

Supplemental Digital Content is Available in the Text.
Neublastin showed some evidence of pain relief among patients with painful lumbosacral radiculopathy; however, there was no clear dose–response relationship.
Keywords: SPRINT, Neublastin, Bayesian adaptive, Painful lumbosacral radiculopathy, Sciatica
Abstract
Neublastin (BG00010) is a first-in-class, glial cell–derived neurotrophic factor shown in preclinical studies and an early clinical trial to have potential for the treatment of neuropathic pain. SPRINT was a phase 2, multicenter, double-blinded, placebo-controlled study to evaluate efficacy/safety of 5 neublastin doses (50, 150, 400, 800, and 1200 μg/kg) administered as an intravenous injection 3 times/week for 1 week in patients with chronic painful lumbosacral radiculopathy, utilizing Bayesian response-adaptive study design. Primary endpoint was change from baseline in mean 24-hour average general pain intensity over a 5-day period (week 1) after the last dose, analyzed using a Bayesian normal dynamic linear model. One hundred seventy-six patients were randomized and received treatment (placebo n = 48, 50 μg/kg n = 38, 150 μg/kg n = 13, 400 μg/kg n = 16, 800 μg/kg n = 20, 1200 μg/kg n = 41). Among the tested neublastin doses, the lowest dose (50 μg/kg) showed the greatest difference from placebo for change from baseline in mean average general pain intensity at week 1 after last dose, followed by the highest dose (1200 μg/kg) (posterior mean difference −1.36 [95% credible interval −2.22 to −0.52] and −0.75 [−1.59 to 0.08], respectively). Similar trends were observed in secondary efficacy endpoints. The most common adverse event in all neublastin dose groups was pruritus (79% vs 10% with placebo). There was no dose–response relationship with respect to primary/secondary efficacy outcomes or incidence of pruritus, despite dose-proportional increases in serum neublastin concentrations. In conclusion, while this study showed some evidence of pain relief with neublastin, particularly at the lowest dose, there was no clear dose–response relationship for pain reduction or the most common adverse event of pruritus.
1. Introduction
Painful lumbosacral radiculopathy (PLSR) is a neuropathic pain condition characterized by pain radiating along one or more lumbar or sacral dermatomes and caused by nerve root irritation, inflammation, or compression. Painful lumbosacral radiculopathy is thought to be the most common form of neuropathic pain, with lifetime prevalence estimates from 12% up to 43% in the general population.20,32,33 While acute PLSR (<6 weeks' duration) will often resolve spontaneously, a significant proportion of patients (estimated at around 40%37) develop chronic PLSR (>12 weeks' duration). Neuropathic pain, including PLSR, imposes a substantial burden on patients' quality of life and has a sizeable economic impact due to health care utilization and loss of work productivity.25,30
Currently, no pharmacologic treatment options are specifically indicated for PLSR. Pharmacologic treatment of chronic PLSR typically follows guideline recommendations for neuropathic pain,10 including treatment with tricyclic antidepressants and anticonvulsants. To date, clinical trials of drug therapies conducted specifically among patients with chronic PLSR have tended to show limited efficacy or inconclusive outcomes, including trials of pregabalin,4 topiramate,19 nortriptyline/morphine,18 and tumor necrosis factor-alpha inhibitors.2,10,17,21–23
Neublastin (BG00010, artemin) is a first-in-class, glial cell line–derived neurotrophic factor family member1,3 that has shown potential for the treatment of neuropathic pain in preclinical studies and an early clinical trial. Neublastin is a selective ligand for the glial cell line–derived neurotrophic factor family receptor alpha-3 coreceptor that is expressed predominantly on small dorsal root ganglion sensory neurons and acts as a survival factor for sensory and sympathetic neurons in cell culture.3 Preclinical data from surgically and chemically induced nerve injury models have shown that neublastin normalizes morphological and neurochemical features of injured small dorsal root ganglion neurons and mitigates behavioral symptoms associated with neuropathic pain states.5,15,36,39 Neublastin thus has a novel mechanism of action with potential to influence underlying neuronal dysfunction in neuropathic pain, in addition to pain and associated symptom relief.
In phase 1 single- and multiple-ascending dose studies in patients with sciatica,26,29 intravenous infusion of neublastin produced dose-dependent increases in serum neublastin concentrations, was generally well tolerated, and in the multiple-ascending dose study,26 tentatively associated with improvements in pain measures. The primary objective of this phase 2 trial (SPRINT) was to further evaluate the safety and efficacy of intravenous neublastin to reduce pain in patients with PLSR. Given the novel mechanism of action of neublastin, there is uncertainty around optimal dosing; therefore, to explore a range of doses, we employed an advanced study design (Bayesian response-adaptive allocation), which has not commonly been used for pain research and analgesics development.
2. Methods
2.1. Study design
The SPRINT trial was a phase 2, multicenter, double-blind, placebo-controlled study utilizing a Bayesian response-adaptive allocation design to evaluate the efficacy and safety of neublastin administered as an intravenous injection 3 times per week for 1 week in patients with PLSR. The study duration was approximately 13 weeks, including a screening visit within 28 days of first dose, a 1-week treatment period, and a 56-day follow-up period (end-of-study visit was on day 61; Fig. 1). The SPRINT trial is registered on ClinicalTrials.gov (NCT01873404).
Figure 1.
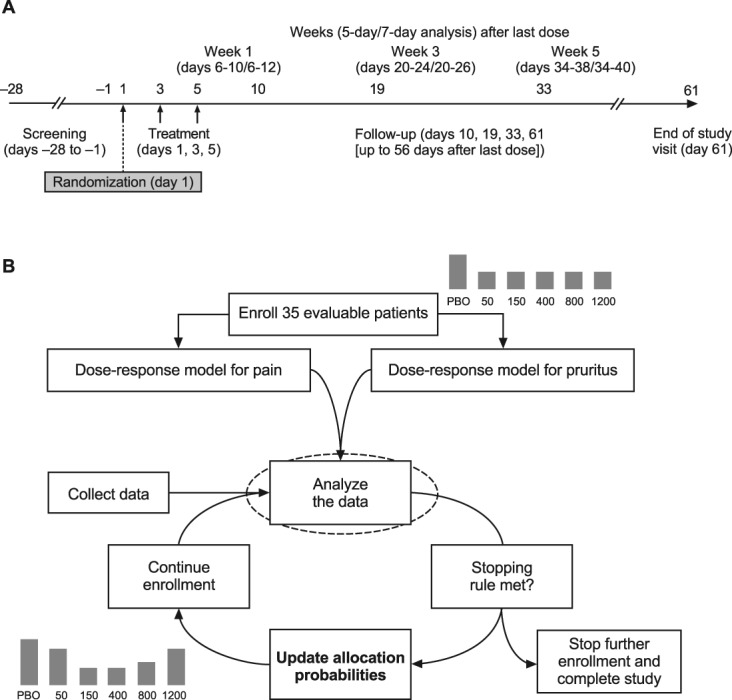
(A) Study design and (B) flow diagram illustrating the Bayesian algorithm used for randomization and dose selection. PBO, placebo.
