

Synopsis
The central role of hormonal 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) is to regulate calcium and phosphorus homeostasis via actions in intestine, kidney and bone. These actions as well as many additional pleiotropic activities in a wide variety of cell types not involved in mineral metabolism are mediated by the vitamin D receptor (VDR), a nuclear protein that functions in virtually all target tissues to regulate the expression of genes and gene networks. Recent studies using genome-wide scale techniques have extended fundamental ideas regarding vitamin D mediated control of gene expression while simultaneously revealing a series of new concepts as well. In this article, we summarize our current view of the biological actions of the vitamin D hormone and then focus on new concepts that drive our understanding of the mechanisms through which vitamin D operates.
Keywords: Mineral homeostasis, calcemic hormones, PTH, FGF23, Cyp27b1, nuclear receptor, transcription, target genes, ChIP-seq analysis, CRISPR/Cas9, IL-17, RANKL, transient receptor potential vanilloid type-6 (TRPV6), Mmp13, genome-wide principles, genetic linkage
I. Introduction/Historical Perspective
Discovered many decades earlier by Mellanby (1), McCollum (2), Steenbock (3), and Windaus (4), vitamin D is now known through ensuing research efforts by many investigators to be converted to 1,25-dihydroxvyvitamin D3 (1,25(OH)2D3), its biologically active form, and to be centrally involved in the regulation of calcium and phosphorus homeostasis in higher vertebrates (5). The “vitamin’s” mode of action involves the regulation of gene expression in specific tissues; this activity is mediated by the nuclear receptor for vitamin D (VDR), a DNA binding protein that interacts directly with regulatory sequences near target genes and which functions to recruit chromatin active complexes that participate genetically and epigenetically in modifying transcriptional output (6–9). Based on these individual features as well as the fact that the VDR itself is a member of the nuclear receptor family of transcription factor genes, the hormonally active form of vitamin D is the key component of a classic steroid hormone endocrine system (10–12). Target genes regulated by vitamin D are not limited to those involved in mineral homeostasis, however, but also include genes that are linked to highly diverse biological processes associated with the cardiovascular and immune systems, the skin, the metabolism of xenobiotics, and numerous additional cellular processes as well (13). The vitamin D hormone also controls cellular proliferation and differentiation, implying that in addition to its broad therapeutic potential in metabolic diseases, it may also be useful in the control of certain cancers (14). This article provides an overview of the vitamin D system and then focuses on details of the mechanism of action of this hormone in specific cell types, illuminating several of the genes that are involved.
II. Vitamin D Production and Metabolism
It is appropriate in this overview to begin with a brief summary of the gene products that function to synthesize and control the levels of 1,25(OH)2D3 in the blood and that serve to degrade the hormone in target tissues. Importantly and consistent with concepts of endocrinology, at least two of these gene products are directly regulated by 1,25(OH)2D3 through important feedback loops. Vitamin D is produced in skin following exposure to sunlight through a process that involves photolysis of cutaneous 7-dehydrocholesterol (provitamin D) to previtamin D followed by isomerization (15). It was soon realized, however, that vitamin D must be metabolically activated further prior to function (see Figure 1). Accordingly, the parent vitamin is first hydroxylated in the liver to 25(OH)D3 by CYP2R1, a 25-hydroxylase (25-OHase) discovered by Russell and colleagues (16,17). This enzyme exhibits the highest affinity for vitamin D and is likely the most important of the 25-OHases based upon genetic evidence that suggests that defects in this gene in humans lead to vitamin D dysfunction (17,18). CYP2R1 knock out mice still produce significant levels of 25(OH)D3, however, suggesting that other hydroxylases may be involved including mitochondrial CYP27A1 as well as microsomal CYP2D11, CYP2D25, CYP2J2/3, and CYP3A4 (19). Thus, additional work will be required to define all the components of this particular modification to vitamin D in vivo.
Figure 1.

Sunlight mediated photolysis of 7-dehydrocholesterol to vitamin D in the skin, and its activation through subsequent sequential hydroxylation in the liver by Cyp2R1 to 25(OH)D3 and by Cyp27b1 to the vitamin D hormone 1,25(OH)2D3 in the kidney. Cyp24a1 initiates the degradation of 25(OH)D3 to 24,25(OH)2D3 in the kidney and the degradation of 1,25(OH)2D3 to 1,24,25(OH)3D3 in all vitamin D targets tissues that results in eventual conversion to calcitroic acid. Alternative degradation pathways in mice also result from Cyp24a1 activity as well.
The second, and most important, hydroxylation of vitamin D occurs in the kidney through the actions of mitochondrial CYP27B1, and results in the synthesis of the active hormone 1,25(OH)2D3 (20) (see Figure 1). The validity of this enzyme as the exclusive source of 1,25(OH)2D3 is supported by the subsequent identification of mutations in the CYP27B1 gene that result in vitamin D-dependency rickets, type 1 (VDDR-1) that accounts for the 1,25(OH)2D3 deficiency first reported in 1973 (21,22). Importantly, the biochemical and skeletal phenotype of this syndrome has been recapitulated more recently through genetic deletion of the Cyp27b1 gene in mice using homologous recombination (23). The activity of renal CYP27B1 is critical to the production and maintenance of physiologic levels of circulating 1,25(OH)2D3 (5). As a consequence, the synthesis and activity of CYP27B1 is tightly regulated by endocrine factors that are elaborated in response to changes in plasma calcium and/or phosphorus, most notably PTH (24) (see Figure 2). In addition to PTH, which is induced in response to hypocalcemia and is the major stimulator of 1,25(OH)2D3 production, FGF23, which requires the transmembrane co-receptor αKlotho, is also an important modulator of vitamin D metabolism (24,25) (see Figure 2). The molecular mechanisms and signaling pathways whereby PTH induces and FGF23 suppresses the expression of renal CYP27B1 remain undefined at present (26). Importantly, completion of the vitamin D endocrine circuit involves a potent feedback mechanism through which 1,25(OH)2D3 acts to suppress CYP27B1 gene expression in the kidney, to downregulate PTH production and secretion by the parathyroid gland and to upregulate FGF23 expressed in bone (27,28). A similar negative feedback mechanism appears to exist for FGF23, which acts not only at the kidney to down-regulate CYP27B1, but which may function in the parathyroid gland to suppress PTH as well (29). A number of additional factors also regulate CYP27B1 expression including the sex and adrenal hormones, prolactin and growth hormone (27). A recent report delineates how prolactin may induce CYP27B1 via activation of STAT5 (30). Interestingly, broad studies also suggest that the expression of CYP27B1 may not be restricted to the kidney, but rather is synthesized in small amounts in other cell types as well (31). If correct, this would suggest that circulating levels of 25(OH)D3 could control the production of 1,25(OH)2D3 in these cells in either a direct or in a paracrine or autocrine manner independently of the kidney. The skin, in contrast, may represent a third example where in addition to 1,25(OH)2D3, 25(OH)D3 is also made internally via expression of CYP2R1 (32,33).
Figure 2.
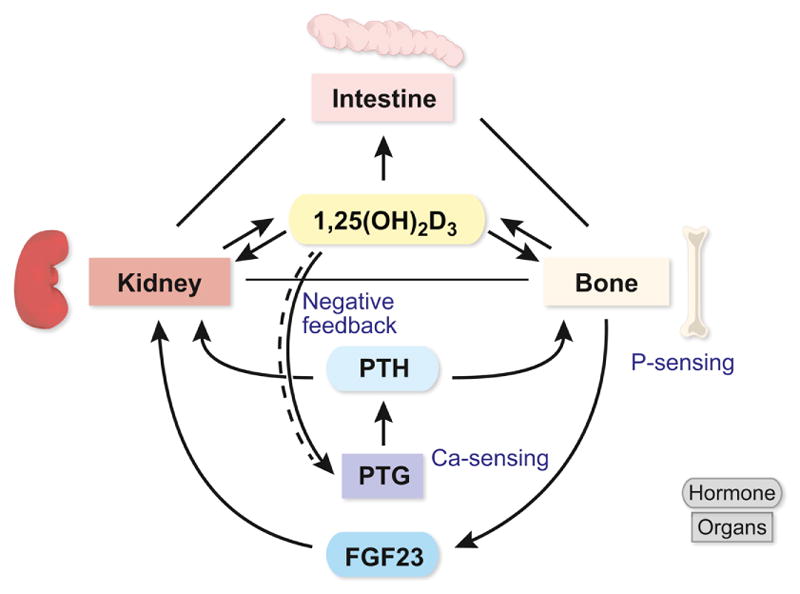
Scheme for the regulation of mineral homeostasis in higher vertebrates. The hormones PTH and FGF23 monitor extracellular calcium and phosphate, respectively, and orchestrate the mineral regulating activities of the intestine, kidney and bone through actions on vitamin D metabolism.
