

Abstract
Protein instability is a major obstacle in the production and delivery of monoclonal antibody–based therapies for cancer. This study presents real-time isothermal differential scanning fluorimetry as an emerging method to evaluate the stability of human immunoglobulin G protein with high sensitivity. The stability of polyclonal human immunoglobulin G against urea-induced denaturation was assessed following: (1) oxidation by the free-radical generator 2,2-Azobis[2-amidinopropane]dihydrochloride and (2) in selected storage buffers. Significant differences in immunoglobulin G stability were detected by real-time isothermal differential scanning fluorimetry when the immunoglobulin G was stored in 1,4-Piperazinediethanesulfonic acid buffer compared to phosphate-buffered saline, with half-maximal rate of denaturation occurring at a higher urea concentration in 1,4-Piperazinediethanesulfonic acid than phosphate-buffered saline (K nd;PIPES = 3.56 ± 0.09 M, K nd;PBS = 2.94 ± 0.08 M; P < .01), but differential scanning fluorimetry did not detect differences in unfolding temperature (T m;PIPES = 70.5 ± 0.3°C, T m;PBS = 69.7 ± 0.2°C). The effects of 2,2-Azobis[2-amidinopropane]dihydrochloride-induced oxidation on immunoglobulin G stability were analyzed by real-time isothermal differential scanning fluorimetry; the oxidized protein showed greater sensitivity to urea (K nd;CNTRL = 3.96 ± 0.19 M, K nd;AAPH = 3.49 ± 0.07 M; P < .05). Similarly, differential scanning fluorimetry indicated greater thermal sensitivity of oxidized immunoglobulin G (T m;CNTRL = 70.5 ± 0.3°C, T m;AAPH = 62.9 ± 0.1°C; P < .001). However, a third method for assessing protein stability, pulse proteolysis, proved to be substantially less sensitive and did not detect significant effects of 2,2-Azobis[2-amidinopropane]dihydrochloride on the half-maximal concentration of urea needed to denature immunoglobulin G (C m;CNTRL= 6.8 ± 0.1 M; C m;AAPH = 6.4 ± 0.7 M). Overall these results demonstrate the merit of using real-time isothermal differential scanning fluorimetry as a rapid and sensitive technique for the evaluation of protein stability in solution using a quantitative real-time thermocycler.
Keywords: protein stability, protein denaturation, antibody formulation, denaturation kinetics
Introduction
Monoclonal antibodies are one of the most important and promising recent advances in the treatment of cancers and now represent a sizable portion of all pharmaceuticals in clinical trials.1 Antibodies are particularly well-suited to the treatment of cancers as they can be designed to recognize and bind particular antigens on malignant cells with high specificity and affinity. This interaction can be exploited using multiple strategies, for example: to agonize proapoptotic receptors and antagonize growth-factor receptors; to induce phagocytosis, complement activation, or antibody-dependent cellular cytotoxicity; as well as for targeted delivery of chemotherapeutic drugs, radioisotopes, cytokines, small interfering RNA, or drug-activating enzymes to malignant cells.2,3 However, beyond the difficulties in identifying therapeutic targets, the development of therapeutic antibodies remains complicated by the stability and storage of these proteins.
The development of antibody-based therapeutics involves particular challenges in maintaining conformational stability, effectiveness, long shelf-life, and low toxicity of the antibodies themselves.4–6 Denaturation is the transition of a protein from the native conformation to an unfolded state and is generally accompanied by a major loss of protein function and, therefore, buffer formulations for protein-based therapeutics must stabilize against even small changes in protein structure that can be induced by common stresses encountered during production, transport, and storage.7,8 External environmental factors such as temperature, pH, ionicity, and concentration can also be individually controlled to achieve maximal protein stability.9,10 Formulation additives such as sugars, amino acids, and surfactants are also routinely employed as stabilizers to maintain solubility and reduce protein aggregation.4,11
In recent years, differential scanning fluorimetry (DSF) has emerged as an efficient means of evaluating protein stability and ligand binding. Importantly, multiple DSF trials can now be run in parallel using either 96 or 384 microplate formats, requiring only low concentrations of purified protein and a real-time polymerase chain reaction thermocycler.12 Recently, a new DSF-based technique was developed to investigate isothermal denaturation kinetics in real time, since referred to as real-time isothermal DSF (RT-iDSF). This method can provide information about the kinetics of protein stability derived from initial rates of denaturation experienced upon protein exposure to varying concentrations of denaturant (ie, urea).13 Importantly, the RT-iDSF technique is orthogonal to DSF as proteins are denatured chemically at a constant temperature rather than thermally as in DSF. This technical difference is significant since the mode of unfolding differs, and each method provides unique information about the conformational stability of the protein.10,14,15 Whereas DSF provides information about the thermodynamics of the protein transition from the native to unfolded state, RT-iDSF assesses the kinetic parameters of denaturation; from this information, RT-iDSF may be able to detect small changes in protein stability at physiological and production-relevant temperatures that cannot be observed by DSF.13 By using the real-time fluorescence measurements generated from RT-iDSF monitoring denaturation reactions, 3 kinetic parameters of protein stability can be derived using the Hill equation: (1) the kinetic constant of denaturation (K nd; defined as the concentration of denaturant that yields the half-maximal rate of denaturation), (2) the maximal rate of denaturation (D max), and (3) the µ-coefficient (indicating the degree of cooperativity of the denaturant molecules).13,16 Such rate determinants can be used to quantify the aspects of protein denaturation kinetics that cannot be revealed by standard thermal denaturation.