The study was conducted at 37 participating sites in the United States, of which 23 enrolled patients. The period of study conduct was between June 2013 and March 2015. Prior to the first patient's enrollment at each site, initiation visits were held for the purpose of training site staff on data collection, procedures, and tests and evaluations to be performed in the study. Three investigator meetings were also held on March 2013, December 2013, and June 2014. Patients also received Accurate Pain Reporting Training (Analgesic Solutions, Natick, MA), and a Placebo Response Reduction Program was provided to all research staff and patients (Analgesic Solutions).
The study design and amendments were approved by the relevant local ethics committees at participating sites. The study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and with International Conference on Harmonisation Good Clinical Practice Guidelines. Written informed consent was obtained from all patients.
2.2. Patient population
Patients (≥18 years of age) were enrolled who had symptoms of pain radiating to or below the knee in a dermatomal pattern that had been present for ≥6 months but no longer than 10 years (at the time of screening), and that on the basis of the patient's history would lead the investigator to conclude that symptoms were due to PLSR. Patients also had objective, documented evidence of PLSR, as evidenced by one of the following assessments, correlating with their symptomatology of PLSR: (1) electromyographic evidence of fourth or fifth lumbar root (L4/L5)/first sacral root (S1) irritation on the affected side; or (2) imaging (magnetic resonance imaging or computed tomography/myelogram) with evidence of nerve root compression in the lower lumbar region on the affected side. In addition, all patients were required, on the basis of physical neurological examination, to have one of the following: evidence, on the affected side, of decreased/absent ankle or patellar reflex, weakness of muscles below the knee, or sensory loss in L5/S1 distribution, or a positive straight leg raising test.
Eligible patients were further required to have lower back pain (the onset of which occurred within ≥6 months of the time of randomization) on an average of 5 days per week for the past 6 months, and a leg pain score of ≥4 on an 11-point numeric rating scale (NRS) at screening. Patients also had to have completed average general pain intensity (AGPI) measurements on ≥5 of the 7 days before randomization, with the average of these AGPI scores ≥4 and <9 on the 11-point NRS, and to have failed to respond to ≥2 standard-of-care therapies for PLSR (as determined by practice standards in the local community).
Patients were ineligible if they had signs and symptoms of peripheral neuropathy affecting the limbs other than those explained by L4/L5/S1 radiculopathy, or if they had >1 surgical treatment for PLSR, >1 neurolytic injection, or treatment with epidural corticosteroids in the 3 months before randomization. Patients were allowed to remain on current treatment for PLSR symptoms, but the dose and regimen of opioid or nonopioid analgesics, multiple anticonvulsants, multiple antidepressants, or combinations of anticonvulsants and antidepressants must have been stable for ≥4 weeks before screening. Doses of other prescription and over-the-counter products also had to have been stable for ≥2 weeks before enrollment.
Rescue medication use was not allowed during the first 2 weeks after the first dose of neublastin. After this period, rescue medication with acetaminophen <4 g/d was permitted for PLSR and for pain and headaches other than PLSR.
2.3. Randomization and stopping criteria
Following screening, eligible patients were randomized on day 1 to treatment with placebo or neublastin 50, 150, 400, 800, or 1200 μg/kg, administered by intravenous injection on days 1, 3, and 5. Randomization was performed centrally using an interactive voice/web response system (IXRS). All patients, investigators, site personnel, and the sponsor's staff were masked to treatment assignment.
The first 35 patients were randomized in a 2:1:1:1:1:1 ratio to placebo and each of the 5 active doses (ie, 10 patients in the placebo group and 5 for each dose of active treatment). Subsequently, 2 of every 7 enrolled patients were assigned to placebo. Interim data evaluations of pain (AGPI) and pruritus questionnaire data (proportion of patients who reported “the itch is severe enough to cause major problems for me” on an Itch Impact Questionnaire) were used to update the allocation probability according to a Bayesian algorithm for adaptive allocation and to assess efficacy and futility criteria for early stopping of enrollment (Fig. 1). Interim evaluations and updates to the allocation probabilities were performed weekly by an independent statistical vendor (further details of the statistical modeling are provided in a supplementary appendix online at http://links.lww.com/PAIN/A433). Enrollment was to be stopped early after ≥50 patients had been followed for 4 weeks if either the efficacy criterion (>80% probability that the maximum utility dose reduces the pain score by ≥1.5 points more than the placebo) or the futility criterion (<45% probability that the maximum utility dose reduces pain more than the placebo) was met.
The study was also to be terminated directly if 3 dose groups were permanently discontinued because of specified safety criteria pertaining to treatment-emergent neurological abnormalities, increases in pain, or higher than anticipated incidence of severe pruritus, severe rash, or serious adverse events (SAEs) of a similar nature. Safety criteria, with the exception of criteria regarding SAEs, were reviewed by an unblinded statistician every 2 weeks after 35 patients had been randomized and received ≥1 dose of study drug to determine whether any of the discontinuation criteria were met. Serious adverse events were reviewed by Safety and Benefit-Risk Management (SABR; Biogen) on an ongoing basis. If any of the discontinuation criteria were met in an active dose group, it would prompt an unblinded review of the relevant results by SABR, who would determine whether to permanently discontinue randomization in that dose group.
2.4. Efficacy assessments
Patients were required to complete an electronic diary (PHT Corporation, Geneva, Switzerland) from the screening visit onwards to rate their 24-hour AGPI daily on an 11-point NRS. Pain data recorded during the 7 days before day 1 were used to determine baseline pain scores.
The primary endpoint was change from baseline in mean 24-hour AGPI, which was calculated over a 5-day period (week 1) after the last dose (ie, days 6-10). Secondary efficacy endpoints were changes from baseline in mean 24-hour AGPI at weeks 3 (days 20-24) and 5 (days 34-38), as well as mean 24-hour average back pain intensity (ABPI) and mean 24-hour average leg pain intensity (ALPI) at weeks 1, 3, and 5 after last dose.
Exploratory efficacy measures included the Daily Sleep Interference Scale (DSIS),34 Patient Global Impression of Change (PGIC),11 Brief Pain Inventory (BPI)9 (including 2 summary scores: BPI Overall Severity Score and BPI Interference Score), and physical activity and sleep parameters measured using a wrist accelerometer.
2.5. Safety assessments
Data pertaining to adverse events (AEs) other than PLSR-related pain were collected throughout the study and coded using the Medical Dictionary for Regulatory Activities version 17. The relationship between study medication and AEs was evaluated by the investigators at the site. All SAEs occurring during the study were followed until the event had resolved, stabilized, or returned to baseline status. Routine clinical laboratory evaluations (hematology, biochemistry, and urinalysis), physical examination, vital signs, 12-lead electrocardiogram, and clinical neurological examination were also performed.
For AEs of special interest, pruritus and rash, additional analyses were conducted, including analyses of maximum severity, resolution at end of study, and number of days with the AE. Patients reporting pruritus were also asked to complete an Itch Impact Questionnaire via the electronic diary and the Itch Quality of Life Questionnaire (ItchyQoL)13 at the end of the study or early termination.