The degradation of 1,25(OH)2D3 is accomplished via the action of CYP24A1, a microsomal enzyme that is expressed basally in the kidney and via 1,25(OH)2D3 induction in this as well as virtually all vitamin D target tissues (34). This degradation pathway involves a third hydroxylation of 1,25(OH)2D3 at carbon 24 to produce 1,24,25-trihydroxyvitamin D3 (1,24,25(OH)3D3) or at carbon 23 to produce 1,25-hydroxyvitamin D3-26,23-lactone both of which proceed through further oxidation to calcitroic acid and CO2 (27). 25(OH)D3, the substrate for 1,25(OH)2D3 production, is also hydroxylated by CYP24A1 in the kidney, leading to the secretion of 24,25-dihydroxyvitamin D3 (24,25(OH)2D3) into the blood. When compared to the regulation of CYP27B1, CYP24A1 is reciprocally regulated (stimulated by 1,25(OH)2D3 and suppressed by PTH), an activity that tends to sustain the systemic levels of 1,25(OH)2D3 (35,36). Together, both FGF23 and α-klotho induce CYP24A1 expression, although the transcription factor is currently unknown (26). Since CYP24A1 is present in all vitamin D target cells as well, CYP24A1 can also modulate the intracellular levels of 1,25(OH)2D3 resulting in the control of cellular response to 1,25(OH)2D3. A biological role for the 24,25(OH)2D3 metabolite has been suggested for several decades, although evidence for this hypothesis has yet to emerge in vivo. Indeed, although deletion of the CYP24A1 gene in the mouse by St. Arnaud and coworkers resulted in a phenotype of hypercalcemia, hypercalciuria, renal calcification and skeletal abnormalities, all of these effects were subsequently attributed to the toxicity induced by high circulating levels of 1,25(OH)2D3 (37). Thus, simultaneous excision of the genes for not only CYP24A1 but for the VDR as well completely abrogated the toxicity that resulted from these high levels of the hormone (38). Interestingly, debilitating mutations within the CYP24A1 gene that potentiate response to 1,25(OH)2D3 have been found recently in very young children with idiopathic infantile hypercalcemia and in adults. In adults, patients were characterized by hypercalcemia, hypercalciuria and recurring kidney stones (39–42). These findings provide evidence for a critical role of CYP24A1 in humans. As indicated above, one of the fundamental actions of 1,25(OH)2D3 in all target cells is to stimulate the expression of CYP24A1, thus initiating the means to its own destruction. Surprisingly, although the general mechanism through which 1,25(OH)2D3 induces CYP24A1 transcription was thought to be understood at the molecular level, more recent studies have revealed a more complex mode of activation by the vitamin D hormone (43). This additional complexity represents a paradigm for how most genes are believed to be regulated through distal genomic modulation (9,44). Interestingly, the molecular mechanisms through which the primary regulators PTH, 1,25(OH)2D3 and FGF23 control the expression of renal Cyp27b1 and Cyp24a1 remain to be determined, although their actions are likely all transcriptional in nature. These mechanisms are critical to understanding the vitamin D system as these components represent the exclusive enzymatic mediators of the production and degradation of the key metabolite of vitamin D and their levels are frequently aberrant as a consequent of numerous disease processes.
III. The Roles of Vitamin D in Classic and Non Classical Target Tissues
Calcium and phosphorus homeostasis is orchestrated via the inter-regulatory actions of PTH and FGF23, respectively (45,46) (see Figure 2). These hormones control in turn the expression of renal CYP27B1 which completes the synthesis of 1,25(OH)2D3, acting directly through intestine, kidney, and bone to control mineral balance (47). Accordingly, these tissues function to acquire calcium and phosphate from the diet, to resorb the ions from glomerular filtrate, and to provide an immediate source of skeletal calcium and phosphate when the diet becomes deficient. Integrating these tissue activities so that plasma calcium and phosphorus concentrations are tightly maintained is central to the function of 1,25(OH)2D3 and to multiple downstream processes linked to normal calcium and phosphate levels such as muscle and nerve function. The intestinal actions of 1,25(OH)2D3 are focused upon regulating the production of proteins essential to the processes of dietary calcium and phosphorus absorption, and include such gene products as calbindin D9K (S100g), NCX1, TRPV6, and ATP2B1 (48) (see Figure 3). The principal action of vitamin D in maintenance of calcium homeostasis is to promote calcium absorption from the intestine. This conclusion is based on the observation that rickets and osteomalacia can be prevented in VDR-null mice fed a prevention diet high in calcium, phosphorus and lactose (49,50). In addition, when patients with hereditary 1,25(OH)2D3-resistant rickets are treated with intravenous or high oral calcium, the skeletal phenotypes of these patients are reversed (51). In the traditional facilitated diffusion model, 1,25(OH)2D3 acts by regulating a) calcium entry through the apical calcium channel TRPV6, b) transcellular movement of calcium by binding to the calcium binding protein calbindin D and c) extrusion of calcium from the cell by the plasma membrane Ca-sensitive ATPase PMCA2b. However studies in TRPV6- and calbindin D-null mice have challenged this traditional view. Indeed, there are no phenotypic differences between calbindin-D9k- or TRPV6-null mice and wildtype mice when dietary calcium is normal (52–54). These findings indicate that under adequate calcium conditions, calbindin-D9k and TRPV6 are redundant for intestinal calcium absorption and suggest compensation by other channels or proteins yet to be identified. On the other hand, intestine specific transgenic expression of TRPV6 has been shown to result in a marked increase in intestinal calcium absorption and bone density in VDR-null mice, indicating a significant role for TRPV6 in the calcium absorptive process (55). Recent studies suggest the possibility that vitamin D-sensitive calcium uptake is achieved via a complex network of active calcium regulating components rather than through a single entity (56). It should also be noted that although the duodenum has been the focus of research related to 1,25(OH)2D3 regulation of calcium absorption over many years, it is the distal intestine where the majority of ingested calcium is absorbed (57). Indeed, studies in which VDR is expressed or deleted specifically in the distal region of the intestine, highlight the importance of both the distal as well as proximal segments of the intestine in vitamin D-mediated calcium homeostasis and bone mineralization (58,59). Studies related to mechanisms involved in 1,25(OH)2D3 regulation of calcium absorption in the distal intestine may suggest new strategies to increase efficiency of calcium absorption in individuals at risk for bone loss due to aging, bariatric surgery or inflammatory bowel disease.
Figure 3.

General mechanisms of action of 1,25(OH)2D3 and its diverse biology in target cells. 1,25(OH)2D3 regulates gene transcription in target cells by binding to VDR. This activated VDR heterodimerizes with RXR and binds to VDREs (vitamin D response elements) in and around target genes. Transcription proceeds through the interaction of the VDR with coactivators and with the transcription machinery. Liganded VDR interacts with steroid receptor coactivator 2 (SRC2; also known as GRIP1), which has histone acetylase activity (HAT), as a primary coactivator. SRC2 can recruit proteins as secondary coactivators such as CBP/p300 which also have HAT activity. VDR also interacts with mediator complex, which facilitates the activation of the RNA polymerase II holoenzyme through its C-terminal domain (CTD), thus promoting formation of the preinitiation complex. The SWI/SNF complex, which remodels chromatin using the energy of ATP hydrolysis, also contributes to activation by VDR. 1,25(OH)2D3 is known to maintain calcium homeostasis and to affect numerous other cell types. Effects on other cells systems include inhibition of proliferation of cancer cells and modulation of the immune system.
1,25(OH)2D3 can also provoke calcium and phosphorus mobilization from the skeleton through a process that involves both the stimulation of bone-resorbing osteoclast activity as well as the induction of new osteoclast formation from cellular precursors (60–62). This mechanism takes advantage of the hormone’s ability to induce expression of the autocrine TNFα-like factor RANKL(Receptor Activator of NF-κB Ligand) from chondrocytes, osteoblasts and osteocytes, as will be discussed later in this review. Recent studies suggest that 1,25(OH)2D3 may also play an active role in modulating the expression of mineralization regulating factors such as Spp1 (osteopontin), MGP (matrix gla protein), ENPP1 (ectonucleotide pyrophosphatase phosphodiesterase 1) and ENPP2, and ANK (progressive ankylosis protein), and ALPL (intestinal alkaline phosphatase), and perhaps others as well (63). This overall mechanism of calcium release from bone has a particularly profound consequence when dietary levels of calcium and phosphate are insufficient to maintain extracellular levels leading to a rise in both PTH and 1,25(OH)2D3. In such cases, maintenance of calcium and phosphorus levels in the blood is prioritized resulting in bone resorption and a corresponding structural weakening of the skeleton, thereby increasing the risk of bone fracture. Interestingly, recent studies also suggest that osteocytes, mature osteoblasts that have become fully encased in bone mineral, not only function to control calcium and phosphate release from bone through the production of RANKL and mineralization regulators (64,65), but may also act to remodel bone directly during certain physiological states such as lactation (66). This process is prompted by both 1,25(OH)2D3 and parathyroid hormone related protein. The actions of these hormones also influence the expression of the FGF23 gene directly from the osteocyte and perhaps other cell types as well, and its cellular processing and liberation into the circulation where it controls renal modulation of phosphate levels (46). It is also important to note that depletion of calcium and phosphorus levels in the blood results in the failure of bone to mineralize via physicochemical principles, resulting in rickets or osteomalacia (47). Thus, these studies support a direct effect of 1,25(OH)2D3 on bone as well as an indirect role through calcium provision to bone via stimulation of intestinal calcium absorption. If normal serum calcium cannot be maintained by intestinal calcium absorption, then 1,25(OH)2D3 acts together with PTH to stimulate osteoclastogenesis and to increase calcium reabsorption from the distal tubules of the kidney (36). Although the actions of 1,25(OH)2D3 are modest at the kidney, the actual amount of calcium recovered through reabsorption is highly significant due to the large daily load of calcium that is filtered by this organ.
Central to the orchestration of intestinal, kidney and bone actions by 1,25(OH)2D3 are the regulatory systems that monitor calcium and phosphorus content in plasma and participate along with the vitamin D hormone in maintaining those levels in the extracellular compartment. In the case of calcium, the level of this ion is continually monitored by the calcium-sensing receptor in the parathyroid gland, which responds by increasing parathyroid gland secretion of PTH when calcium content decreases. While the consequence of calcium liberation from the skeleton increases phosphate levels as well, both PTH and FGF23 function collectively in the kidney to promote phosphate diuresis through a mechanism involving cellular relocation of multiple sodium-phosphate transporters (29,67). FGF23 represents the long sought-after phosphate regulating hormone or phosphatonin that is not only induced by phosphate when it is in abundance but by 1,25(OH)2D3 as well, although neither of these mechanisms are currently understood (28,68,69). Linkage of FGF23 to the regulation of phosphate was initially derived from phenotypes associated with tumor-induced osteomalacia (TIO), autosomal dominant hypophosphatemic rickets (ADHR), and X-linked hypophosphatemic (XLH) syndromes (70). More recently, studies in which the Fgf23 gene has been either deleted or overexpressed in the mouse have emerged to strongly support this linkage. With regard to FGF23, a novel mechanism has now been identified whereby FGF23 promotes the redistribution of the phosphate transporters NaPi2a (Slc34a1) and NaPi2c (Slc34a3) such that proximal tubular reabsorption of phosphate is reduced (29). FGF23’s ability to suppress circulating 1,25(OH)2D3 also results in a reduction in intestinal phosphate uptake. These and additional actions in kidney and bone as well as in intestine restore calcium and phosphorus concentrations to their appropriate levels in plasma. Feedback mechanisms via FGF23 described earlier then act to prevent phosphorus levels from increasing beyond acceptable limits thereby maintaining calcium and phosphorus levels within narrow boundaries. The molecular mechanism(s) whereby 1,25(OH)2D3 orchestrates the expression of several of the genes whose functions are integral to the maintenance of calcium and phosphorus homeostasis are being defined.