The goal of this study was to demonstrate and evaluate the use of RT-iDSF for the assessment of human polyclonal immunoglobulin G (IgG) protein stability. Whereas monoclonal antibodies have a homogenous protein structure among the population, polyclonal IgG contains multiple subclasses of the IgG protein, which are highly conserved but polymorphic in their constant regions and are of variable composition in their antigen-binding regions. As a proof of principle, urea-induced denaturation of polyclonal IgG was utilized and quantified by RT-iDSF to assess protein stability in the presence of select physiological buffers and following the incubation of IgG with the free-radical generator 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH) to model the effect of protein oxidation. 2,2′-azobis(2-amidinopropane) dihydrochloride generating alkyl, alkoxyl, and alkyl peroxy radicals in a predictable fashion and at a constant rate.17 These radicals can cause oxidation of Met, Cys, Trp, Lys, Tyr, and His residues of proteins.18,19 Although Azo compounds such as AAPH are not found in vivo, AAPH provides a simple and reliable model to simulate differential effects of oxidation on protein stability in aqueous solutions and has been employed previously.20,21 To evaluate the accuracy and sensitivity of the technique, RT-iDSF results were compared to those obtained from DSF and an older, widely used method for studying protein stability called pulse proteolysis (PP).22,23
Materials and Methods
Immunoglobulin G Samples and Reagents
Lyophilized powder of human polyclonal IgG was obtained from Equitech-Bio Inc (Cat# SLH66; Kerrville, Texas). A stock solution of IgG was prepared to 10 mg/mL in phosphate-buffered saline solution (PBS; 100 mM monopotassium phosphate, 150 mM NaCl [pH 7.0]), or in 100 mM 1,4-Piperazinediethanesulfonic acid [PIPES] with 150 mM NaCl [pH 7.0], where indicated; this solution was then further diluted for RT-iDSF and PP assays at their specified concentrations. For DSF assays, IgG solutions were prepared in 100 mM buffer (as indicated) with 150 mM NaCl (at various pH, as indicated). Spectrophotometric analysis was used to ensure that aggregation was not occurring over the time course of the experiments. All reagents were obtained from Sigma-Aldrich (St. Louis, Missouri), with the exception of SYPRO Orange fluorescent dye (Cat# S-6650; LifeTechnologies, Carlsbad, California) and PIPES buffer (Cat# 528132; CalBioChem, La Jolla, Californai). SYPRO Orange was diluted to a 40× stock in PBS (from the 5000× stock solution supplied by LifeTechnologies) and further diluted to a final concentration of 5× in DSF and RT-iDSF experiments.
Equipment
All DSF and RT-iDSF assays were carried out using a BioRad iCycler real-time quantitative polymerase chain reaction (RT-qPCR) thermocycler (BioRad, Hercules, Californai). Assays were carried out in 96-well, unskirted PCR microplates (BioRad, Cat# MLP-9601), and thermocyclers were equipped with filters for both excitation (495 ± 30 nm) and emission (625 ± 30 nm) wavelengths of SYPRO Orange.
Differential Scanning Fluorimetry
Thermal DSF assays were performed according to the protocol described by Niesen et al12 For assays of different pH and buffer conditions, DSF incubations contained 1.0 mg/mL human IgG, 100 mM buffer (see Table 1 for list), 150 mM NaCl, 5× SYPRO Orange. For DSF assays of IgG thermal stability following oxidation by AAPH (2,2-Azobis[2-amidinopropane]dihydrochloride; Cat# 440914; Sigma-Aldrich), samples of IgG (2.0 mg/mL in PBS, pH 7.0) with various concentrations (0, 2.5, 5, 10, 25, or 50 mM) of AAPH were incubated for 2 hours at 4°C. All samples were then subjected to spun-column chromatography on a Sephadex G-25 size-exclusion matrix (Cat# G2580; Sigma-Aldrich) equilibrated in PBS; this removed the AAPH from treated samples and returned all samples into PBS buffer alone. Following desalting, DSF assays were performed using 20 µL of 1.0 mg/mL human IgG, 100 mM monobasic potassium phosphate (pH 7.0), 150 mM NaCl, and 5× SYPRO Orange. In all DSF experiments, samples were heated from 15°C to 90°C by increments of 0.5°C at intervals of 30 seconds, with fluorescence intensity measured at each interval.
Table 1.
Unfolding Temperatures (T m) of Human IgG Over a Range of pH Conditions, as Determined From DSF Analysis, Along With Corresponding Buffers Used and Their pKa.a
| pH | Buffer | pKa at 25°C | IgG T m (°C) |
|---|---|---|---|
| 3.0 | PBS | 2.2 | 39 ± 1b |
| 4.0 | Sodium Acetate | 4.8 | 50.1 ± 0.4* |
| 5.0 | Sodium Acetate | 4.8 | 66.1 ± 0.3c |
| 6.0 | PIPES | 6.8 | 69.3 ± 0.2d, f |
| 7.0 | PIPES | 6.8 | 70.5 ± 0.2e |
| 7.0 | PBS | 7.2 | 69.7 ± 0.3d, e |
| 8.0 | CHES | 9.5 | 68.8 ± 0.1f |
| 9.0 | CHES | 9.5 | 67.4 ± 0.3c |
| 10.0 | CAPS | 10.4 | 63.4 ± 0.1* |
| 12.0 | PBS | 12.4 | 37 ± 2b |
Abbreviations: ANOVA, analysis of variance; CHES, 2-(Cyclohexylamino)ethane sulfonic acid CAPS, 3-(Cyclohexylamino)-1-propanesulfonic acid; DSF, differential scanning fluorimetry; IgG, immunoglobulin G; PBS, phosphate-buffered saline; PIPES, 1,4-Piperazinediethanesulfonic acid; SEM, standard error of mean.
aData are presented as means ± SEM (n = 4). One-way ANOVA with post hoc Tukey test showed that T m results were all significantly different from each other (P < .05), except for results labeled with the same superscript letter, which was not significantly different. Results marked with an asterisk (*) are significantly different from all other unfolding temperatures (P < .05).