Other safety evaluations included skin punch biopsy for determination of intraepidermal nerve fiber density (performed for first 50 randomized patients on days 1 and 33, with biopsies obtained from the distal lateral thigh), clinical neurological examinations (motor, deep tendon reflex, and sensory examinations), and the Columbia Suicide Severity Rating Scale (C-SSRS).27
2.6. Pharmacokinetics and immunogenicity assessments
Blood samples were collected before dose and within 1 minute of neublastin injection at days 1, 3, and 5 for analysis of serum neublastin concentrations. Following the day 5 dose, samples were also taken at 1 hour after dose for all patients and at 2 hours after dose for the first 35 patients. Serum concentrations of neublastin were measured using a chemiluminescent enzyme-linked immunosorbent assay.
Serum samples for evaluation of neublastin-binding and -neutralizing antibodies were collected at baseline, day 33, and end of study (day 61). The presence of anti-neublastin antibodies was determined using a tiered assay approach involving a screening assay and a confirmation assay, followed by titration of positive samples. Titer values were determined for confirmed positive samples, which were then evaluated in the neutralizing antibody assay.
2.7. Other assessments
Baseline data for neuropathic pain were collected using the PainDETECT questionnaire14 and the Neuropathic Pain Symptom Inventory (NPSI).7 A Patient Expectation Questionnaire was also completed at baseline and at the end of the study to determine expectations for pain reduction and beliefs with regard to treatment received.
2.8. Statistical analyses
The target enrollment was 165 patients in order to provide 150 evaluable patients in the efficacy population for the primary endpoint. The maximum sample size was determined by simulation, so that the study would provide approximately 75% power to detect a treatment difference of 1.5 points between the most efficacious dose and placebo in change from baseline in the mean AGPI score over the 5 days (week 1) after last dose if the final data were analyzed using traditional analysis of variance tests. This simulation assumed a common SD of 2.2 in all arms and a dropout rate of 10%. The maximum sample size could be increased if the dropout rate was more than expected or the SD was >2.2.
The primary efficacy analysis of change from baseline in mean AGPI over the 5 days (week 1) after last dose was analyzed using a Bayesian normal dynamic linear model (NDLM), with adjustment for treatment group and baseline AGPI score. A sensitivity analysis was performed on the primary endpoint using an analysis of covariance (ANCOVA) model with treatment group as a factor and baseline AGPI score as a covariate. Subsequent 5-day averages for AGPI scores were calculated for weeks 2 to 8 after last dose. If a patient had fewer than 5 of 7 nonmissing baseline pain scores or fewer than 3 of 5 nonmissing scores for any of the posttreatment weeks, mean weekly score for those weeks was set to missing. Analysis of covariance was also used to analyze change from baseline in mean AGPI at weeks 3 and 5 and mean ABPI (back pain) and ALPI (leg pain) at weeks 1, 3, and 5 after last dose.
The following populations were defined for analyses: (1) efficacy population: all patients who were randomized, received ≥1 dose of study treatment, and had ≥1 after baseline assessment of the parameter being analyzed; (2) per-protocol population: the subset of the efficacy population without any major protocol deviations that could impact efficacy assessments; (3) safety population: all patients who were randomized and received ≥1 dose of study treatment; (4) pharmacokinetic population: patients who were randomized, received ≥1 dose of study treatment, and had ≥1 measurable neublastin concentration in the samples collected; and (5) immunogenicity population: patients who were randomized, received ≥1 dose of study treatment, and had immunogenicity data collected after dose.
3. Results
3.1. Patient population
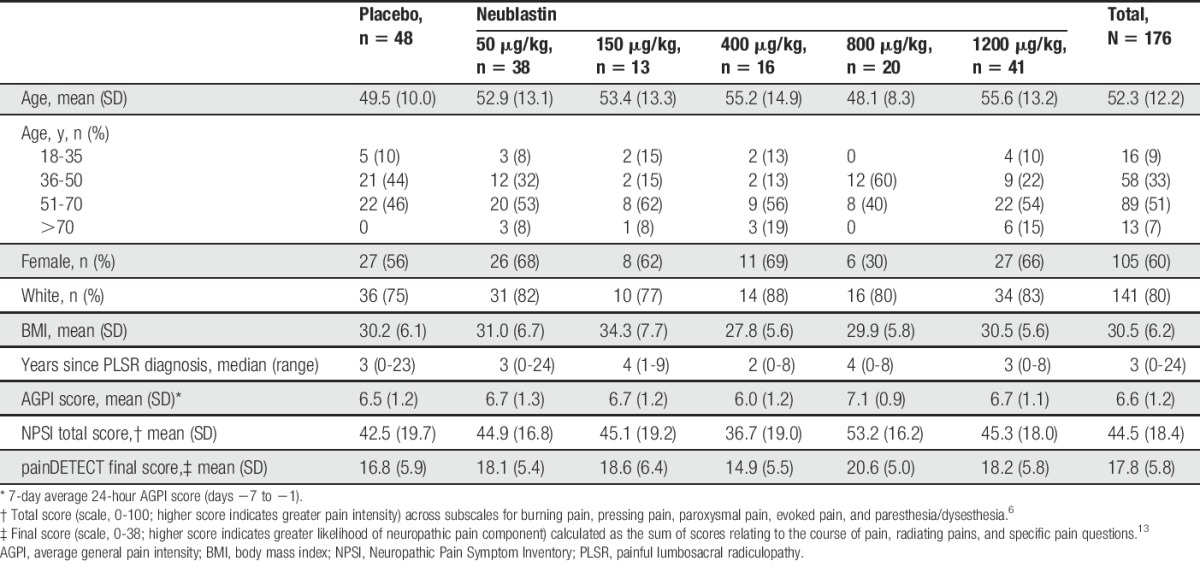
A total of 183 patients with PLSR were randomized (rapid recruitment at the end of the study led to over-enrollment relative to the target of 165), of whom 176 received treatment: 48 received placebo, 38 neublastin 50 μg/kg, 13 neublastin 150 μg/kg, 16 neublastin 400 μg/kg, 20 neublastin 800 μg/kg, and 41 neublastin 1200 μg/kg (Fig. 2). Baseline demographic and disease characteristics were reasonably well balanced between the treatment groups (Table 1). The mean age across treatment groups was 52.3 years, and 60% of the treated population was female. Median time since diagnosis of PLSR was 3 years. The mean NPSI total score at baseline was 44.5 (scale, 0-100; higher score indicates greater pain intensity), and mean painDETECT score was 17.8 (scale, 0-38; higher scores indicate a greater neuropathic pain component; Table 1).
Figure 2.

Flow of patients through the study. *n is for the primary efficacy analysis population; n may differ for the efficacy population for other outcome measures. AE, adverse event; PK, pharmacokinetics.
Table 1.
Baseline demographics and clinical characteristics (safety population).
Most treated patients (165 of 176; 94%) received all 3 doses of study medication. During the study, 89% of patients took concomitant pain medications (98% in the placebo group and 69%-92% in the active treatment groups). The most frequently reported concomitant pain medications were acetaminophen (35%), ibuprofen (20%), and gabapentin (19%). Rescue medication (acetaminophen) was used by 36% of the study population (42% in the placebo group and 25%-44% in the active treatment groups). Twenty-two percent of patients used rescue medication within 2 weeks of the first dose of study medication, which was classified as a major protocol deviation. Overall there was a relatively high number of protocol deviations during the study, with major deviations reported for 54% of patients overall and major deviations leading to exclusion from the per-protocol population in 32% of patients overall. Most deviations related to concomitant medication use (rescue medication use within 2 weeks of the first dose or disallowed medications) despite clear instructions about the importance of adherence to the protocol.