1,25(OH)2D3 is also a regulator of cellular proliferation and differentiation, activities not unlike those manifested by many of the steroid hormones (see Figure 3). These growth-regulating actions of 1,25(OH)2D3, as well as many additional biological processes that are regulated by 1,25(OH)2D3 highlight not only important physiologic activities of the hormone but potential therapeutic roles for both the hormone and for synthetic vitamin D analogs. These include treatments for cancer, control of skin function, regulation of the immune system and autoimmune diseases and control of cardiovascular disease. Studies in VDR-null mice, for example provide direct evidence in vivo that in the absence of VDR there is increased sensitivity in response to a chemical carcinogen to the development of a variety of tumors including skin tumors, tumors of the lymph nodes and estrogen receptor negative tumors (71,72). VDR-null mice also develop hypertension and cardiac hypertrophy (73). Moreover, VDR-null mice also develop more severe conditions of inflammatory bowel disease which is associated with increased numbers of inflammatory cytokine (IL-17 and IFNγ) -secreting T cells in experimental models of colitis (74,75) and exhibit alterations in innate and adaptive immunity as well. The biological actions of the vitamin D hormone in several of these non-classical target tissues will be considered in other sections in this issue. Importantly, the underlying key feature of vitamin D response in all of these “non-classical” tissues is expression of the VDR.
IV. The Vitamin D Receptor and Genomic Mechanisms of Action
A. The Vitamin D Receptor (VDR)
A binding protein eventually designated the VDR was first discovered in the chicken intestine and then in other tissues including the parathyroid glands, kidney, and bone (6,7). This protein’s biochemical features, including its retention in chromatin (76) and its ability to bind to DNA (8) identified several years later suggested that it was similar to that of other receptors for known steroid hormones and that it likely played a role in transcriptional regulation. Despite much effort that preceded this event, it was the cloning of the chicken VDR gene (10) and the human (11) and rat (77) versions of this receptor that followed shortly thereafter, which ushered in a new era in vitamin D research. The availability of VDR cDNA clones enabled the direct detection of VDR RNA in cells, and following the introduction of RT-PCR analysis in the late 1980’s, made possible the development of the most sensitive assay for the VDR that is now currently in use. The cloning of the VDR and the domain structure of the receptor that was eventually revealed also confirmed that the VDR was a true steroid receptor and a bona fide member of the steroid receptor gene family (12,78,79). Equally important, the structural cloning and sequence analysis of the human chromosomal gene (80,81) for the VDR that followed in short order led ultimately to the identification of a series of mutations within the gene itself that was responsible for the syndrome of hereditary 1,25(OH)2D3 resistant rickets (HVDRR) (82–86). This syndrome had been identified earlier by Bell and colleagues in 1978 (86) and its etiology suggested it to be due to defects in the VDR gene by several groups of investigators (87–90), an hypothesis that was confirmed during the intervening years (91). The discovery of mutations in the VDR gene, the first for any member of the nuclear receptor family, solidified the integral and essential role of the VDR as the sole mediator of the activities of the vitamin D hormone which was eventually confirmed and extended through recapitulation of the disease phenotype observed in the mouse following deletion of the VDR from this model organism’s genome.
B. General Features of VDR Action
1. Sites of DNA binding
Early studies suggested that 1,25(OH)2D3 activated gene expression programs in a wide variety of cells and identified numerous gene candidates for further investigation. Most prominent among these were tissues that expressed the vitamin D-dependent calcium binding proteins (calbindins) (92,93), and the osteocalcin (94,95), osteopontin (96) and CYP24A1 (97) proteins, although a number of others emerged during the following several decades as well. The cloning of many of the genes for these proteins and identification of their structural organization prompted exploration of the mechanisms through which 1,25(OH)2D3 and its receptor could promote their regulation (see Figure 3). These studies, first with human BGLP (osteocalcin) (98,99) and subsequently with Spp1 (osteopontin) (100), Cyp24a1 (101–104) and others (105), suggested that the VDR bound to a 15 bp vitamin D responsive DNA element (VDRE) comprised of two directly repeated consensus AGGTCA hexanucleotide half-sites separated by three bp that was generally located within a kilobase or so of the promoters for these genes (99). These features were similar, but not identical, to those for other nuclear receptors. 1,25(OH)2D3 also strongly suppressed the expression of numerous genes most notably those for PTH and CYP27B1 but also in more recent studies of the IL-17 gene. Repression of IL-17 transcription by 1,25(OH)2D3 has been reported to involve, in part, dissociation of histone acetylase activity, recruitment of deacetylase and VDR/RXR binding to NFAT sites (106) (see Figure 4). While some progress has been made, the mechanisms of suppression for these and other down-regulated genes, have yet to be fully understood but are almost certain to be highly diverse. The presence of unique “negative VDREs” has been suggested for some negatively regulated genes, although this mechanism has yet to be substantiated (107).
Figure 4.
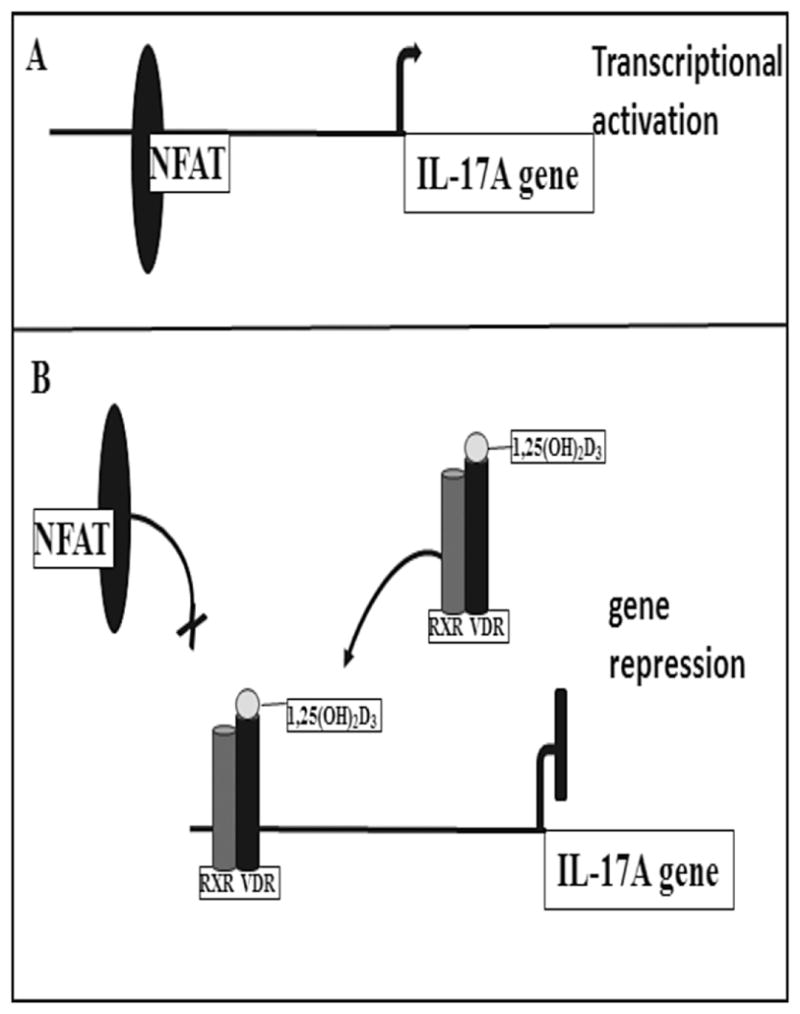
Mechanism of suppression of the IL-17 gene by 1,25(OH)2D3. Mechanism of repression of IL-17A activated transcription by 1,25(OH)2D3. The negative effect of 1,25(OH)2D3 on IL-17A involves blocking NFAT (an essential regulator of IL-17A gene transcription) from binding to its sites on the IL-17 gene and 1,25(OH)2D3 dependent association of RXR/VDR with the NFAT elements. 1,25(OH)2D3 repression of IL-17A also involves 1,25(OH)2D3 mediated recruitment of histone deacetylase and sequestration of Runx1 (also an important T cell receptor mediated transcriptional regulator of IL-17A) by 1,25(OH)2D3/VDR (not shown). From Joshi S, Pantalena LC, Liu XK, et al. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol. 2011;31(17):3653–3669, with permission.
2. Heterodimer formation with retinoid X receptors (RXR)
Accompanying the discovery of the first VDREs was the important finding that VDR binding to these specific DNA sequences was dependent upon an unknown nuclear factor (108–110). The identity of this protein was subsequently revealed when it was discovered that specific members of the steroid receptor family termed retinoid X receptors (RXR) were capable of forming heterodimeric complexes with the VDR and other members of this class of steroid receptors (111) (see Figure 3). Importantly, 1,25(OH)2D3 was found to promote heterodimer formation between VDR and RXR, although the cellular location of this interaction remains undefined currently (110,112). It has been suggested that in addition to its contribution to DNA binding, RXR may participate in the transcriptional activation process as well (113,114). Recent structural studies, however, suggest that this complex appears capable of recruiting only a single co-regulatory molecule (115).
