : Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol 28:1337-1344, 2010 [DOI] [PubMed] [Google Scholar]
- 6. Esgueva R, Park K, Kim R, et al: Next-generation prostate cancer biobanking: Toward a processing protocol amenable for the International Cancer Genome Consortium. Diagn Mol Pathol 21:61-68, 2012. [DOI] [PMC free article] [PubMed]
- 7.Udager AM, Shi Y, Tomlins SA, et al. : Frequent discordance between ERG gene rearrangement and ERG protein expression in a rapid autopsy cohort of patients with lethal, metastatic, castration-resistant prostate cancer. Prostate 74:1199-1208, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Juric D, Castel P, Griffith M, et al. : Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518:240-244, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kambhampati M, Perez JP, Yadavilli S, et al. : A standardized autopsy procurement allows for the comprehensive study of DIPG biology. Oncotarget 6:12740-12747, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie T, Musteanu M, Lopez-Casas PP, et al. : Whole exome sequencing of rapid autopsy tumors and xenograft models reveals possible driver mutations underlying tumor progression. PLoS One 10:e0142631, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beltran H, Prandi D, Mosquera JM, et al. : Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22:298-305, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffman LM, DeWire M, Ryall S, et al. : Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: Implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4:1, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rennert H, Eng K, Zhang T, et al. : Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genom Med 1:16019, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754-1760, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xi R, Hadjipanayis AG, Luquette LJ, et al. : Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA 108:E1128-E1136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quail MA, Kozarewa I, Smith F, et al. : A large genome center’s improvements to the Illumina sequencing system. Nat Methods 5:1005-1010, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romanel A, Lago S, Prandi D, et al. : ASEQ: Fast allele-specific studies from next-generation sequencing data. BMC Med Genomics 8:9, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramos AH, Lichtenstein L, Gupta M, et al. : Oncotator: Cancer variant annotation tool. Hum Mutat 36:E2423-E2429, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A, Hanna M, Banks E, et al. : The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297-1303, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Mullighan CG, Easton J, et al. : CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8:652-654, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faltas BM, Prandi D, Tagawa ST, et al. : Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet 48:1490-1499, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nixon KC: The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 15:407-414, 1999 [DOI] [PubMed] [Google Scholar]
- 23.Dobin A, Davis CA, Schlesinger F, et al. : STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15-21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stransky N, Cerami E, Schalm S, et al. : The landscape of kinase fusions in cancer. Nat Commun 5:4846, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. GitHub: STAR-Fusion/STAR-Fusion. https://github.com/STAR-Fusion/STAR-Fusion.
- 26. doi: 10.1101/011650. Nicorici D, Satalan M, Edgren H, et al: FusionCatcher: A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv . [DOI] [Google Scholar]
- 27.Sboner A, Habegger L, Pflueger D, et al. : FusionSeq: A modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol 11:R104, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Handsaker B, Wysoker A, et al. : The sequence alignment/map format and SAMtools. Bioinformatics 25:2078-2079, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Roberts A, Goff L, et al. : Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562-578, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Derrien T, Johnson R, Bussotti G, et al. : The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 22:1775-1789, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shugay M, Ortiz de Mendíbil I, Vizmanos JL, et al. : Oncofuse: A computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29:2539-2546, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Chung S, Tamura K, Furihata M, et al. : Overexpression of the potential kinase serine/threonine/tyrosine kinase 1 (STYK 1) in castration-resistant prostate cancer. Cancer Sci 100:2109-2114, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao D, Chen Y: Organoid development in cancer genome discovery. Curr Opin Genet Dev 30:42-48, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fatehullah A, Tan SH, Barker N: Organoids as an in vitro model of human development and disease. Nat Cell Biol 18:246-254, 2016 [DOI] [PubMed] [Google Scholar]
- 35.Prandi D, Baca SC, Romanel A, et al. : Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol 15:439, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demichelis F, Greulich H, Macoska JA, et al. : SNP panel identification assay (SPIA): A genetic-based assay for the identification of cell lines. Nucleic Acids Res 36:2446-2456, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Pei XH, Yan J, et al. : CUL9 mediates the functions of the 3M complex and ubiquitylates survivin to maintain genome integrity. Mol Cell 54:805-819, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almeida AM, Murakami Y, Layton DM, et al. : Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med 12:846-851, 2006 [DOI] [PubMed] [Google Scholar]
- 39. doi: 10.1158/2159-8290.CD-16-1154. Pauli C, Hopkins BD, Prandi D, et al: Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov 10.1158/2159-8290.CD-16-1154 [epub ahead of print on March 22, 2017] [DOI] [PMC free article] [PubMed] [Google Scholar]