3.2. Average general pain intensity
Figure 3A shows the change from baseline in the mean AGPI in each treatment group across the course of the study (5-day average; weeks 1-8 after last dose). The temporal profile shows that 3 of the 5 neublastin dose levels (50, 800, and 1200 μg/kg) were associated with greater numeric reductions in mean AGPI scores than placebo throughout the study duration (no statistical tests were performed for these analyses).
Figure 3.
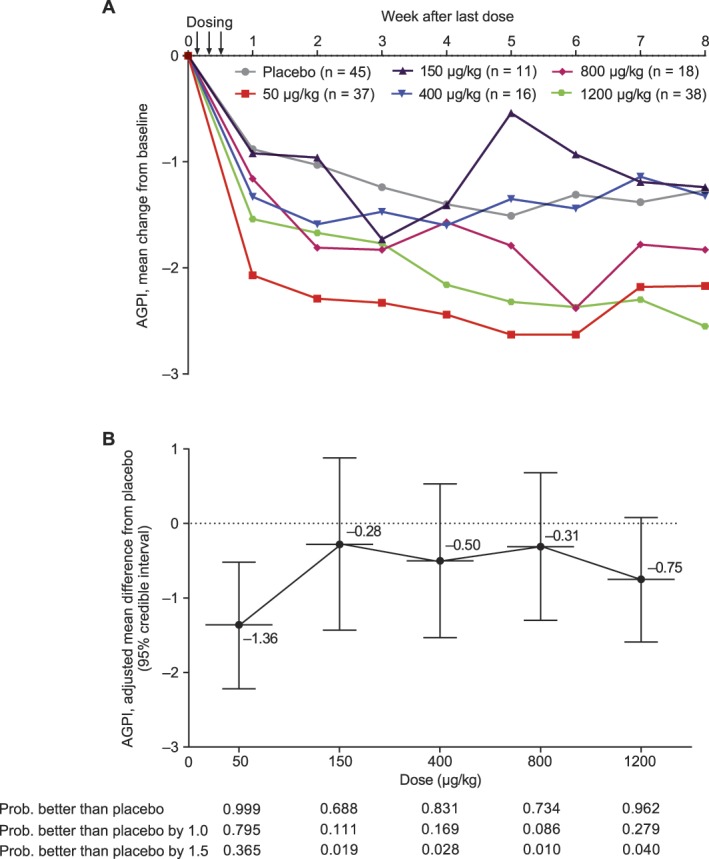
(A) Change from baseline in mean average general pain intensity (AGPI) by week of follow-up (5-day average) and (B) mean (standard deviation) placebo-adjusted change from baseline in mean AGPI at week 1 after last dose (5-day average; days 6-10) based on a Bayesian normal dynamic linear model (efficacy population).
At week 1 after last dose, the mean AGPI score decreased in the placebo group (−0.88) and all active treatment groups (50 μg/kg, −2.07; 150 μg/kg, −0.92; 400 μg/kg, −1.33; 800 μg/kg, −1.16; and 1200 μg/kg, −1.54) relative to baseline, with the greatest reduction observed in the neublastin 50-μg/kg dose group and the smallest reduction in the neublastin 150-μg/kg dose group. The primary efficacy analysis of change from baseline in mean AGPI score at week 1 after last dose (NDLM) revealed that the lowest neublastin dose of 50 μg/kg showed the greatest difference from placebo (posterior mean difference −1.36, 95% credible interval −2.22 to −0.52), followed by the highest dose of 1200 μg/kg (posterior mean difference from placebo −0.75, 95% credible interval −1.59, 0.08; Fig. 3B). No clear dose response was observed for the primary endpoint. In the sensitivity analysis for the primary efficacy endpoint, a statistically significant difference from placebo was reported for only the 50-μg/kg dose group by ANCOVA (P = 0.007; Table 2).
Table 2.
Change from baseline in mean 24-hour AGPI, ABPI, and ALPI at weeks 1, 3, and 5 after last dose (5-day average; ANCOVA) (efficacy population).
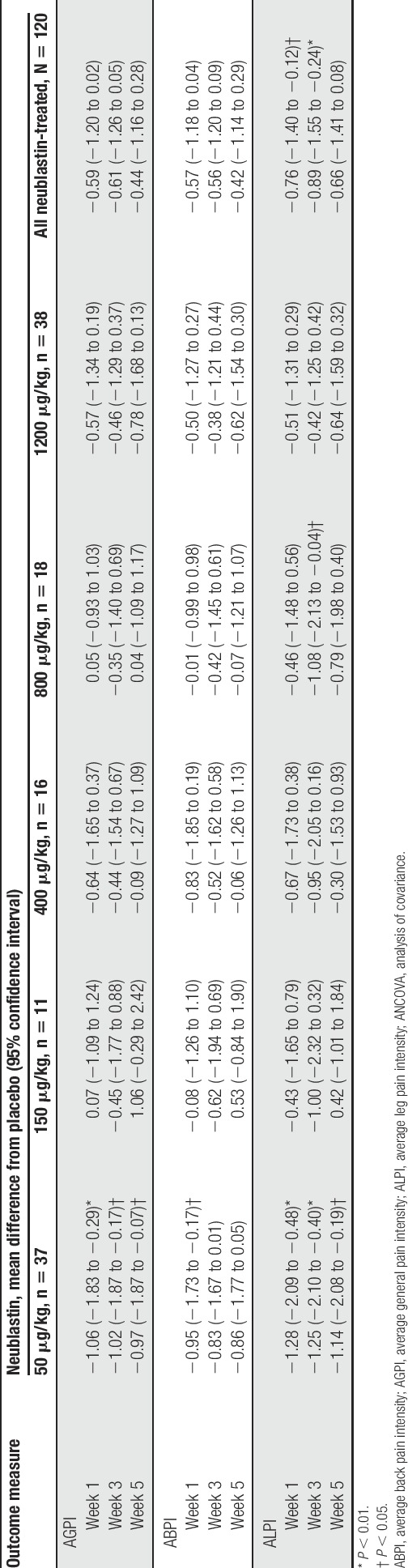
Analysis of mean AGPI at week 1 after last dose in the per-protocol population (n = 120) by ANCOVA showed a similar trend, although the magnitude of effect was slightly greater, and statistical significance was reached in both the 50-μg/kg dose group (P = 0.001) and the 1200-μg/kg dose group (P = 0.048).
Similarly, at week 1 after last dose, the proportion of patients achieving a ≥30% reduction from baseline in mean AGPI score (based on 5-day average) was highest in the neublastin 50-μg/kg group (43.2%) compared with 9.1%-26.3% in the other dose groups, and 19.1% with placebo. The proportion of patients achieving a ≥50% reduction was 27.0% in the 50-μg/kg group, 0%-17.9% in the other dose groups, and 8.5% with placebo.