Real-Time Isothermal DSF
Real-time isothermal differential scanning fluorimetry experiments were performed according to the protocol described by Biggar et al.13 For all RT-iDSF assays, incubations containing 1.0 mg/mL human IgG, 100 mM monobasic potassium phosphate (or PIPES, where specified; pH 7), 150 mM NaCl, and 5× SYPRO Orange, with the addition of various concentrations of urea (0-6 M in PBS, pH 7), were carried out at 4°C with fluorescence intensity measured at 5 seconds intervals over 50 minutes. Fluorescence measurements were initiated ∼5 seconds after the addition of protein to the urea solution. To assay the effect of buffer species on IgG stability, incubations contained either PBS buffer or PIPES, as described above. For RT-iDSF analysis of the effect of oxidation by AAPH-generated radicals, IgG samples (5.0 mg/mL in PBS, pH 7.0) were first incubated at 4°C for 2 hours with 50 mM AAPH; control samples containing no AAPH were incubated in parallel with the experimental samples. Immediately following incubation, samples were subjected to spun-column chromatography (as above) and then assayed by RT-iDSF. For all experiments, fluorescence intensities were normalized relative to the 0 M Urea condition, and a series of assays containing reaction buffer with no IgG were performed to ensure that the rate of fluorescence increase was not due to the increased concentration of urea alone.
Pulse Proteolysis
Samples of IgG were diluted to 10 mg/mL in PBS (pH 7.0) and then incubated with or without the presence of 50 mM AAPH for 2 hours at 4°C. All samples were then subjected to spun-column chromatography (as above) and were then incubated with various concentrations of urea (0-8 M; made up in PBS) for 17 hours at 4°C. Following urea incubation, samples were again returned to PBS by spun-column chromatography. Samples were then digested by incubation with thermolysin from Bacillus thermoproteolyticus rokko for 5 minutes at room temperature (0.4 mg/mL final thermolysin concentration; Cat# 88303, Sigma-Aldrich). Proteolysis was halted by the addition of excess EDTA in PBS to a final concentration 15 mM.
Digested IgG solutions were mixed in equal parts with 2× SDS buffer (100 mM Tris [pH 7.4], 0.4% wt:vol SDS, 20% vol:vol glycerol, 0.2% wt:vol bromophenol blue, and 10% vol:vol 2-mercaptoethanol) and boiled for 5 minutes. Samples were then separated under reducing conditions on SDS-polyacrylamide gel electrophoresis (10% acrylamide resolving gel, 4% stacking gel) for 50 minutes at 180 V in running buffer (25 mM Tris-base [pH 8.3], 250 mM glycine, and 0.1% wt:vol SDS) in a Mini-Protean III apparatus (BioRad). Gels were fixed for 30 minutes in fixing solution (10% vol:vol acetic acid, 25% vol:vol methanol) under gentle agitation. Gels were stained overnight with Coomassie Brilliant Blue (0.025% wt:vol Coomassie Blue, 50% vol:vol methanol, 16.7% vol:vol glacial acetic acid) and then destained (50% vol:vol methanol, 16.7% vol:vol glacial acetic acid). Gels were imaged under light using a Chemi-Genius BioImager with GeneSnap software, and band densitometry of the IgG heavy chain was performed with GeneTools software (v4.02; Syngene, Frederick, Maryland). Optical densities of 3 replicate gels were normalized to the mean density of the 0 M urea condition for each treatment group (control or oxidized), allowing the fraction of folded protein remaining to be plotted as a function of urea concentration.
Statistics
For DSF, unfolding temperatures (T m) were defined as the inflection point of the increased fluorescence measured at 625 nm during protein denaturation. T m values were determined from fluorescence measurements using the Boltzmann function of Origin software (v.8.5; OriginLab, North Hampton, Massachusetts). For RT-iDSF, denaturation rates (in units of FU/s) were determined from the slope of a plot of fluorescence intensity over time. Kinetic parameters of denaturation including K nd (half-maximal rate of native denaturation), D max (maximum rate of denaturation), and Hill coefficient (µ, the cooperativity of individual denaturants in protein unfolding) were calculated for IgG samples by fitting a plot of mean initial denaturation rates over the urea gradient to the Hill equation using Kinetics software (v.3.5.1).24 For PP, the kinetic constant C m of IgG heavy chain was derived from the optical densities using the I50 (constant of inhibition) equation from Kinetics software (v.3.5.1).24 All results are presented as mean ± standard error of mean (SEM; n = 4 trials). Statistical testing was performed by Student t test, except for the results of DSF assays that were tested by analysis of variance with post hoc Tukey test.
Results
Stability Effects of Buffer Composition
Human IgG stability, assessed in terms of unfolding transition midpoint temperature (T m), was determined by DSF for a range of pH values and buffer types (Figure 1A; Table 1). Immunoglobulin G was found to be most resistant to thermal denaturation over the range of pH 5.0 to 10.0, with denaturation occurring in a single transition. Unfolding temperatures dropped greatly outside of this range. Maximal IgG thermal stability was observed at pH 7.0 in PIPES buffer (T m = 70.5 ± 0.2°C), but this was not significantly greater than for IgG in PBS (T m = 69.7 ± 0.3°C).
Figure 1.