3. The VDR functions to recruit co-regulatory complexes that mediate gene regulation
Early studies revealed that transcription factor binding near promoters leads to an interaction with basal transcriptional machinery that enhances gene output. It is now known, however, that transcriptional modulation by most DNA binding transactivators is far more complex and involves the receptor-mediated recruitment of different co-regulatory complexes, each with unique functions. Overcoming and/or restoring the inherent repressive state of chromatin requires the presence of regulatory machinery able to shift and/or displace nucleosomes (chromatin remodeling), alter the condensation state and therefore the local architecture of chromatin (histone modifications), create or restrict novel binding sites for additional co-regulatory complexes (epigenetic sites), and/or facilitate the entry of RNA polymerase II (RNA pol II) at appropriate times and sites (see Figure 3). Three complexes that participate in these activities that are known to be recruited by the VDR include 1) vertebrate ATPase-containing homologs of the yeast SWI/SNF complex that utilize the energy of ATP to remodel and reposition nucleosomes (116), 2) complexes that contain either histone acetyltransferases (HATs) or methyltransferases (HMTs), deacetyltransferases (HDACs) or histone demethylases (DMTs) which function to modify the lysine or arginine containing tails of histone 3 and/or histone 4 at specific locations (117,118), and 3) Mediator complex, believed to facilitate the entry of RNA pol II into the general transcriptional apparatus and perhaps to play a role in transcriptional re-initiation (119). Other regulatory complexes are also apparent. Several classes of regulatory complexes comprise components of dynamic and highly active mechanisms that are epigenetic in nature, and involve the coordinated expression of gene networks across the genome (120). These programs are widely responsible for development, differentiation and mature cell function (121,122). HATs and their reciprocal HDACs, for example, regulate the level of epigenetic histone H3 and H4 marks, controlling the degree to which chromatin is condensed and therefore the DNA accessible for transcription factor binding (123). The recruitment of these large chromatin regulatory complexes are frequently coordinated by factors such as the p160 family SRC-1, SRC-2 and SRC-3, the HATs CBP and p300, and the corepressors SMRT or NCoR as well as any one of the many HDACs that interact directly with transcription factors such as the VDR (117). Importantly, activation of the VDR with 1,25(OH)2D3 leads to the creation of a binding site on the VDR protein that mediates the link between the receptor and these co-regulatory complexes (124–126). Recent studies show that the ability of the VDR to recruit several of these co-regulatory factors results in striking changes in epigenetic histone marks that facilitate altered gene output (127–129). Thus, it is clear that like other DNA binding proteins, the function of the VDR in a dynamic way is simply to focus the recruitment of transcriptionally active complexes to gene subsets that are integral in a cell-specific manner to vitamin D hormone response. Considerable crystallographic information has now accrued to support not only the structural organization of the VDR/RXR heterodimer, but its association with DNA and its recruitment of coregulators (115). Many of these interactions are prompted by 1,25(OH)2D3, and perhaps differentially affected by analogues of the vitamin D hormone. These latter differential interactions form the basis for the concept of analogue selectivity in vivo, although this remains controversial for the vitamin D system.
C. Applying New Methodological Approaches to Study VDR Action
The recent coupling of chromatin immunoprecipitation initially to tiled microarray analysis (ChIP-chip) and subsequently to Next Generation DNA sequencing (ChIP-seq) analysis has revitalized the study of transcription both in vitro and in vivo, providing new methodologies capable of revealing unprecedented detail on a genome-wide scale (130–132). These techniques permit the site-specific crosslinking of structural chromatin proteins, regulatory transcription factors and nucleosomes to DNA, and their detection as well as the detection of functional modifications to these components via antibodies at nucleotide level resolution across the genomes of all organisms for which genomic sequence is available. This method and others that focus on direct DNA methylation, detection of presumed structural/functional DNA bound proteins complexes via DNase1 hypersensitivity analysis (DHS) and a multitude of others provide genome-wide annotation that was not previously attainable, have strikingly advanced both the field of factor-based gene regulation, the emerging field of epigenetics and evaluation of the impact of chromatin epigenetic states on gene regulation as well (133–137). Interestingly, while numerous transcriptional principles obtained through earlier studies have been confirmed, others have required extensive revision or radical alteration due to the highly biased and potentially misleading nature of many of the previously employed techniques. Perhaps more important, however, are the many new principles which have emerged as well, particularly when paired with transcriptomic measurements using RNA-seq analysis (138). A final methodology which deserves special mention is that which has enabled the selective editing of the genomes of both cells in culture and model organisms such as mice, rats, and even primates in vivo. This approach, particularly as it relates to the use of the RNA sequence-directed CRISPR/Cas9 method, now permits rapid and inexpensive deletion of not only genes, but of the regulatory elements of genes, thereby enabling loss-of-function assessment of their regulatory activities in cells (139–141). Due to their importance, we discuss the results of some of these collective studies which have provided important new insight into the transcriptional mechanism of action of 1,25(OH)2D3 as well as other hormones later in this review.
1. Overarching principles of VDR interaction at target cell genomes
As indicated earlier, studies using traditional methods coupled in later years to direct ChIP analysis pinpointed the regulatory regions of a number of vitamin D target genes near their promoters, including Bglap, Spp1, Cyp24a1 and others (142). In subsequent studies, we used ChIP-chip and then ChIP-seq analyses not only to confirm these findings but to obtain a broader overarching, genome-wide perspective on binding sites for the VDR in cells (see Box 1). Focusing on bone osteoblasts, we discovered using ChIP-chip analysis that we could detect approximately 1000 residual binding sites for the VDR in the absence of 1,25(OH)2D3, which was increased following hormonal treatment to approximately 7000–8000 (143). A collection of binding sites such as these have been termed a cellular cistrome. This observation supports the idea that VDR DNA binding is largely hormone-dependent (144). Importantly, a similar analysis of binding sites for RXR revealed a more extensive collection of sites that was only modestly increased through 1,25(OH)2D3 activation, emphasizing our understanding that RXR is a heterodimer partner for not only the VDR but several additional nuclear receptors as well (111). These findings have been confirmed in similar studies in MSCs, osteoblasts, osteocytes and adipocytes which also revealed that while overlap was present, binding sites for the VDR differ significantly across the genome depending upon the cell type examined (144–146). Subsequent studies of VDR binding sites in EB-immortalized human B cells, primary B cells and monocytes (147), and THP-1 monocytes (148) confirmed each of these findings. Importantly, although genome-wide ChIP-seq analyses have confirmed binding sites for the VDR near the promoters of genes such as Bglap, Spp1, Cyp24a1 and a few others (144), this technique has often been unable to confirm promoter-proximal VDR elements for many genes. These observations highlight the problems inherent to traditional biased approaches such as plasmid-based transfection methods and the potential for this approach to yield frequent false positives. Despite this, many of these genes have been shown to retain transcription factors such as C/EBPβ and others through traditional means that have been confirmed via unbiased methods. Thus, the presence of many of these factors at promoter-proximal regions cannot be discounted at present, but rather must simply be confirmed. In the case of binding sites, de novo motif finding analyses of the most common DNA sequence elements found in these diverse genome-wide collections of VDR binding sites has confirmed that a high percentage contain the originally postulated VDRE motif comprised of AGGTCA xxg AGGTCA (144,149). Thus, the consensus VDRE developed initially from that found in the human osteocalcin gene (98,99) is most representative of the sequence with which the VDR can interact at all genomes thus far examined.
Box 1. Overarching Principles of Vitamin D Action in Target Cells.
VDR Binding Sites (The Cistrome): 2000–8000 1,25(OH)2D3-sensitive binding sites/genome whose number and location are determined as a function of cell-type
Active Transcription Unit for Induction: The VDR/RXR heterodimer
Distal Binding Site Locations: Dispersed in cis-regulatory modules (CRMs or enhancers) across the genome; located in a cell-type specific manner near promoters, but predominantly within introns and distal intergenic regions; frequently located in clusters of elements
VDR/RXR Binding Site Sequence (VDRE): Induction mediated by classic hexameric half-sites (AGGTCA) separated by 3 base pairs; Repression mediated by divergent sites
Mode of DNA Binding: Predominantly, but not exclusively, 1,25(OH)2D3-dependent
Modular Features: CRMs contain binding sites for multiple transcription factors that facilitate either independent or synergistic interaction
Epigenetic CRM Signatures: Defined by the dynamically regulated post-translational histone H3 and H4 modifications
VDR Cistromes are highly dynamic: Cistromes change during cell differentiation, maturation, and disease activation and thus have consequential effects on gene expression
Interestingly, the unbiased nature of ChIP-seq analysis has also provided a number of surprising and perhaps unexpected insights of major significance (150) (see Box 1). Perhaps most important, although traditional studies of 1,25(OH)2D3 action identified regions immediately upstream of the transcriptional start sites (TSS) of genes, unbiased ChIP-seq analyses have revealed that regulatory regions for the vitamin D hormone and its receptor are more commonly located in clusters within introns or in intergenic regions 10’s if not 100’s of kilobases upstream or downstream of regulated genes (144,145,149). Examples of such distal elements for the VDR abound, but can be found in many of the genes whose putative promoter proximal elements were undetectable by ChIP-seq analysis. They include the mouse Tnfsf11 (RANKL) gene in bone cells where at least five intergenic regulatory regions for the VDR are located (62), the Cyp24a1 gene in numerous cell types where in addition to the well-known promoter proximal element discussed above, a complex downstream cluster of regulatory elements exists in both the mouse and the human genes (43), the Vdr gene in bone cells where both upstream regulatory regions and several intronic elements are present (127,151), the TRPV6 gene in intestinal cells which contains multiple upstream elements (56,152), the S100g gene in the intestine which also contains multiple upstream elements (56), and the many target genes such as c-FOS and c-MYC in human colorectal cancer cells as well (149). Indeed, enhancers for other transcription factors have been identified more than a megabase from the genes they are known to regulate, although at present the most distal VDR binding site is 335 kb upstream of the human c-MYC promoter. It is important to note, however, that the linear/distal nature of regulatory elements for genes is illusionary, as these distances do not take into account the looping of DNA that brings key regulatory segments into proximity of a gene’s promoter region (153–157). It is also worth a comment that of the tens of thousands of “putative” VDREs that naturally occur across the genome based purely on in silico analyses alone, only a very small proportion of these putative elements are functional due to chromatin restriction (133,135). On the other hand, a peak of activity defined by high quality ChIP-seq analysis is almost certain to represent a true transcription factor binding site and provides far more assurance than that developed via direct qPCR-ChIP analysis. An additional observation, as indicated above, is the finding through ChIP-seq analysis that most genes are regulated by more than one distal regulatory enhancer and, in some cases, by multiple enhancer regions. Recent ENCODE estimates suggest that genes are regulated by an average of 10 separate enhancers (135). Spp1 and Cyp24a1 represent classic examples, where additional elements located upstream of the former gene and downstream in the latter mouse and human genes (43,144) have been defined using these unbiased assays. Unfortunately, the presence of multiple enhancers located at distal sites complicates the studies of gene regulation enormously, as will be discussed below.