For the secondary endpoint of change from baseline in mean AGPI at weeks 3 and 5 after last dose, neublastin 50 μg/kg was again the only dose that demonstrated a statistically significant difference from placebo (P = 0.019 and P = 0.034, respectively; Table 2). In a post hoc pooled analysis across all neublastin doses, the difference from placebo in reduction in AGPI was slightly greater at weeks 1 and 3 than at week 5, although statistical significance was not reached at any time point (Table 2).
3.3. Average back pain intensity and average leg pain intensity
For the secondary endpoints ABPI (back pain) and ALPI (leg pain), the greatest mean difference from placebo in change from baseline was observed in the neublastin 50-μg/kg dose group at each of weeks 1, 3, and 5 after last dose (Table 2). The comparisons of neublastin 50 μg/kg vs placebo were statistically significant at all 3 time points for ALPI but only at week 1 for ABPI. For ALPI, the reduction from baseline was also statistically significant vs placebo for the 800-μg/kg dose group at week 3. In a pooled analysis across all neublastin doses (post hoc), the difference from placebo in reduction in ALPI was statistically significant at weeks 1 and 3 (P = 0.020 and P = 0.008, respectively) but not at week 5. Placebo-adjusted differences for reduction in ABPI were also greater at weeks 1 and 3 than at week 5, but the difference vs placebo did not reach statistical significance at any time point (Table 2).
3.4. Exploratory efficacy endpoints
On the PGIC, the proportion of responders (reporting “much improved” or “very much improved”) at day 19 ranged from 25% to 50% in the active treatment groups compared with 21% in the placebo group (Table 3). At day 19, the highest proportion of responders was in the 800-μg/kg group (50%), followed by the 150-μg/kg (42%) and 50-μg/kg groups (38%). A similar trend was observed at day 33.
Table 3.
Proportions of Patient Global Impression of Change (PGIC) responders at days 19 and 33 (efficacy population).

All neublastin dose levels were associated with greater mean reductions (better sleep) from baseline in daily sleep interference (DSIS; 7-day average) than placebo throughout the course of the study (Fig. 4). Trends across dose levels were generally similar to AGPI, with the greatest sustained reduction in the 50-μg/kg dose group.
Figure 4.
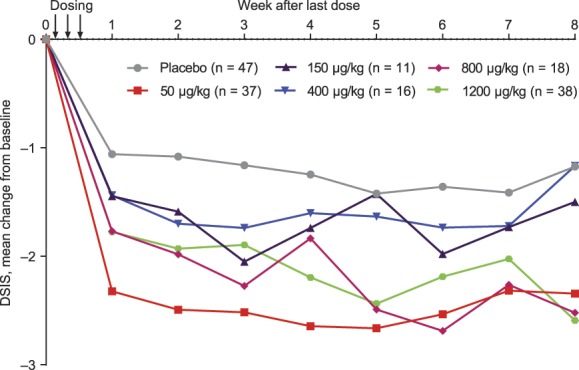
Mean change from baseline in Daily Sleep Interference Scale (DSIS) scores (7-day average) (efficacy population).
Evaluation of BPI scores also showed similar trends, with the greatest reductions from baseline in both overall severity of general pain and pain interference scores in the 50-μg/kg dose group. In this group, reduction in overall severity of general pain score with neublastin was −2.5 vs −1.0 with placebo at day 19, and −2.5 vs −1.4, respectively, at day 33 (0-10 scale); reduction in pain interference score with neublastin was −2.0 vs −1.1 with placebo at day 19, and −2.4 vs −1.5, respectively, at day 33 (0-10 scale). No clear trends were identified in physical or sleep parameters measured by wrist accelerometer.
Assessment of patient beliefs at the end of the study showed that 66% in the placebo group correctly believed that they had received placebo, while 71% across the neublastin treatment groups correctly believed they had received active drug.
3.5. Safety
Overall, 115 of 128 (90%) neublastin-treated patients experienced ≥1 treatment-emergent AE compared with 25 of 48 (52%) patients in the placebo group. The most common AEs were pruritus, headache, rash, and feeling hot (Table 4). Most AEs were mild or moderate in severity, with only 4% (5 of 128) of neublastin-treated patients reporting a severe AE (no patients in the placebo group reported severe AEs). More neublastin-than placebo-treated patients had an AE considered to be related to treatment (79% vs 19%). Four patients (2 each in the neublastin 50-μg/kg and 1200-μg/kg dose groups) experienced SAEs, which were worsening of depression, pneumonia, failed right hip prosthesis, and worsening of hypertension; none were considered related to treatment and none led to discontinuation of study drug. Six patients had a total of 7 AEs that led to discontinuation of the study drug (2 AEs of moderate pruritus, 3 of severe pruritus, 1 of mild decreased light brush sensation, and 1 of moderate headache), all of whom received neublastin.
Table 4.
Summary of treatment-emergent adverse events (safety population).
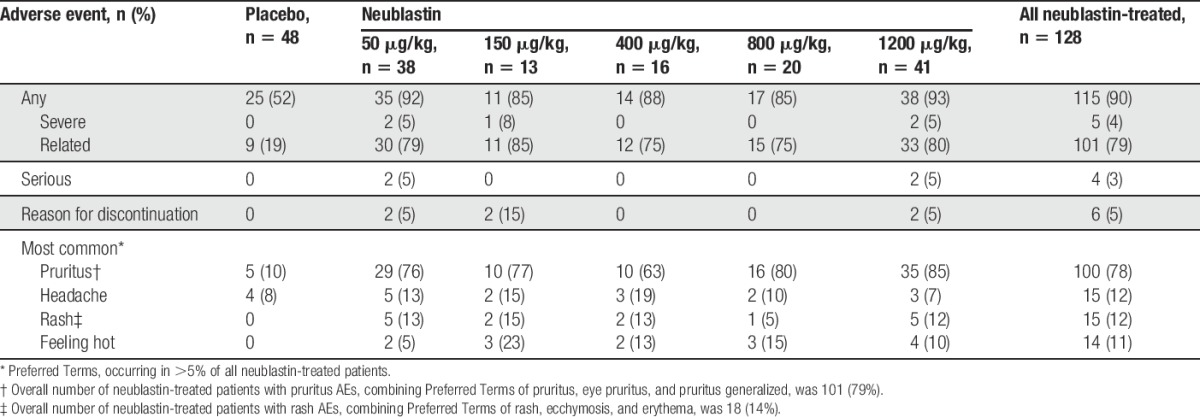
Pruritus AEs (includes Preferred Terms of pruritus, eye pruritus, and pruritus generalized) occurred in 79% of neublastin-treated patients compared with 10% of placebo-treated patients. Most AEs of pruritus were mild (for 73% of neublastin-treated patients and 100% of the placebo group), and most resolved by study end (94% of neublastin-treated patients and 60% of the placebo group). The median number of study days with pruritus was 16 among neublastin-treated patients and 5 in the placebo group. On the Itch Impact Questionnaire (0-4 scale; 0 = no itch), 21% of neublastin-treated patients reported a score of 3 (“the itch is severe enough to cause problems for me”) and 3% a score of 4 (“the itch is severe enough to cause major problems for me”) over the treatment week (days 1-7), decreasing in subsequent weeks to 4% and 0.8%, respectively, over days 22 to 28 (no placebo patients reported scores of 3 or 4 at any time point). The overall scores on the ItchyQoL, completed for patients experiencing pruritus, were generally low, with a mean overall score of 1.392 (SD, 0.5706; scale for impact on symptoms/functioning/emotions, 1-5; 1 = never, 5 = all the time); dose-dependent trends were not observed.