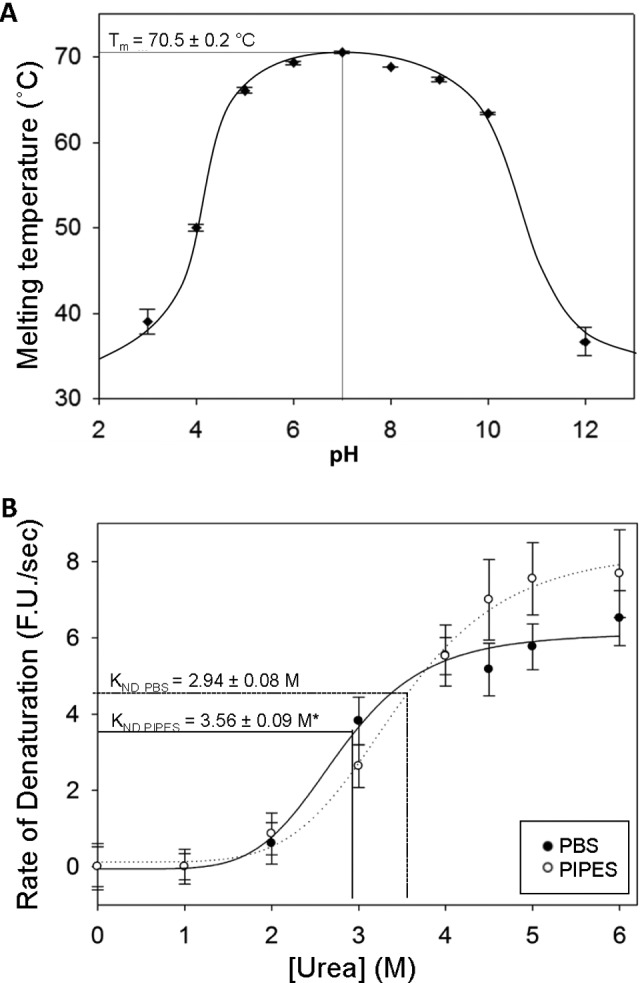
The effect of buffer species and environmental pH on immunoglobulin G (IgG) stability against denaturation by heat and urea. A, Unfolding temperatures (T m) of human IgG over a range of pH conditions in various buffers (see Table 1 for list) as determined by differential scanning fluorimetry (DSF) with a heating rate of 0.5°C/30 s. Optimal thermal stability of IgG was observed at pH 7.0, with a stable range of pH 5.0 to 10.0. B, Effect of buffer species on the velocity of denaturation of IgG as determined by the rate of SYPRO Orange fluorescence increases at various concentrations of urea. Immunoglobulin G samples were incubated at 4°C in either phosphate-buffered saline (PBS) or 1,4-Piperazinediethanesulfonic acid (PIPES) buffer (both pH 7.0), with various concentrations of urea. An asterisk (*) indicates significantly different from the PBS value (P < .01). Data for plots (A) and (B) represent mean ± standard error of mean (SEM; n = 4 trials). For both experiments, fluorescence (λex 485 nm; λem 625 nm) was measured using a modified BioRad iCycler thermocycler.
Subsequently, RT-iDSF was employed to ascertain the relative stability of IgG in both PIPES and PBS-buffered solutions at pH 7.0. Real-time isothermal differential scanning fluorimetry analysis showed that IgG in PIPES solution had a significantly greater constant of denaturation by urea (K nd;PIPES = 3.56 ± 0.09 M) as compared to PBS-buffered IgG (K nd;PBS = 2.94 ± 0.08; P < .01; Table 2; Figure 1B). However, PIPES-buffered samples also showed a greater maximal rate of denaturation than those in PBS (D max;PIPES = 8.9 ± 0.4 FU/s; D max;PBS = 5.4 ± 0.2 FU/s; P < .001). The cooperativity of urea-mediated protein unfolding was not significantly different in PBS (µPBS = 9.2 ± 3.5) than PIPES buffer (µPIPES = 4.8 ± 0.5).
Table 2.
Unfolding Temperatures and Kinetic Constants of Denaturation for Human IgG in Either PIPES or PBS Buffers at pH 7.0, as Determined by DSF and RT-iDSF.a
| DSF | RT-iDSF | |||
|---|---|---|---|---|
| Buffer | IgG T m (°C) | K ND (M Urea) | μ | D max (FU/s) |
| PBS | 69.7 ± 0.3 | 2.94 ± 0.08 | 9.2 ± 3.5 | 5.4 ± 0.2 |
| PIPES | 70.5 ± 0.3 | 3.56 ± 0.09* | 4.8 ± 0.5 | 8.9 ± 0.4** |
Abbreviations: DSF, differential scanning fluorimetry; PBS, phosphate-buffered saline; PIPES, 1,4-Piperazinediethanesulfonic acid; RT-iDSF, real-time isothermal differential-scanning fluorimetry.
aData are presented as means ± SEM (n = 4). Results that are significantly different from the corresponding value for PBS by a Student t test are indicated with a single asterisk (*) for P < .01 or a double asterisk (**) for P <.001.
Effect of AAPH-Induced Oxidation
The free-radical generator AAPH was employed to evaluate the effect of protein oxidation on IgG stability in PBS at pH 7.0 measured by 3 different methods: DSF, RT-iDSF, and PP (Table 3). Differential scanning fluorimetry analysis showed that incubation with AAPH caused a concentration-dependent decrease in the unfolding temperature (T m) of IgG, with 50 mM AAPH causing a large T m decrease of nearly 8°C as compared to the untreated protein (P < .001; Table 3, Figure 2A). Real-time isothermal differential scanning fluorimetry showed a significant decrease in the stability of IgG to urea denaturation when the protein was preincubated with 50 mM AAPH (K nd = 3.49 ± 0.09 M urea) compared to control IgG (K nd = 3.96 ± 0.07 M urea; P < .05; Figure 2B). Incubation of IgG with AAPH also significantly increased denaturant cooperativity (µControl = 4.3 ± 0.9 µAAPH = 9.0 ± 1.0; P < .05), while the maximal rate of denaturation did not change significantly (D max Control = 4.5 ± 0.4; D max AAPH = 6.0 ± 2.0).
Table 3.
A Comparison of DSF, RT-iDSF, and Pulse Proteolysis (PP) to Determine the Effect of Incubation With 50 mM AAPH on the Stability of Human Polyclonal IgG in PBS (pH 7.0).a
| DSF | RT-iDSF | PP | |||
|---|---|---|---|---|---|
| Condition | Tm (°C) | K ND (M Urea) | μ | D max (FU/s) | Cm (M Urea) |
| Control IgG | 70.5 ± 0.3 | 3.96 ± 0.07 | 9.0 ± 1.0 | 4.5 ± 0.4 | 6.8 ± 0.1 |
| +50 mM AAPH | 62.9 ± 0.1** | 3.49 ± 0.09* | 4.3 ± 0.9* | 6.0 ± 2.0 | 6.4 ± 0.7 |
Abbreviations: AAPH, 2,2-Azobis[2-amidinopropane]dihydrochloride; DSF, differential scanning fluorimetry; PP, pulse proteolysis; RT-iDSF, real-time isothermal differential-scanning fluorimetry; SEM, standard error of mean.
aData are means ± SEM (n = 4). Experimental results that are significantly different from the control value (for IgG in PBS [pH 7.0] not treated with AAPH) according to a Student t test are indicated by an asterisk (*) for P < .05 or a double asterisk (**) for P < .001.
Figure 2.