An additional regulatory feature of genes that has been identified is that of modularity. Thus, individual enhancers generally contain organized arrays of linear DNA sequences capable of assembling distinct, non-random transcription factor complexes that can function uniquely to regulate the gene with which they are linked. Numerous examples abound, but it is interesting that over 42% of VDR binding sites in bone cells are located in enhancers that contain prebound C/EBPβ and the master regulator RUNX2 (144,158). Indeed, these factors assemble in a highly organized fashion relative to each other, a nucleoprotein structure we have termed an Osteoblast Enhancer Complex. Not surprisingly, both RUNX2 and C/EBPβ in this configuration can positively and perhaps negatively influence the overall regulatory activity of 1,25(OH)2D3 and its receptor in very unique ways. An alternative arrangement has also been identified in bone cells as found in the Mmp13 gene where binding sites for the VDR, C/EBPβ and RUNX2 are dispersed across three separate upstream enhancers (159,160). The activities of these three regions are not independent, however, but rather can influence each other’s activity via looping in an overall hierarchical manner to modulate the expression of Mmp13. It is likely that many other genes contain this or similar arrangements as well. A collective summary of many of the newly acquired features of vitamin D-mediated gene regulation obtained via genome-wide analyses is provided in Box 1).
2. Genome-wide coregulatory recruitment to target genes via the VDR
The function of the VDR is to recruit chromatin-active coregulatory complexes that facilitate modulation of gene output as indicated earlier. Numerous studies at single gene levels support the capacity of the VDR to recruit these complexes, and recent ChIP-seq analyses support the presence of these complexes on a genome-wide scale as well. Thus, for example, the VDR was found to recruit coactivators such as SRC1, CBP, and MED1 as well as the corepressors NCoR and SMRT in colorectal LS180 cells (161). This recruitment correlated most profoundly with VDR binding sites that are linked to genes that are modulated directly by 1,25(OH)2D3. This correlation was not preferentially linked to either up or downregulated genes, as might be expected, suggesting that the roles of these coregulators are not limited specifically to activation or repression and that their activities are likely to be gene-context driven. On the other hand, recent studies in liver stellate cells suggested that 1,25(OH)2D3-mediated repression of a profibrotic gene expression program induced by TGFβ does not involve the apparent recruitment of corepressors SMRT and NCoR (162). In addition to SRC1, CBP and MED1, Brahma-related gene 1 (BRG1), an ATPase that is a component of the SWI/SNF chromatin remodeling complex, has been reported to play a fundamental role in 1,25(OH)2D3 induced transcription (163). C/EBP and BRG1 are components of the same complex and are recruited to the C/EBP site of the CYP24A1 gene by 1,25(OH)2D3. PRMT5, a type II protein arginine methyltransferase which interacts with BRG1, represses 1,25(OH)2D3 induced CYP24A1 transcription via its methylation of H3r8 and H4R3. Thus, the SWI/SNF complex can play a role in the silencing as well as in the activation of VDR mediated transcription.
3. Identifying underlying early mechanistic outcomes in response to VDR/RXR binding
The ability of the VDR to recruit epigenetically active coregulatory complexes such as histone acetyltransferases (HATs), histone deacetylatransferases (HDACs) and a variety of histone methyltransferases that regulate chromatin structure, as just discussed suggests that 1,25(OH)2D3 may influence the levels of distinct epigenetic marks imposed by these chromatin modifiers as a means of regulating gene output. Importantly, many such epigenetic marks on histones H3 and H4 are enriched in regions within gene loci that are uniquely active (131,132,164,165). Perhaps of most importance are changes in the levels of acetylation at H4K5 (H4K5ac), H3K9 (H3K9ac), and H3K27 (H3K27ac) that reflect alterations in the transcriptional activity of the genes with which they are linked; these modifications generally occur within enhancers that regulate these genes, although they can also occur at locations within genes as well. Regulatory regions that are marked both by genetic and epigenetic information at gene loci are frequently termed variable chromatin modulators (166). An increase in several of these acetylation marks occurs at specific sites of VDR binding in genes such as Spp1 and Cyp24a11, Lrp5, Tnfsf11, and the Vdr following 1,25(OH)2D3 stimulation (9,165,167) and can be used to define sites of action of 1,25(OH)2D3 even in the absence of evidence for VDR occupancy. These and other findings stimulated a recent assessment of the consequence of VDR binding following 1,25(OH)2D3 treatment on generalized H3 and H4 acetylation at a genome-wide scale in colorectal cells and at more specific histone sites in differentiating bone osteoblasts and osteocytes. All of these studies have revealed a striking increase in the level of H3 and H4 acetylation in response to 1,25(OH)2D3. Although 1,25(OH)2D3 can also provoke enrichment of enhancer methylation marks, these changes are generally gene-specific, suggesting that the VDR may retain gene selective functions as well. Overall, histone modification analyses suggest that 1,25(OH)2D3 promotes VDR/RXR binding at sites on cellular genomes that are marked by acetylated H3K9, H3K27 and H4K5, and that these interactions frequently result in an upregulation of acetylation that facilitates enhanced levels of gene expression. These studies provide a global perspective on the actions of vitamin D in several cell types, indicating that the primary role of the VDR is to facilitate the recruitment of chromatin modifiers such as acetyltransferases and deacetyltransferases that function to impose epigenetic histone changes within the enhancers of some but not all vitamin D sensitive target genes.
4. The dynamic impact of cellular differentiation and disease on VDR cistromes and transcriptional outcomes
Perhaps the most important observation made on a genome-wide scale has been the discovery that cellular differentiation exerts a dramatic quantitative and qualitative impact on genomic VDR binding, an effect that correlates directly with the hormone’s ability to regulate the differentiating cell’s transcriptome in a highly dynamic manner (144,146). This process is likely responsible for the cell type-specific nature of VDR binding and thus transcriptional outcomes at diverse sets of genes that can be measured in different tissues. A general change in the cellular RNA profile in response to 1,25(OH)2D3 is perhaps not surprising, given the fact that the overall effects of 1,25(OH)2D3 on osteoblast-lineage cells are known to differ significantly depending upon the state of bone cell differentiation. This concept of differentiation-induced changes in VDR binding and transcriptional integrity is aptly illustrated through a detailed examination of the differential expression of several genes including Mmp13 in osteoblast precursors and mature mineralizing osteoblasts (159).
Evans and colleagues have recently demonstrated that disease processes can affect VDR cistromes as well (162). The activation of hepatic stellate cells via the upregulation of TGFβ in the liver induces the expression of a collagen program that causes hepatic fibrosis and can induce cirrhosis of the liver. This disease progression can be ameliorated by simultaneous treatment in vivo with an analogue of vitamin D, and presumably by 1,25(OH)2D3 itself, although this was not tested. The authors show that the VDR cistrome which functions normally to suppress the program of collagen expression is altered as a result of TGFβ action, redirecting VDR binding to alternative sites of action away from collagen genes, thereby blunting opposing sites of vitamin D action. Interestingly, while these findings identify an important action of the VDR to prevent liver fibrosis, they also highlight the role of the VDR in the disease-potentiating activation of stellate cells, in a process that could be considered analogous to that of differentiation. Further studies of this system identify the role of the chromatin regulator BRD4 in this activity, and suggest that direct inhibition of this downstream factor by a small molecular regulator can bypass the positive effects of a vitamin D analogue (168).
D. Linking VDR Binding to the Expression of Specific Genes
1. The problem
ChIP-seq analyses can identify sites of occupancy for transcription factors and confirm that these sites retain epigenetic histone signatures that are consistent with the presence of enhancers. They cannot, however, identify the genes to which enhancers are functionally linked largely because the regulatory regions of genes are frequently located significant distances from gene promoters, and in many cases not contiguous with their target genes (157,169). These issues are of major concern for studies of mechanisms of gene regulation and of particular relevance to the myriad of GWAS studies that have been reported over the past decade. In these studies, single nucleotide variants can be correlated with a particular biologic phenotype or disease risk, and yet the majority of the genes whose activities are influenced by these SNPs and represent determinants of the phenotype remain to be identified. Much research is currently focused on bioinformatic approaches to establishing linkage between enhancers and the genes they modulate although this has not yet been accomplished with any certainty.