Rash occurred only in the neublastin groups and had an incidence of 14% (includes Preferred Terms of rash, ecchymosis, and erythema). Most AEs of rash (78%) were of a mild nature, and all AEs of rash resolved by study end for all patients. The median number of study days with rash was 17 among neublastin-treated patients.
No dose response was observed in the incidence of AEs overall or common AEs, including pruritus and rash. There were no clinically significant differences in intraepidermal nerve fiber density, clinical neurological examination abnormalities, or suicide-related events based on C-SSRS across treatment groups including placebo.
3.6. Pharmacokinetics and immunogenicity
Serum neublastin concentrations were shown to increase in a dose-proportional manner, and between-subject variability in neublastin pharmacokinetics was small (Fig. 5). No neutralizing antibodies (at clinically relevant levels) were detected in any patient during the course of the study.
Figure 5.
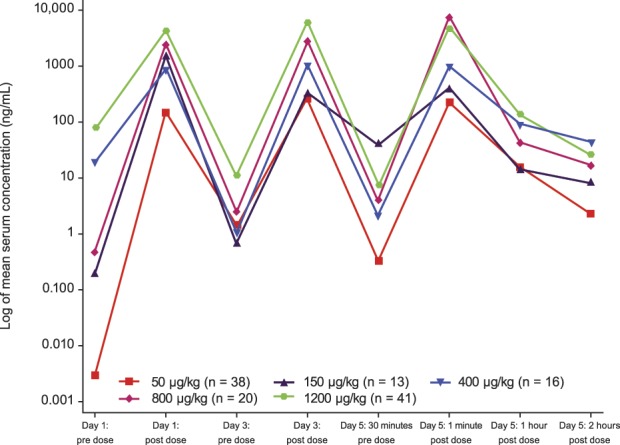
Mean serum concentration of neublastin over time (pharmacokinetic population).
4. Discussion
The SPRINT trial demonstrated statistically significant differences from placebo in AGPI (general pain) change from baseline with a 50-μg/kg dose of neublastin at weeks 1, 3, and 5 after dosing, in patients with chronic PLSR. The 50-μg/kg dose of neublastin also showed statistically significant differences from placebo in ALPI (leg pain) at weeks 1, 3, and 5, and in ABPI (back pain) at week 1. A pooled analysis across all neublastin doses suggested that the greatest reductions overall were for ALPI, with significant differences from placebo at weeks 1 and 3. However, statistically significant differences from placebo were not observed consistently across all doses or endpoints, and a dose–response relationship was not apparent, with the next most effective dose after 50 μg/kg being the highest tested of 1200 μg/kg.
Dosing in this trial was with 3 neublastin injections over 1 week, with the primary endpoint evaluation the week after last dose. However, overall follow-up extended for 56 days after last dose to evaluate neutralizing antibodies, and to ensure that potential cumulative, longer term treatment effects were captured and that safety was maintained during this time period. It is of note that a reduction in pain scores was observed across the 8-week study period, despite dosing only during the first week of the study. Improvements in sleep interference (DSIS) mirrored reductions in pain across the dose levels. The safety profile of neublastin was similar across all doses; pruritus occurred in a majority of neublastin-treated patients, but most cases were mild and resolved by the end of the study.
The reason for the lack of a dose–response relationship across the tested neublastin doses of 50 μg/kg to 1200 μg/kg is unclear. We considered whether the higher than expected incidence of protocol violations may have contributed to this lack of dose response; however, the trend in results remained the same for the per-protocol population as for the overall population. Serum concentrations of neublastin were dose proportional, so the apparent lack of dose response could not be explained either by variations in neublastin exposure in the vasculature, although it remains a possibility that serum neublastin exposure does not correlate with concentrations at the nerve root. In follow-up to the trial, we also conducted simulation analyses to explore whether the lack of dose response could be a consequence of the Bayesian adaptive design (eg, the different number of subjects allocated to each dose); these analyses indicated that the design was unlikely to have been a contributing factor (data not shown). There are several examples of “U-shaped” dose–response curves of efficacious drugs, including in trials with a fixed randomization design (eg, trials of aripiprazole,24 belimumab,35 and crofelemer8); thus a U-shaped dose–response curve does not invalidate a finding of efficacy.
The efficacy, safety, pharmacokinetic, and immunogenic profiles of neublastin in this study were generally consistent with those previously reported in patients with sciatica participating in phase 1 single- and multiple-ascending dose studies of neublastin.26,29 The single-ascending dose study (first time in human) evaluated doses ranging from 0.3 to 800 μg/kg (selected based on preclinical toxicology results). Based on the observed tolerability in the single-ascending dose study, the multiple-ascending dose study evaluated a higher dose range from 50 to 1200 μg/kg (50-800 μg/kg, one dose per week; 400-1200 μg/kg, one dose per 48 hours). Similar to the current study, neither phase 1 trial demonstrated a clear dose-dependent trend in numerical rating scales for pain.26,29 Although neublastin appears to be generally well tolerated, pruritus has been consistently reported in all studies as a common AE.26,29 The underlying mechanism of pruritus and the relationship between severity, onset, and duration of pruritus and neublastin dosage regimens remain to be elucidated.
Innovative steps were taken in the design and conduct of this trial, most notably the use of the Bayesian adaptive design. Neublastin is a first-in-class compound with a novel mechanism of action, and optimal dosing for reduction of neuropathic pain had not been established in earlier trials. We selected the Bayesian adaptive allocation design, because compared with a fixed-dose design, this enables efficient evaluation of multiple doses while maintaining an achievable sample size, as it permits the most patients to be randomized to the most efficacious and safe dose level. The Bayesian NDLM was selected for the primary efficacy analysis as a flexible model to analyze dose–response.6,31,38 Dose selection for this trial was based on the observed safety profile (no treatment-related SAEs or dose-limiting toxicities up to the highest dose of 1200 μg/kg) and efficacy signals in the prior single- and multiple-ascending dose studies.26,29 Adaptation in SPRINT was based on key efficacy and safety criteria of AGPI (general pain) reduction and the proportion of patients who reported “the itch is severe enough to cause major problems for me.” Given the low proportion of patients reporting itch of this severity, adaptation was ultimately driven primarily by pain reduction. Since more patients were allocated to the neublastin doses with the largest pain reductions, the algorithm for adaptive allocation was considered to have been effective. The Bayesian design also allows for the discontinuation of enrollment if evidence suggests a high probability of success or failure; however, for SPRINT, early stopping criteria were not met.
Pain trials have historically noted high rates of placebo response (particularly trials in lower back pain28) and frequent failure to replicate results across studies. To address this issue, the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has established recommendations for improving assay sensitivity in chronic pain trials.12 We aimed to follow these recommendations closely in the design of the SPRINT trial, in particular, incorporating standardized procedures for physical examination, diagnosis and monitoring, and extensive training of investigators and other site staff in all procedures and assessments. Of particular note, a Placebo Response Reduction Program (Analgesic Solutions), in which all research staff and patients were trained to have appropriate levels of expectation of treatment response, was provided. Relative to other pain trials, we feel that SPRINT has achieved a relatively low placebo response, which may be attributable to these efforts.