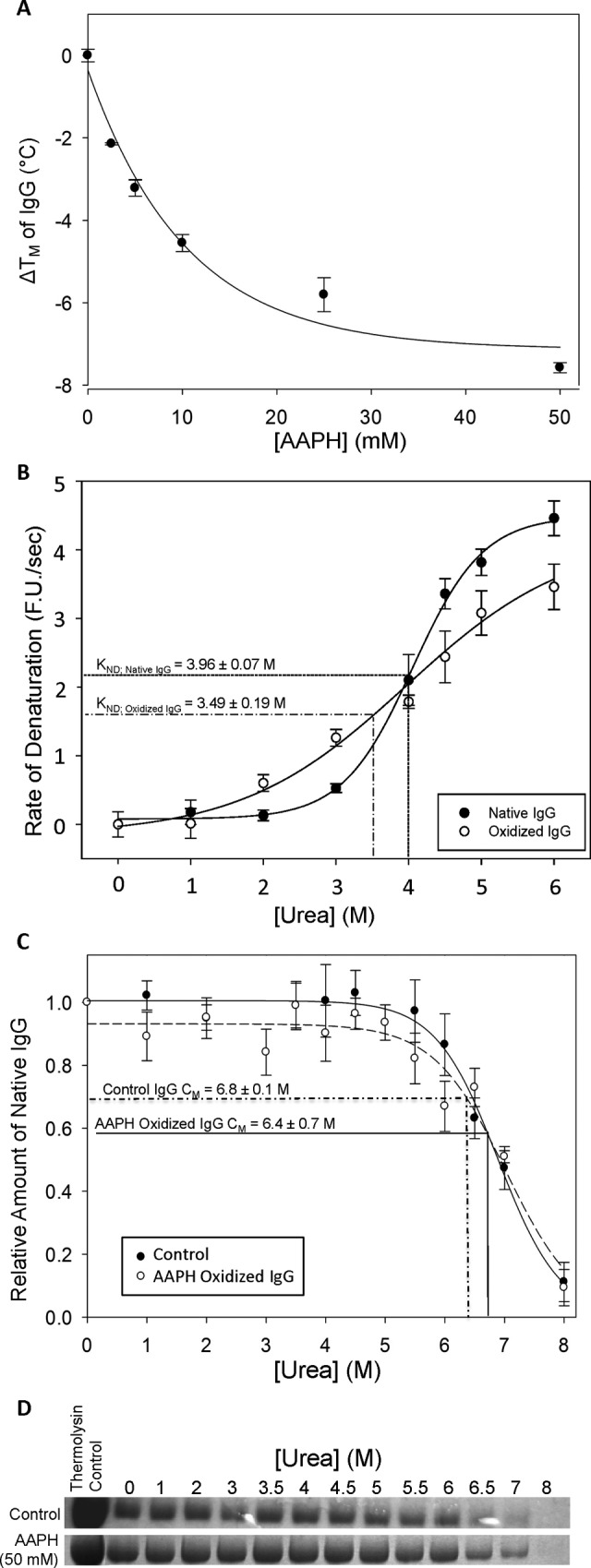
The influence of oxidation by 2,2-Azobis[2-amidinopropane]dihydrochloride (AAPH)-generated free radicals on immunoglobulin G (IgG) stability was investigated using differential scanning fluorimetry (DSF), real-time isothermal differential scanning fluorimetry (RT-iDSF), and pulse proteolysis. A, Thermal shift changes (ie, change in T m) determined by DSF for human IgG (1 mg/mL) incubated with {0, 2.5, 5, 10, 25, or 50} mM AAPH for 1 hour at 4°C. B, The effect of oxidation on the velocity of denaturation of IgG by urea as determined by RT-iDSF. Immunoglobulin G samples were incubated with or without 50 mM AAPH for 2 hours at 4°C immediately prior to assaying. An asterisk (*) indicates significantly different from the control value (P < .05). C, Plot showing the effect of preincubation with 50 mM AAPH on the rate of cleavage of human IgG in pulse proteolysis with thermolysin. The decreasing optical density of the IgG heavy chain band at high concentrations of urea indicates an increasing rate of proteolysis during the “pulse” step. Prior to proteolysis, IgG samples were preincubated with or without 50 mM AAPH for 2 hours at 4°C, as well as in urea solutions in various concentrations from 0 to 8 M. D, Representative SDS-PAGE gels from PP of human IgG, preincubated with or without 50 mM AAPH for 2 hours at 4°C, followed by the treatment as described in (C). The “no pulse’” lane depicts a control IgG sample that was not subjected to thermolysin treatment. Data for plots (A), (B), and (C) represent means ± standard error of the mean (SEM; n = 4 trials).
Immunoglobulin G samples with and without preincubation in 50 mM AAPH were also subjected to PP (Figure 2C and D). SDS-PAGE followed by densitometry analysis of the IgG heavy chain band (∼50 kDa) showed that when IgG was incubated with varying concentrations of urea to unfold the protein, the susceptibility of IgG to thermolysin cleavage was not significantly different between control and AAPH-incubated samples (C m;AAPH= 6.4 ± 0.7 M, C m;CNTRL= 6.8 ± 0.1 M). A comparison of samples containing no urea, with or without AAPH preincubation, showed that roughly two-thirds of the heavy chain IgG in a 10 mg/mL IgG solution was cleaved during the 5 minutes thermolysin “pulse”.
Discussion
The ability to sensitively monitor the conformational stability of a protein is critical since protein-based therapeutics, such as antibodies, may have reduced efficacy or safety if degradation occurs.6,25 Various modes of protein damage may be encountered during the stages of antibody production including purification, transport, storage, and administration7,8,10; this may be compounded in vivo by redox imbalances associated with disease states.26,27 As such, understanding protein susceptibility to the various stresses that may be encountered is crucial to the development of any new protein-based therapeutic.4,11 In this study, human polyclonal IgG protein was used as a model antibody for conformational stability assays using 3 techniques: DSF, RT-iDSF, and PP. The polymorphic nature of polyclonal IgG could lead to some variability in protein stability, increasing the error of measurements by these techniques.