2. The solution
We and others have taken several approaches in attempts to link distal enhancers to the genes they regulate and to explore the mechanism through which they control the expression of the gene of interest. A preliminary approach, for example, is to explore the relationship between the putative target gene’s promoter and an identified enhancer using a proximity assay (chromatin conformation capture (3C) or more complex versions thereof) which can provide evidence that the distal element is in physical contact with the candidate gene’s promoter, perhaps in a hormone-dependent manner (156). Importantly, proximity assays have been extended technically to encompass multiple interactions using DNA sequencing and analysis on genome-wide scales as well (155,170,171). This approach with respect to gene-linkage is largely correlative, however, and must generally be confirmed via direct assessment. One approach is to create large minigenes that contain both the potential regulatory regions of genes in the vicinity identified by ChIP-seq analysis and the transcription unit of the putative target itself (43). A second and perhaps more robust strategy is to create individual enhancer deletions within the context of the genome itself, an approach that is most appropriately conducted in the mouse in vivo (172). A final approach that has emerged most recently involves direct editing of the genome in either cell lines or in the mouse genome using the RNA-directed CRISPR/Cas9 nuclease method to provoke precise genomic deletions, insertions, and/or mutations at specific gene locations (159,160). The former two approaches have been used to examine several genes of relevance to vitamin D biology including the Cyp24a1 and Vdr genes as well as the more complex Tnfsf11 gene and the latter has been used to explore the Mmp13 and several other genes. A brief summary of our studies of the Tnfsf11 and Mmp13 genes are considered in the following two sections.
3. Characterizing the regulatory elements of the Tnfsf11 (RANKL) gene
RANKL is a membrane-bound and sometimes soluble TNFα-like factor derived from the Tnfsf11 gene that strongly induces osteoclast differentiation from hematopoietic precursors (173,174). Indeed, the signal transduction pathways that mediate activation of this complex differentiation pathway are now well described (175). Importantly, the actions of RANKL over the intervening years have been dramatically extended to include immune regulation, mammary gland maturation, thermogenesis, and cardiovascular calcification, to name a few. Despite considerable effort, however, early attempts using traditional methods to identify regions mediating the regulation of the Tnfsf11 gene by 1,25(OH)2D3 as well as PTH and cytokines such as IL-6 and OSM in bone cells were largely unsuccessful (176,177), suggesting that the regulation of this gene might be mediated through more distal sites. As the mouse and human TNFSF11 genes are located in gene deserts and bounded on each side by nearly 200 kb of intergenic DNA, we explored this gene in osteoblasts for regulation by the vitamin D hormone. Importantly, we used ChIP-chip analysis of both VDR and RXR, and extended our query of mouse Tnfsf11 to over 500 kb of DNA surrounding the gene’s transcription unit (62). This initial study, now fully confirmed by ChIP-seq analysis, revealed that while neither VDR nor RXR were present near the Tnfsf11 promoter region following administration of 1,25(OH)2D3, both were strongly detected at five distal regions -16 (termed D1), -22 (D2), -60 (D3), -69 (D4) and -75/76 (D5) kb upstream of the gene’s promoter. Similar follow-on studies revealed that PTH-induced CREB binding at several of these regulatory regions and cytokine factors such as IL-6 induced transcription factor STAT3 binding at one of these enhancers (D5) as well as at a new enhancer at −88 kb. The binding of these and other factors identified across the Tnfsf11 gene is summarized schematically in Figure 5. These studies suggested that the regulation of Tnfsf11 expression by 1,25(OH)2D3, PTH and other hormonal factors is mediated by multiple independent enhancers located at significant distances from the gene’s TSS. Interestingly, additional unbiased analysis in T cells revealed a second set of three regulatory enhancers located intergenically even further upstream of the Tnfsf11 TSS between −123 and −155 kb (termed T1–T3) (178,179) (see Figure 5). These enhancers together with the enhancer at D5 mediate the expression of Tnfsf11 exclusively in hematopoietic B and T cells. Thus, a set of at least 10 independent enhancers have been identified to regulate Tnfsf11 expression in osteoblast- and hematopoietic-lineage cells when assessed in cultured cell lines (167). Interestingly, it is these regions that contain single nucleotide variants (SNPs) of genome-wide significance that appear to influence TNFSF11 expression and bone mineral density in human populations.
Figure 5.

The Tnfsf11 gene locus and its osteoblastic and hematopoietic regulatory regions. Arrows indicate the CTCF/RAD21-defined boundaries of the locus that includes the transcription unit and its upstream non-coding regulatory control regions. Enhancers that mediate osteoblast lineage regulation (D regions) and hematopoietic regulation (T regions) are numbered and indicated in orange ovals. Their distance from the Tnfsf11 TSS in kb is indicated below the oval. The D5 enhancer is active in both cell lineage types. Factors that have been shown to bind to each enhancer by ChIP-chip and/or ChIP seq analysis are indicated below the gene locus in orange blocks.
To confirm that these enhancers were responsible for the regulation of Tnfsf11, we and others individually deleted three of the key osteoblast specific enhancers D2, D5 and D6 as well as a single hematopoietic enhancer at −123 kb (T1) from the mouse genome (180,181). Mice bearing these deletions were then subjected to extensive regulatory and biological phenotyping. The results of these analyses indicate that 1) the D2 enhancer mediates PTH action in osteoblasts (180), 2) the D5 enhancer reduces basal expression of RANKL in both osteoblasts and hematopoietic cells and mediates both PTH and 1,25(OH)2D3 action in the former tissue (172,181), 3) the enhancer at D6 limits the regulation of RANKL by inflammatory cytokines, and 4) the enhancer at T1 reduces the basal expression of RANKL exclusively in hematopoietic cells (181). Biologically, the osteoblast-active enhancers exert profound effects on the skeleton whereas the hematopoietic cell-active enhancer T1 has no effect. Numerous additional phenotypic responses were identified as well. We conclude from these studies that the Tnfsf11 gene is regulated congruently both in vitro and in vivo by multiple distal enhancers that retain features that mediate the regulation of Tnfsf11 expression in unique temporal, hormonal and tissue-specific fashions.
4. Studies of the Mmp13 gene
Recent studies have examined the mechanisms through which Mmp13 expression is regulated in osteoblastic cells in culture by 1,25(OH)2D3 and other hormones (159). In studies of these cells, 3 regions located upstream of the Mmp13 gene and bearing epigenetic histone enhancer signatures H3K4me1 and H3K27ac were identified by ChIP-seq analysis. The most proximal of these enhancers located −10 kb upstream of the Mmp13 gene bound the VDR, mediating induction by 1,25(OH)2D3, whereas the other two −20 kb and −30 kb upstream bound the osteoblast master regulators C/EBPβ and RUNX2, respectively. We utilized CRISPR/Cas9 methods to create a series of homozygous daughter osteoblast cell lines derived from a parental UAMS source that contained deletions of the promoter proximal region, the −10 kb and −30 kb enhancers, and either VDR or RUNX2, and then examined these lines for both basal as well as 1,25(OH)2D3 inducible expression of Mmp13 transcripts. Loss of the −10 kb enhancer as well as loss of the VDR fully compromised the ability of 1,25(OH)2D3 to promote VDR binding and to induce Mmp13 expression. Deletion of the promoter-proximal region, the −30 kb enhancer and RUNX2 expression each dramatically reduced basal expression of Mmp13, but surprisingly also reduced the ability of 1,25(OH)2D3 to induce Mmp13 RNA. Further studies revealed that loss of the −30 kb enhancer or of RUNX2 binding activity affected the actions of the remaining enhancers in a hierarchical manner. These studies employing CRISPR/Cas9 methodology disclosed that Mmp13 enhancers strongly interact with each other as well as with the Mmp13 gene promoter, and that one enhancer mediates a hierarchical, master regulatory action on Mmp13 expression. Interestingly, PTH response was mediated through RUNX2 binding activity near the Mmp13 promoter (160), as suggested by Partridge and colleagues (182–184), although the enhancer at −30 kb also exerted dominance over this proximal element as well.
5. CRISPR/Cas9 mediated deletion of the 1,25(OH)2D3-inducible Mmp13 enhancer in vivo
To examine the role of the hormone inducible regulatory enhancer in the Mmp13 gene in vivo, we utilized the CRISPR/Cas9 approach to delete the −10 kb enhancer region in the Mmp13 locus in the mouse, obtaining homozygous mice through subsequent cross-breeding (160). Bone marrow cells from the −10 kb enhancer-deleted mice were then isolated, cultured in osteogenic medium and then examined for response to 1,25(OH)2D3 as compared to control mice. While wildtype cells responded strongly to 1,25(OH)2D3, those derived from the enhancer-deleted mice were resistant. These results confirm the regulatory role of this specific Mmp13 enhancer in vivo and highlight the utility of the CRISPR/Cas9 approach in defining enhancer activities in vivo.
V. Summary
In this review, we have discussed the metabolic activation of vitamin D to its hormonal form, the mechanism of its action to regulate genes through a specific nuclear receptor, its central homeostatic function to regulate mineral metabolism through genes that include S100G, ATP2B1 and TRPV6, and the regulation of additional genes in the skeleton that include TNFSF11 (RANKL) and others. We also discuss the actions of vitamin D to regulate genes such as IL-17 in the immune system. New methodologies have emerged in the past few years that now enable detailed studies of these transcriptional mechanisms in unprecedented detail both in cells in culture and in animal models in vivo. These approaches are leading to profound new insights into the mechanisms through which the vitamin D hormone operates to control not only mineral metabolism but to modulate unique cell-specific biology in numerous extra-skeletal systems as well. These molecular details have the potential to illuminate novel mechanisms that may be sensitive to newly designed vitamin D therapeutics.
Key Points.
The vitamin D hormone functions to regulate calcium and phosphorus metabolism in higher vertebrates and to control a multitude of additional biological activities linked to the immune and cardiovascular systems, skin and muscle function, cellular growth control and numerous additional biological processes as well.
Virtually all the biological activities of the vitamin D hormone are mediated by the vitamin D receptor, a nuclear receptor protein that functions to control the expression of genes and gene networks in a cell-type selective manner.
Recent unbiased genome-wide approaches have revealed a series of additional principles through which the VDR acts that have provided new insight into vitamin D hormone action.