There are no universally accepted diagnostic criteria for PLSR, and definitions vary within clinical trials and the broader literature.16 We were therefore careful to establish specific and standardized criteria to ensure enrollment of a homogenous population with documented PLSR, including both symptoms judged by the investigator to be because of PLSR, and objective evidence of PLSR correlating with the patient's symptomatology. Again, in line with IMMPACT recommendations to improve assay sensitivity,12 patients were enrolled with a minimum pain duration of ≥6 months (to reduce the rate of spontaneous resolution) and minimum pain intensity of ≥4 at baseline. Patients additionally had failed to respond to at least 2 standard-of-care therapies. A limitation of this approach is that patients enrolled in SPRINT were generally representative of a refractory PLSR population, which may have made it more difficult to achieve substantial reductions in pain, and which limits generalizability to the broader PLSR population.
Despite efforts to optimize patient selection and assay sensitivity, it is possible that other unidentified factors unique to investigations among the PLSR population contributed to the observed lack of dose response. Historically, studies of drug therapies in PLSR patients have frequently shown inconclusive outcomes,2,4,10,17–19,21–23 and it may be that further endeavors are required to establish a reliable framework for evaluation of therapeutics in this population. Additional limitations of this study are the small sample size in some of the dose groups, adding to uncertainty around the lack of dose response, and the relatively short duration; it is possible that a longer series of neublastin doses are necessary to consistently alleviate pain, particularly among patients with chronic pain refractory to other treatments. The side effects of pruritus and rash may also have contributed to partial unblinding during the study.
In conclusion, this study showed some evidence of a biological effect of neublastin for the reduction of pain in patients with PLSR, particularly at the lowest tested dose of 50 μg/kg. However, statistically significant differences from placebo were not observed consistently across all doses or endpoints, and there was no clear dose–response relationship for either pain reduction or common AEs. Further exploration of neurotrophic factors for the treatment of neuropathic pain and nerve injury is warranted to establish whether the promising mechanistic effects observed in preclinical studies will translate to clinically relevant benefits.
Conflict of interest statement
This study was funded by Biogen. M. Backonja is an employee of PRA Health Sciences and has served as a consultant to Biogen, Pfizer, Scilex, Wex Pharma, and Zynerb. L. Williams and X. Miao are employees of and hold stock/stock options in Biogen. C. Chen was an employee of Biogen at the time of the study and holds stock in Biogen. N. Katz is an employee of Analgesic Solutions, paid consultants to Biogen in relation to the study design and other services for this study.
Supplementary Material
Acknowledgments
Biogen provided funding for medical writing support in the development of this article. Elizabeth Harvey, PhD, and Malcolm J. M. Darkes, PhD, from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript as per journal requirements. Biogen reviewed and provided feedback on the paper to the authors. The authors had full editorial control of the paper and provided their final approval of all content.
Appendix A. Supplemental Digital Content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/A433.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002;3:383–94. [DOI] [PubMed] [Google Scholar]
- [2].Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen TS, Nurmikko T. European Federation of Neurological Societies. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010;17:1113–e88. [DOI] [PubMed] [Google Scholar]
- [3].Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Jr, Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3–RET receptor complex. Neuron 1998;21:1291–302. [DOI] [PubMed] [Google Scholar]
- [4].Baron R, Freynhagen R, Tölle TR, Cloutier C, Leon T, Murphy TK, Phillips K; A0081007 Investigators. The efficacy and safety of pregabalin in the treatment of neuropathic pain associated with chronic lumbosacral radiculopathy. PAIN 2010;150:420–7. [DOI] [PubMed] [Google Scholar]
- [5].Bennett DL, Boucher TJ, Michael GJ, Popat RJ, Malcangio M, Averill SA, Poulsen KT, Priestley JV, Shelton DL, McMahon SB. Artemin has potent neurotrophic actions on injured C-fibres. J Peripher Nerv Syst 2006;11:330–45. [DOI] [PubMed] [Google Scholar]
- [6].Berry DA, Muller P, Grieve AP, Smith M, Parke T, Blazek R, Mitchard R, Krams M. Adaptive Bayesian designs for dose-ranging drug trials. In: Gatsonis C, Kass RE, Carlin B, Carriquiry A, Gelman A, Verdinelli I, West M, editors. Case studies in Bayesian statistics V. New York: Springer-Verlag, 2001:99–181. [Google Scholar]
- [7].Bouhassira D, Attal N, Fermanian J, Alchaar H, Gautron M, Masquelier E, Rostaing S, Lanteri-Minet M, Collin E, Grisart J, Boureau F. Development and validation of the neuropathic pain symptom inventory. PAIN 2004;108:248–57. [DOI] [PubMed] [Google Scholar]
- [8].Center for Drug Evaluation and Research. Statistical review and evaluation: crofelemer tablers (125 mg). Available at: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202292Orig1s000StatR.pdf. Accessed July 25, 2016.
- [9].Cleeland CS. The Brief pain inventory: user guide. Available at: http://www.mdanderson.org/education-and-research/departments-programs-and-labs/departments-and-divisions/symptom-research/symptom-assessment-tools/BPI_UserGuide.pdf. Accessed January 16, 2016.
- [10].Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, Kalso EA, Loeser JD, Miaskowski C, Nurmikko TJ, Portenoy RK, Rice AS, Stacey BR, Treede RD, Turk DC, Wallace MS. Pharmacologic management of neuropathic pain: evidence-based recommendations. PAIN 2007;132:237–51. [DOI] [PubMed] [Google Scholar]
- [11].Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, Carr DB, Chandler J, Cowan P, Dionne R, Galer BS, Hertz S, Jadad AR, Kramer LD, Manning DC, Martin S, McCormick CG, McDermott MP, McGrath P, Quessy S, Rappaport BA, Robbins W, Robinson JP, Rothman M, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Wernicke J, Witter J; IMMPACT. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. PAIN 2005;113:9–19. [DOI] [PubMed] [Google Scholar]
- [12].Dworkin RH, Turk DC, Peirce-Sandner S, Burke LB, Farrar JT, Gilron I, Jensen MP, Katz NP, Raja SN, Rappaport BA, Rowbotham MC, Backonja MM, Baron R, Bellamy N, Bhagwagar Z, Costello A, Cowan P, Fang WC, Hertz S, Jay GW, Junor R, Kerns RD, Kerwin R, Kopecky EA, Lissin D, Malamut R, Markman JD, McDermott MP, Munera C, Porter L, Rauschkolb C, Rice AS, Sampaio C, Skljarevski V, Sommerville K, Stacey BR, Steigerwald I, Tobias J, Trentacosti AM, Wasan AD, Wells GA, Williams J, Witter J, Ziegler D. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. PAIN 2012;153:1148–58. [DOI] [PubMed] [Google Scholar]
- [13].Emory University Office of Technology Transfer: Research Administration. ItchyQol: a pruritus-specific quality of life instrument. Available at: http://emoryott.technologypublisher.com/tech?title=ItchyQol%3A_A_Pruritus-Specific_Quality_of_Life_Instrument. Accessed July 25, 2016.