Antibodies have relatively high thermal stability, with a T m of roughly 70°C.7 The thermal stability of IgG was initially assayed by DSF to screen for optimum pH conditions for further study (Figure 1A; Table 1). Optimal thermal stability of human IgG was observed in the range pH 5.0 to 10.0, with the greatest resistance to denaturation occurring at pH 7.0. However, there was no significant difference in thermal stability of IgG with PBS or PIPES as a buffer at pH 7.0 (Table 2), despite PIPES having a slightly greater sensitivity of pKa to temperature changes than PBS. Findings from DSF assays were in agreement with IgG transition temperatures from DSC as reported by Szenczi et al11; however, DSF analysis did not allow us to distinguish unfolding transitions of individual domains as has been previously characterized by DSC and circular dichroism.28
Real-time isothermal differential scanning fluorimetry was employed to compare the kinetic parameters of IgG denaturation with PBS or PIPES as a buffer. Real-time isothermal differential scanning fluorimetry results demonstrated that IgG incubated in PIPES buffer had significantly improved conformational stability against urea-induced denaturation (in terms of K ND) when compared to samples incubated in PBS (Figure 1B; Table 2); this indicates that PIPES has a more favorable molecular interaction than phosphate with IgG. The increased maximal rate of denaturation of IgG in PIPES buffer indicates that denaturation occurs more rapidly in this formulation when a large concentration of denaturant is present. We assume that the difference in IgG stability is not due to buffer capacity given that PIPES and PBS both have similar pKa values, all solutions contained ample buffer (100 mM), and IgG was generally stable over the pH range 5 to 10 in DSF assays (Figure 1A). Although phosphate is a widely utilized physiological buffer system, PIPES is a synthetic zwitterionic buffer introduced by Good et al29 as a solution to issues of reactivity, toxicity, and inefficiency associated with biological buffers. The increased stabilization of IgG by PIPES can be explained by the Hofmeister series, where sulfonate anions are predicted to have a greater protein-stabilizing (ie, “salting in”) effects than phosphate. Given that PIPES contains 2 sulfonate groups, PIPES may increase IgG stability through this mechanism.29–33 However, the biochemical system in question is complicated by the presence of other ions (urea, sodium, potassium, and chloride) that influence these effects. Furthermore, the stabilization effect is exaggerated by the higher ionic strength and buffer concentration of formulations in this study than is generally used in antibody oncology products. A previous study has described a similar effect of ethane sulfonic acid buffers (including PIPES) on protein conformational stability, where PIPES decreased Na+/K+-adenosine triphosphatase (ATPase) activity by stabilizing the protein and preventing the conformational change that is required for ATPase activity.30 Although not studied in the current work, other buffers commonly used in antibody formulations (such as acetate, histidine, or tris) may also have differential effects on antibody stability following the Hofmeister series, though would need to be determined empirically as protein-specific factors are also involved and exceptions to the series have been described.31
Oxidation damage is a major concern in the production and storage of protein-based therapeutics.32,33 As such, the ability to design buffers to maximize protein stability against oxidative stress is of interest for the formulation of protein-based therapies.34 In this study, AAPH was used as a free-radical generator to cause oxidation of the protein, such that the effect of oxidation on the conformational stability of polyclonal IgG could be evaluated. Following 2 hours of incubation with 50 mM AAPH, RT-iDSF analysis of IgG stability against urea denaturation was assessed and showed a decrease in K nd when compared to untreated control samples (Figure 2B, Table 2). Similar results were noted through DSF analysis, with an AAPH concentration–dependent decrease in IgG thermal stability (Figure 2A). These results are in agreement with a previous study that found changes in the native conformation and decreasing thermal stability of IgG1 constant fragment (Fc) following methionine oxidation.34 Conversely, the results of PP did not indicate any significant change to IgG stability as a result of AAPH oxidation (Figure 2C and D). This finding is consistent with a previous study that found that oxidation alone did not cause IgG cleavage and that digestion by trypsin and chymotrypsin were not significantly altered following oxidation.35 These results suggest that RT-iDSF is a more sensitive method for determining protein stability than PP and may provide alternate mechanistic insights into protein degradation as compared with DSF.
As a generator of alkyl, alkoxy, and alkyl peroxy radicals, AAPH could potentially oxidize sulfur-containing and aromatic amino acid side chains.18–20 Previous studies have shown that 2 conserved Met residues in IgG1 (M252 and M458) are particularly susceptible to oxidation, leading to changes in antibody structure and stability34,36–38 (Figure 3). In contrast, other nonconserved Met residues found in some alleles of IgG1 are more resistant to oxidation (eg, Met358).35 The modified residue, methionine sulfoxide, might alter protein structure due to decreased hydrophobicity.32 A recent study found that AAPH treatment led to the oxidation of the aforementioned Met residues, as well as 2 Trp residues located in the Fab fragment of the heavy chains of an IgG1 monoclonal antibody; consequently, the oxidized antibody displayed a greater tendency to aggregation.39 Although IgG contains many intra- and interchain disulfide bonds, some free cysteine residues have been found in both serum and recombinant IgG40,41; while rare, these thiols are highly reactive and may also play a role in the oxidative degradation of IgG. Overall, these results highlight the need to evaluate the vulnerability of protein therapeutics to oxidative damage.
Figure 3.
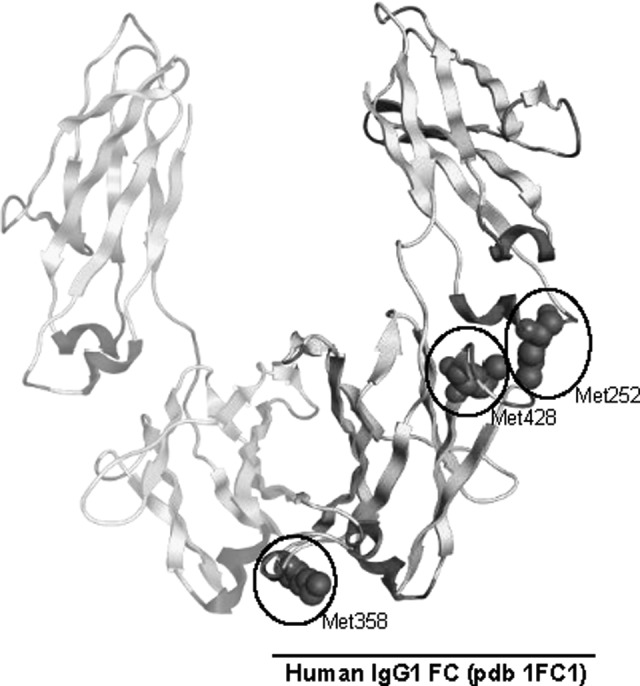
The human IgG1 heavy chain contains several methionine residues (M252, M358, and M428) that are prone to oxidation. A, Model of the IgG1 constant fragment with oxidizable Met residues circled on 1 chain of the dimer. The 2 most reactive residues, M252 and M428, are found in close proximity, located at the CH2–CH3 domain interface. Image was produced with MOE software using the available human IgG1 structure deposited within the protein data bank (accession 1FC1).