Mechanistic studies of specific genes including vitamin D targets such as Cyp24a1, Spp1, IL-17, Tnfsf11 and Mmp13 have yielded novel genetic and epigenetic templates for understanding the additional complexity associated with vitamin D action.
Acknowledgments
The work of the authors cited in this article was supported by numerous grants from the NIH to JWP and SC. The authors wish to thank past and current members of their laboratories for their contributions to the individual work described here and the numerous senior collaborators that have also been involved in specific aspects of this research. We also acknowledge Laura Vanderploeg and Puneet Dhawan for artistic contributions to this review as well.
Footnotes
Disclosure: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Melanby E. An experimental investigation on rickets. Lancet. 1919;1:407–412. [Google Scholar]
- 2.McCollum E, Simmonds N, Becker J, Shipley P. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J Biol Chem. 1922;1922:293–298. [PubMed] [Google Scholar]
- 3.HSHB Fat soluble vitamins. XVII. The induction of growth promoting and calcifying properties in a ration by exposure to ultraviolet light. J Biol Chem. 1924;61:405–422. [Google Scholar]
- 4.Windaus A, Schenck F, von Werden F. Uber das antirachitisch wirksame bestrahlungs-produkt aus 7-dehydrocholesterin. Hoppe-Seyler’s Z Physiol Chem. 1936;241:100–103. [Google Scholar]
- 5.DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(6 Suppl):1689S–1696S. doi: 10.1093/ajcn/80.6.1689S. [DOI] [PubMed] [Google Scholar]
- 6.Brumbaugh P, Haussler M. 1 Alpha,25-dihydroxycholecalciferol receptors in intestine. I. Association of 1 alpha,25-dihydroxycholecalciferol with intestinal mucosa chromatin. J Biol Chem. 1974;249(4):1251–1257. [PubMed] [Google Scholar]
- 7.Brumbaugh PF, Haussler MR. 1a,25-dihydroxycholecalciferol receptors in intestine. II. Temperature-dependent transfer of the hormone to chromatin via a specific cytosol receptor. J Biol Chem. 1974;249(4):1258–1262. [PubMed] [Google Scholar]
- 8.Pike JW, Haussler MR. Purification of chicken intestinal receptor for 1,25-dihydroxyvitamin D. Proc Natl Acad Sci U S A. 1979;76(11):5485–5489. doi: 10.1073/pnas.76.11.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pike JW, Meyer MB, Benkusky NA, Lee SM, St John H, Carlson A, Onal M, Shamsuzzaman S. Genomic Determinants of Vitamin D-Regulated Gene Expression. Vitam Horm. 2016;100:21–44. doi: 10.1016/bs.vh.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonnell DP, Mangelsdorf DJ, Pike JW, Haussler MR, O’Malley BW. Molecular cloning of complementary DNA encoding the avian receptor for vitamin D. Science. 1987;235(4793):1214–1217. doi: 10.1126/science.3029866. [DOI] [PubMed] [Google Scholar]
- 11.Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, Shine J, O’Malley BW. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85(10):3294–3298. doi: 10.1073/pnas.85.10.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240(4854):889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plum LA, DeLuca HF. Vitamin D, disease and therapeutic opportunities. Nat Rev Drug Discov. 2010;9(12):941–955. doi: 10.1038/nrd3318. [DOI] [PubMed] [Google Scholar]
- 14.Feldman D, Krishnan AV, Swami S, Giovannucci E, Feldman BJ. The role of vitamin D in reducing cancer risk and progression. Nat Rev Cancer. 2014;14(5):342–357. doi: 10.1038/nrc3691. [DOI] [PubMed] [Google Scholar]
- 15.DeLuca HF. The vitamin D story: a collaborative effort of basic science and clinical medicine. Faseb J. 1988;2(3):224–236. [PubMed] [Google Scholar]
- 16.Cheng JB, Motola DL, Mangelsdorf DJ, Russell DW. De-orphanization of cytochrome P450 2R1: a microsomal vitamin D 25-hydroxilase. J Biol Chem. 2003;278(39):38084–38093. doi: 10.1074/jbc.M307028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng JB, Levine MA, Bell NH, Mangelsdorf DJ, Russell DW. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101(20):7711–7715. doi: 10.1073/pnas.0402490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al Mutair AN, Nasrat GH, Russell DW. Mutation of the CYP2R1 vitamin D 25-hydroxylase in a Saudi Arabian family with severe vitamin D deficiency. J Clin Endocrinol Metab. 2012;97(10):E2022–2025. doi: 10.1210/jc.2012-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu JG, Ochalek JT, Kaufmann M, Jones G, Deluca HF. CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A. 2013;110(39):15650–15655. doi: 10.1073/pnas.1315006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holick MF, Schnoes HK, DeLuca HF. Identification of 1,25-dihydroxycholecalciferol, a form of vitamin D3 metabolically active in the intestine. Proc Natl Acad Sci U S A. 1971;68(4):803–804. doi: 10.1073/pnas.68.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu GK, Lin D, Zhang MY, Bikle DD, Shackleton CH, Miller WL, Portale AA. Cloning of human 25-hydroxyvitamin D-1 alpha-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961–1970. doi: 10.1210/mend.11.13.0035. [DOI] [PubMed] [Google Scholar]
- 22.Fraser D, Kooh SW, Kind HP, Holick MF, Tanaka Y, DeLuca HF. Pathogenesis of hereditary vitamin-D-dependent rickets. An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1 alpha,25-dihydroxyvitamin D. N Engl J Med. 1973;289(16):817–822. doi: 10.1056/NEJM197310182891601. [DOI] [PubMed] [Google Scholar]
- 23.Dardenne O, Prud’homme J, Arabian A, Glorieux F, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142(7):3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 24.Garabedian M, Holick MF, Deluca HF, Boyle IT. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc Natl Acad Sci U S A. 1972;69(7):1673–1676. doi: 10.1073/pnas.69.7.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quarles LD. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012;8(5):276–286. doi: 10.1038/nrendo.2011.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–533. doi: 10.1146/annurev-physiol-030212-183727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones G, Prosser DE, Kaufmann M. Cytochrome P450-mediated metabolism of vitamin D. J Lipid Res. 2014;55(1):13–31. doi: 10.1194/jlr.R031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 29.Clinkenbeard EL, White KE. Systemic Control of Bone Homeostasis by FGF23 Signaling. Curr Mol Biol Rep. 2016;2(1):62–71. doi: 10.1007/s40610-016-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ajibade D, Dhawan P, Fechner A, Meyer M, Pike J, Christakos S. Evidence for a role of prolactin in calcium homeostasis: regulation of intestinal transient receptor potential vanilloid type 6, intestinal calcium absorption, and the 25-hydroxyvitamin D(3) 1alpha hydroxylase gene by prolactin. Endocrinology. 2010;151(7):2974–2984. doi: 10.1210/en.2010-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewison M, Burke F, Evans KN, Lammas DA, Sansom DM, Liu P, Modlin RL, Adams JS. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Steroid Biochem Mol Biol. 2007;103(3–5):316–321. doi: 10.1016/j.jsbmb.2006.12.078. [DOI] [PubMed] [Google Scholar]
- 32.Bikle DD, Pillai S. Vitamin D, calcium, and epidermal differentiation. Endocr Rev. 1993;14(1):3–19. doi: 10.1210/edrv-14-1-3. [DOI] [PubMed] [Google Scholar]
- 33.Bikle DD. Vitamin D and the skin. J Bone Miner Metab. 2010;28(2):117–130. doi: 10.1007/s00774-009-0153-8. [DOI] [PubMed] [Google Scholar]
- 34.Omdahl JL, Morris HA, May BK. Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr. 2002;22:139–166. doi: 10.1146/annurev.nutr.22.120501.150216. [DOI] [PubMed] [Google Scholar]
- 35.Veldurthy V, Wei R, Campbell M, Lupicki K, Dhawan P, Christakos S. 25-Hydroxyvitamin D2 24-Hydroxylase: A Key Regulator of 1,25(OH)2 D3 Catabolism and Calcium Homeostasis. Vitam Horm. 2016;100:137–150. doi: 10.1016/bs.vh.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol Rev. 2016;96(1):365–408. doi: 10.1152/physrev.00014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.St-Arnaud R. Targeted inactivation of vitamin D hydroxylases in mice. Bone. 1999;25(1):127–129. doi: 10.1016/s8756-3282(99)00118-0. [DOI] [PubMed] [Google Scholar]
- 38.St-Arnaud R, Glorieux FH. 24,25-Dihydroxyvitamin D--active metabolite or inactive catabolite? Endocrinology. 1998;139(8):3371–3374. doi: 10.1210/endo.139.8.6185. [DOI] [PubMed] [Google Scholar]
- 39.Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U, Misselwitz J, Klaus G, Kuwertz-Bröking E, Fehrenbach H, Wingen AM, Güran T, Hoenderop JG, Bindels RJ, Prosser DE, Jones G, Konrad M. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(5):410–421. doi: 10.1056/NEJMoa1103864. [DOI] [PubMed] [Google Scholar]
- 40.Tebben PJ, Milliner DS, Horst RL, Harris PC, Singh RJ, Wu Y, Foreman JW, Chelminski PR, Kumar R. Hypercalcemia, hypercalciuria, and elevated calcitriol concentrations with autosomal dominant transmission due to CYP24A1 mutations: effects of ketoconazole therapy. J Clin Endocrinol Metab. 2012;97(3):E423–427. doi: 10.1210/jc.2011-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Streeten EA, Zarbalian K, Damcott CM. CYP24A1 mutations in idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(18):1741–1742. doi: 10.1056/NEJMc1110226. author reply 1742–1743. [DOI] [PubMed] [Google Scholar]
- 42.Dinour D, Beckerman P, Ganon L, Tordjman K, Eisenstein Z, Holtzman EJ. Loss-of-function mutations of CYP24A1, the vitamin D 24-hydroxylase gene, cause longstanding hypercalciuric nephrolithiasis and nephrocalcinosis. J Urol. 2013;190(2):552–557. doi: 10.1016/j.juro.2013.02.3188. [DOI] [PubMed] [Google Scholar]