- [14].Freynhagen R, Baron R, Gockel U, Tolle TR. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin 2006;22:1911–20. [DOI] [PubMed] [Google Scholar]
- [15].Gardell LR, Wang R, Ehrenfels C, Ossipov MH, Rossomando AJ, Miller S, Buckley C, Cai AK, Tse A, Foley SF, Gong B, Walus L, Carmillo P, Worley D, Huang C, Engber T, Pepinsky B, Cate RL, Vanderah TW, Lai J, Sah DW, Porreca F. Multiple actions of systemic artemin in experimental neuropathy. Nat Med 2003;9:1383–9. [DOI] [PubMed] [Google Scholar]
- [16].Genevay S, Atlas SJ, Katz JN. Variation in eligibility criteria from studies of radiculopathy due to a herniated disc and of neurogenic claudication due to lumbar spinal stenosis: a structured literature review. Spine (Phila Pa 1976) 2010;35:803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Genevay S, Viatte S, Finckh A, Zufferey P, Balague F, Gabay C. Adalimumab in severe and acute sciatica: a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2010;62:2339–46. [DOI] [PubMed] [Google Scholar]
- [18].Khoromi S, Cui L, Nackers L, Max MB. Morphine, nortriptyline and their combination vs. placebo in patients with chronic lumbar root pain. PAIN 2007;130:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Khoromi S, Patsalides A, Parada S, Salehi V, Meegan JM, Max MB. Topiramate in chronic lumbar radicular pain. J Pain 2005;6:829–36. [DOI] [PubMed] [Google Scholar]
- [20].Konstantinou K, Dunn KM. Sciatica: review of epidemiological studies and prevalence estimates. Spine (Phila Pa 1976) 2008;33:2464–72. [DOI] [PubMed] [Google Scholar]
- [21].Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Bowman C, Hammond A, Kirkham B, Järvinen S, Niinimäki J, Veeger N, Haapea M, Torkki M, Tervonen O, Seitsalo S, Hurri H. The treatment of disc herniation-induced sciatica with infliximab: one-year follow-up results of FIRST II, a randomized controlled trial. Spine (Phila Pa 1976) 2006;31:2759–66. [DOI] [PubMed] [Google Scholar]
- [22].Korhonen T, Karppinen J, Paimela L, Malmivaara A, Lindgren KA, Järvinen S, Niinimäki J, Veeger N, Seitsalo S, Hurri H. The treatment of disc herniation-induced sciatica with infliximab: results of a randomized, controlled, 3-month follow-up study. Spine (Phila Pa 1976) 2005;30:2724–8. [DOI] [PubMed] [Google Scholar]
- [23].Kreiner DS, Hwang SW, Easa JE, Resnick DK, Baisden JL, Bess S, Cho CH, DePalma MJ, Dougherty P, II, Fernand R, Ghiselli G, Hanna AS, Lamer T, Lisi AJ, Mazanec DJ, Meagher RJ, Nucci RC, Patel RD, Sembrano JN, Sharma AK, Summers JT, Taleghani CK, Tontz WL, Jr, Toton JF. North American Spine Society. An evidence-based clinical guideline for the diagnosis and treatment of lumbar disc herniation with radiculopathy. Spine J 2014;14:180–91. [DOI] [PubMed] [Google Scholar]
- [24].McEvoy JP, Daniel DG, Carson WH, Jr, McQuade RD, Marcus RN. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res 2007;41:895–905. [DOI] [PubMed] [Google Scholar]
- [25].O'Connor AB. Neuropathic pain: quality-of-life impact, costs and cost effectiveness of therapy. Pharmacoeconomics 2009;27:95–112. [DOI] [PubMed] [Google Scholar]
- [26].Okkerse P, Hay JL, Versage E, Tang Y, Galluppi G, Ravina B, Verma A, Williams L, Aycardi E, Groeneveld GJ. Pharmacokinetics and pharmacodynamics of multiple doses of BG00010, a neurotrophic factor with anti-hyperalgesic effects, in patients with sciatica. Br J Clin Pharmacol 2016;82:108–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, Currier GW, Melvin GA, Greenhill L, Shen S, Mann JJ. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 2011;168:1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Puhl AA, Reinhart CJ, Rok ER, Injeyan HS. An examination of the observed placebo effect associated with the treatment of low back pain—a systematic review. Pain Res Manag 2011;16:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rolan PE, O'Neill G, Versage E, Rana J, Tang Y, Galluppi G, Aycardi E. First-in-human, double-blind, placebo-controlled, randomized, dose-escalation study of BG00010, a glial cell line-derived neurotrophic factor family member, in subjects with unilateral sciatica. PLoS One 2015;10:e0125034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith BH, Torrance N. Epidemiology of neuropathic pain and its impact on quality of life. Curr Pain Headache Rep 2012;16:191–8. [DOI] [PubMed] [Google Scholar]
- [31].Smith MK, Jones I, Morris MF, Grieve AP, Tan K. Implementation of a Bayesian adaptive design in a proof of concept study. Pharm Stat 2006;5:39–50. [DOI] [PubMed] [Google Scholar]
- [32].Tarulli AW, Raynor EM. Lumbosacral radiculopathy. Neurol Clin 2007;25:387–405. [DOI] [PubMed] [Google Scholar]
- [33].Van Boxem K, Cheng J, Patijn J, van Kleef M, Lataster A, Mekhail N, Van Zundert J. 11. Lumbosacral radicular pain. Pain Pract 2010;10:339–58. [DOI] [PubMed] [Google Scholar]
- [34].Vernon MK, Brandenburg NA, Alvir JM, Griesing T, Revicki DA. Reliability, validity, and responsiveness of the daily sleep interference scale among diabetic peripheral neuropathy and postherpetic neuralgia patients. J Pain Symptom Manage 2008;36:54–68. [DOI] [PubMed] [Google Scholar]
- [35].Wallace DJ, Stohl W, Furie RA, Lisse JR, McKay JD, Merrill JT, Petri MA, Ginzler EM, Chatham WW, McCune WJ, Fernandez V, Chevrier MR, Zhong ZJ, Freimuth WW. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum 2009;61:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang R, King T, Ossipov MH, Rossomando AJ, Vanderah TW, Harvey P, Cariani P, Frank E, Sah DW, Porreca F. Persistent restoration of sensory function by immediate or delayed systemic artemin after dorsal root injury. Nat Neurosci 2008;11:488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weber H, Holme I, Amlie E. The natural course of acute sciatica with nerve root symptoms in a double-blind placebo-controlled trial evaluating the effect of piroxicam. Spine (Phila Pa 1976) 1993;18:1433–8. [PubMed] [Google Scholar]
- [38].West M, Harrison PJ. Bayesian forecasting and dynamic models. 2nd ed New York: Springer, 1997. [Google Scholar]
- [39].Wong LE, Gibson ME, Arnold HM, Pepinsky B, Frank E. Artemin promotes functional long-distance axonal regeneration to the brainstem after dorsal root crush. Proc Natl Acad Sci U S A 2015;112:6170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.