Conclusion
We initially hypothesized that RT-iDSF (using urea as the denaturant under isothermal conditions) may reveal small changes in antibody conformational stability that were within or below the limits of detection of either thermal denaturation DSF or PP. It was found that RT-iDSF determination of Knd was a more sensitive measure of IgG conformational stability than T m when assaying the stabilizing effect of the buffers PBS and PIPES (Figure 1). However, in experiments on IgG destabilization after AAPH oxidation treatment, both DSF and RT-iDSF were more sensitive than PP (Figure 2). In addition to greater sensitivity, RT-iDSF and DSF experiments are less laborious and time-consuming than PP.22,23 The RT-iDSF method is a logical extension of existing DSF and isothermal denaturation methods and provides alternate insights into protein stability.42–44 With the emergence of RT-iDSF to complement both DSF and other DSF-based isothermal denaturation techniques, there are now a variety of different methods for assessing protein stability in solution using a modified qRT-PCR instrument.12,45,46 Although this study examined the stability of polyclonal IgG, these techniques should prove equally suitable for assessing monoclonal antibodies. A combination of these techniques could be employed as a highly effective, low-cost means to evaluate antibody stability for the development of cancer immunotherapies.10,47,48
Acknowledgments
Thanks to J.M. Storey for editorial review of the manuscript. .
Abbreviations
- AAPH
2,2-Azobis[2-amidinopropane]dihydrochloride
- CAPS
3-(Cyclohexylamino)-1-propanesulfonic acid
- CHES
2-(Cyclohexylamino)ethane sulfonic acid
- DSF
differential scanning fluorimetry
- Fab
fragment antigen-binding region
- FC
fragment crystallizable region
- IgG
immunoglobulin G
- PIPES
1,4-Piperazinediethanesulfonic acid
- PP
pulse proteolysis
- RT-iDSF
real-time isothermal differential-scanning fluorimetry.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research was supported by a discovery grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada to K.B.S. and the Canada Research Chairs program. K.B. held an NSERC postdoctoral fellowship. N.D. was supported by an Ontario Graduate Scholarship. J.M. held an NSERC undergraduate student research award
References
- 1. Pillay V, Gan HK, Scott AM. Antibodies in oncology. N Biotechnol. 2011;28(5):518–529. doi:10.1016/j.nbt.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 2. Bouchard H, Viskov C, Garcia-Echeverria C. Antibody-drug conjugates—a new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24(23):5357–5363. doi:10.1016/j.bmcl.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 3. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–287. doi:10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 4. Falconer RJ, Chan C, Hughes K, Munro TP. Stabilization of a monoclonal antibody during purification and formulation by addition of basic amino acid excipients. J Chem Technol Biotechnol. 2011;86(7):942–948. doi:10.1002/jctb.2657. [Google Scholar]
- 5. Goswami S, Wang W, Arakawa T, Ohtake S. Developments and challenges for mAb-based therapeutics. Antibodies. 2013;2(3):452–500. doi:10.3390/antib2030452. [Google Scholar]
- 6. Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. doi:10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 7. Wang W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int J Pharm. 1999;185(2):129–188. doi:10.1016/S0378-5173(99)00152-0. [DOI] [PubMed] [Google Scholar]
- 8. Daugherty AL, Mrsny RJ. Formulation and delivery issues for monoclonal antibody therapeutics. Adv Drug Deliv Rev. 2006;58(5-6):686–706. doi:10.1016/j.addr.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 9. Kameoka D, Masuzaki E, Ueda T, Imoto T. Effect of buffer species on the unfolding and the aggregation of humanized IgG. J Biochem. 2007;142(3):383–391. doi:10.1093/jb/mvm145. [DOI] [PubMed] [Google Scholar]
- 10. Jiskoot W, Beuvery EC, de Koning AA, Herron JN, Crommelin DJ. Analytical approaches to the study of monoclonal antibody stability. Pharm Res. 1990;7(12):1234–1241. [DOI] [PubMed] [Google Scholar]
- 11. Szenczi Á, Kardos J, Medgyesi GA, Závodszky P. The effect of solvent environment on the conformation and stability of human polyclonal IgG in solution. Biologicals. 2006;34(1):5–14. doi:10.1016/j.biologicals.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 12. Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. doi:10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 13. Biggar KK, Dawson NJ, Storey KB. Real-time protein unfolding: a method for determining the kinetics of native protein denaturation using a quantitative real-time thermocycler. Biotechniques. 2012;53(4):231–238. doi:10.2144/0000113922. [DOI] [PubMed] [Google Scholar]
- 14. Kishore D, Kundu S, Kayastha AM. Thermal, chemical and pH induced denaturation of a multimeric β-galactosidase reveals multiple unfolding pathways. PLoS One. 2012;7(11):e50380 doi:10.1371/journal.pone.0050380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramprakash J, Doseeva V, Galkin A, et al. Comparison of the chemical and thermal denaturation of proteins by a two-state transition model. Anal Biochem. 2008;374(1):221–230. doi:10.1016/j.ab.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 16. Hill AV. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol. 1910;40:iv-vii. [Google Scholar]
- 17. Betigeri S, Thakur A, Raghavan K. Use of 2,2-Azobis(2-amidinopropane) dihydrochloride as a reagent tool for evaluation of oxidative stability of drugs. Pharm Res. 2005;22(2):310–317. doi:10.1007/s11095-004-1199-x. [DOI] [PubMed] [Google Scholar]
- 18. Ji JA, Zhang B, Cheng W, Wang YJ. Methionine, tryptophan, and histidine oxidation in a model protein, PTH: mechanisms and stabilization. J Pharm Sci. 2009;98(12):4485–5000. doi:10.1002/jps.21746. [DOI] [PubMed] [Google Scholar]
- 19. Kang JH, Kim KS, Choi SY, Kwon YH, Won MH. Oxidative modification of human ceruloplasmin by peroxyl radicals. Biochim Biophys Acta. 2001;1568(1):30–36. doi:10.1016/S0304-4165(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 20. Chepelev NL, Bennitz JD, Wright JS, Smith JC, Willmore WG. Oxidative modification of citrate synthase by peroxyl radicals and protection with novel antioxidants. J Enzyme Inhib Med Chem. 2009;24(6):1319–1331. doi:10.3109/14756360902852586. [DOI] [PubMed] [Google Scholar]
- 21. Dean RT, Hunt JV, Grant AJ, Yamamoto Y, Niki E. Free radical damage to proteins: the influence of the relative localization of radical generation, antioxidants, and target proteins. Free Radic Biol Med. 1991;11(2):161–168. doi:10.1016/0891-5849(91)90167-2. [DOI] [PubMed] [Google Scholar]
- 22. Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2(3):207–212. doi:10.1038/nmeth740. [DOI] [PubMed] [Google Scholar]
- 23. Na YR, Park C. Investigating protein unfolding kinetics by pulse proteolysis. Protein Sci. 2009;18(2):268–276. doi:10.1002/pro.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brooks SP. A simple computer program with statistical tests for the analysis of enzyme kinetics. Biotechniques. 1992;13(6):906–911. [PubMed] [Google Scholar]
- 25. Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4(4):298–306. doi:10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 26. Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53(3):209–219. doi:10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 27. Chinta SJ, Andersen JK. Redox imbalance in Parkinson’s disease. Biochim Biophys Acta. 2008;1780(11):1362–1367. doi:doi:10.1016/j.bbagen.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vermeer AW, Norde W. The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys J. 2000;78(1):394–404. doi:10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Good NE, Winget GD, Winter W, Connolly TN, Izawa S, Sing RM. Hydrogen ion buffers for biological research. Biochemistry. 1966;5(2):467–477. doi:10.1021/bi00866a011. [DOI] [PubMed] [Google Scholar]
- 30. Robinson JD, Davis RL. Buffer, pH, and ionic strength effects on the (Na+ + K+)-ATPase. Biochim Biophys Acta. 1987;912(3):343–347. [DOI] [PubMed] [Google Scholar]
- 31. Jungwirth P, Cremer PS. Beyond Hofmeister. Nat Chem. 2014;6(4):261–263. doi:10.1038/nchem.1899. [DOI] [PubMed] [Google Scholar]
- 32. Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009;18(2):424–433. doi:10.1002/pro.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo Q, Joubert MK, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem. 2011;286(28):25134–25144. doi:10.1074/jbc.M110.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu D, Ren D, Huang H. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47(18):5088–5100. doi:10.1021/bi702238b [DOI] [PubMed] [Google Scholar]
- 35. Liu H, Gaza-Bulseco G, Xiang T, Chumsae C. Structural effect of deglycosylation and methionine oxidation on a recombinant monoclonal antibody. Mol Immunol. 2008;45(3):701–708. doi:10.1016/j.molimm.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 36. Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;870(1):55–62. doi:10.1016/j.jchromb.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 37. Chumsae C, Gaza-Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;850(1-2):285–294. doi:10.1016/j.jchromb.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 38. Li S, Schöneich C, Borchardt RT. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol Bioeng. 1995;48(5):490–500. doi:10.1002/bit.260480511. [DOI] [PubMed] [Google Scholar]
- 39. Folzer E, Diepold K, Bomans K, et al. Selective oxidation of methionine and tryptophan residues in a therapeutic IgG1 molecule. J Pharm Sci. 2015;104(9):2824–2831. doi:10.1002/jps.24509. [DOI] [PubMed] [Google Scholar]
- 40. Chumsae C, Gaza-Bulseco G, Liu H. Identification and localization of unpaired cysteine residues in monoclonal antibodies by fluorescence labeling and mass spectrometry. Anal Chem. 2009;81(15):6449–6457. doi:10.1021/ac900815z. [DOI] [PubMed] [Google Scholar]
- 41. Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 2012;4(1):17–23. doi:10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lo MC, Aulabaugh A, Jin G, et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332(1):153–159. doi:10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 43. Pantaliano MW, Petrella EC, Kwasnoski JD, et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001:6(6):429–440. doi:0803973233 [DOI] [PubMed] [Google Scholar]
- 44. Sarver RW, Rogers JM, Epps DE. Determination of ligand-MurB interactions by isothermal denaturation: application as a secondary assay to complement high throughput screening. J Biomol Screen. 2002;7(1):21–28. doi:10.1177/108705710200700104. [DOI] [PubMed] [Google Scholar]
- 45. Vedadi M, Niesen FH, Allali-Hassani A, et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103(43):15835–15840. doi:10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Senisterra GA, Soo Hong B, Park HW, Vedadi M. Application of high-throughput isothermal denaturation to assess protein stability and screen for ligands. J Biomol Screen. 2008;13(5):337–342. doi:10.1177/1087057108317825. [DOI] [PubMed] [Google Scholar]
- 47. Mashalidis EH, Śledź P, Lang S, Abell C. A three-stage biophysical screening cascade for fragment-based drug discovery. Nat Protoc. 2013;8(11):2309–2324. doi:10.1038/nprot.2013.130. [DOI] [PubMed] [Google Scholar]
- 48. Senisterra GA, Finerty PJ. High throughput methods of assessing protein stability and aggregation. Mol Biosyst. 2009;5(3):217–223. doi:10.1039/b814377c. [DOI] [PubMed] [Google Scholar]