- 43.Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. J Biol Chem. 2010;285(20):15599–15610. doi: 10.1074/jbc.M110.119958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gheldof N, Smith E, Tabuchi T, Koch C, Dunham I, Stamatoyannopoulos J, Dekker J. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res. 2010;38(13):4325–4336. doi: 10.1093/nar/gkq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Omdahl JL. Interaction of the parathyroid and 1,25-dihydroxyvitamin D3 in the control of renal 25-hydroxyvitamin D3 metabolism. J Biol Chem. 1978;253(23):8474–8478. [PubMed] [Google Scholar]
- 46.Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. 2012;92(1):131–155. doi: 10.1152/physrev.00002.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLuca HF, Krisinger J, Darwish H. The vitamin D system: 1990. Kidney Int Suppl. 1990;29:S2–8. [PubMed] [Google Scholar]
- 48.Hoenderop JGJ, Nilius B, Bindels RJM. Calcium Absorption Across Epithelia. Physiol Rev. 2005;85(1):373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- 49.Amling M, Priemel M, Holzmann T, Chapin K, Rueger JM, Baron R, Demay MB. Rescue of the skeletal phenotype of vitamin D receptor-ablated mice in the setting of normal mineral ion homeostasis: formal histomorphometric and biomechanical analyses. Endocrinology. 1999;140(11):4982–4987. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 50.Masuyama R, Nakaya Y, Katsumata S, Kajita Y, Uehara M, Tanaka S, Sakai A, Kato S, Nakamura T, Suzuki K. Dietary calcium and phosphorus ratio regulates bone mineralization and turnover in vitamin D receptor knockout mice by affecting intestinal calcium and phosphorus absorption. J Bone Miner Res. 2003;18(7):1217–1226. doi: 10.1359/jbmr.2003.18.7.1217. [DOI] [PubMed] [Google Scholar]
- 51.Hochberg Z, Tiosano D, Even L. Calcium therapy for calcitriol-resistant rickets. J Pediatr. 1992;121(5 Pt 1):803–808. doi: 10.1016/s0022-3476(05)81919-5. [DOI] [PubMed] [Google Scholar]
- 52.Benn BS, Ajibade D, Porta A, Dhawan P, Hediger M, Peng JB, Jiang Y, Oh GT, Jeung EB, Lieben L, Bouillon R, Carmeliet G, Christakos S. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology. 2008;149(6):3196–3205. doi: 10.1210/en.2007-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kutuzova GD, Akhter S, Christakos S, Vanhooke J, Kimmel-Jehan C, Deluca HF. Calbindin D(9k) knockout mice are indistinguishable from wild-type mice in phenotype and serum calcium level. Proc Natl Acad Sci U S A. 2006;103(33):12377–12381. doi: 10.1073/pnas.0605252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kutuzova GD, Sundersingh F, Vaughan J, Tadi BP, Ansay SE, Christakos S, Deluca HF. TRPV6 is not required for 1alpha,25-dihydroxyvitamin D3-induced intestinal calcium absorption in vivo. Proc Natl Acad Sci U S A. 2008;105(50):19655–19659. doi: 10.1073/pnas.0810761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui M, Li Q, Johnson R, Fleet JC. Villin promoter-mediated transgenic expression of transient receptor potential cation channel, subfamily V, member 6 (TRPV6) increases intestinal calcium absorption in wild-type and vitamin D receptor knockout mice. J Bone Miner Res. 2012;27(10):2097–2107. doi: 10.1002/jbmr.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee SM, Riley EM, Meyer MB, Benkusky NA, Plum LA, DeLuca HF, Pike JW. 1,25-Dihydroxyvitamin D3 Controls a Cohort of Vitamin D Receptor Target Genes in the Proximal Intestine That Is Enriched for Calcium-regulating Components. J Biol Chem. 2015;290(29):18199–18215. doi: 10.1074/jbc.M115.665794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wasserman RH. Vitamin D and the dual processes of intestinal calcium absorption. J Nutr. 2004;134(11):3137–3139. doi: 10.1093/jn/134.11.3137. [DOI] [PubMed] [Google Scholar]
- 58.Reyes-Fernandez PC, Fleet JC. Compensatory Changes in Calcium Metabolism Accompany the Loss of Vitamin D Receptor (VDR) From the Distal Intestine and Kidney of Mice. J Bone Miner Res. 2016;31(1):143–151. doi: 10.1002/jbmr.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christakos S, Seth T, Hirsch J, Porta A, Moulas A, Dhawan P. Vitamin D biology revealed through the study of knockout and transgenic mouse models. Annu Rev Nutr. 2013;33:71–85. doi: 10.1146/annurev-nutr-071812-161249. [DOI] [PubMed] [Google Scholar]
- 60.Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, Kanno T, Murakami A, Tsuda E, Morinaga T, Higashio K. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139(3):1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 61.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim S, Yamazaki M, Zella L, Shevde N, Pike J. Activation of receptor activator of NF-kappaB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol. 2006;26(17):6469–6486. doi: 10.1128/MCB.00353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, Lafage-Proust MH, Dresselaers T, Feng JQ, Bonewald LF, Meyer MB, Pike JW, Bouillon R, Carmeliet G. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest. 2012;122(5):1803–1815. doi: 10.1172/JCI45890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonewald LF. Osteocytes as dynamic multifunctional cells. Ann N Y Acad Sci. 2007;1116:281–290. doi: 10.1196/annals.1402.018. [DOI] [PubMed] [Google Scholar]
- 65.Bonewald LF, Johnson ML. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42(4):606–615. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jähn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27(5):1018–1029. doi: 10.1002/jbmr.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murali SK, Andrukhova O, Clinkenbeard EL, White KE, Erben RG. Excessive Osteocytic Fgf23 Secretion Contributes to Pyrophosphate Accumulation and Mineralization Defect in Hyp Mice. PLoS Biol. 2016;14(4):e1002427. doi: 10.1371/journal.pbio.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98(11):6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36(6):971–977. doi: 10.1016/j.bone.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 70.Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16(2):221–232. doi: 10.1016/j.cytogfr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 71.Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103–2109. doi: 10.1093/carcin/23.12.2103. [DOI] [PubMed] [Google Scholar]
- 72.Zinser G, Welsh J. Effect of Vitamin D3 receptor ablation on murine mammary gland development and tumorigenesis. J Steroid Biochem Mol Biol. 2004;89–90(1–5):433–436. doi: 10.1016/j.jsbmb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 73.Xiang W, Kong J, Chen S, Cao LP, Qiao G, Zheng W, Liu W, Li X, Gardner DG, Li YC. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288(1):E125–132. doi: 10.1152/ajpendo.00224.2004. [DOI] [PubMed] [Google Scholar]
- 74.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. 2003;17(12):2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 75.Cantorna MT. Mechanisms underlying the effect of vitamin D on the immune system. Proc Nutr Soc. 2010;69(3):286–289. doi: 10.1017/S0029665110001722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haussler MR, Myrtle JF, Norman AW. The association of a metabolite of vitamin D3 with intestinal mucosa chromatin in vivo. J Biol Chem. 1968;243(15):4055–4064. [PubMed] [Google Scholar]
- 77.Burmester JK, Maeda N, DeLuca HF. Isolation and expression of rat 1,25-dihydroxyvitamin D3 receptor cDNA. Proc Natl Acad Sci U S A. 1988;85(4):1005–1009. doi: 10.1073/pnas.85.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin CH, Kerner SA, Hong MH, Pike JW. Transcriptional activation and dimerization functions in the human vitamin D receptor. Mol Endocrinol. 1996;10(8):945–957. doi: 10.1210/mend.10.8.8843411. [DOI] [PubMed] [Google Scholar]
- 79.McDonnell D, Scott R, Kerner S, O’Malley B, Pike J. Functional domains of the human vitamin D3 receptor regulate osteocalcin gene expression. Mol Endocrinol. 1989;3(4):635–644. doi: 10.1210/mend-3-4-635. [DOI] [PubMed] [Google Scholar]
- 80.Hughes MR, Malloy PJ, Kieback DG, Kesterson RA, Pike JW, Feldman D, O’Malley BW. Point mutations in the human vitamin D receptor gene associated with hypocalcemic rickets. Science. 1988;242(4886):1702–1705. doi: 10.1126/science.2849209. [DOI] [PubMed] [Google Scholar]
- 81.Miyamoto K, Kesterson R, Yamamoto H, Taketani Y, Nishiwaki E, Tatsumi S, Inoue Y, Morita K, Takeda E, Pike J. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol Endocrinol. 1997;11(8):1165–1179. doi: 10.1210/mend.11.8.9951. [DOI] [PubMed] [Google Scholar]
- 82.Hughes M, Malloy P, O’Malley B, Pike J, Feldman D. Genetic defects of the 1,25-dihydroxyvitamin D3 receptor. J Recept Res. 1991;11(1–4):699–716. doi: 10.3109/10799899109066437. [DOI] [PubMed] [Google Scholar]
- 83.Malloy P, Hochberg Z, Tiosano D, Pike J, Hughes M, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest. 1990;86(6):2071–2079. doi: 10.1172/JCI114944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ritchie H, Hughes M, Thompson E, Malloy P, Hochberg Z, Feldman D, Pike J, O’Malley B. An ochre mutation in the vitamin D receptor gene causes hereditary 1,25-dihydroxyvitamin D3-resistant rickets in three families. Proc Natl Acad Sci U S A. 1989;86(24):9783–9787. doi: 10.1073/pnas.86.24.9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sone T, Scott R, Hughes M, Malloy P, Feldman D, O’Malley B, Pike J. Mutant vitamin D receptors which confer hereditary resistance to 1,25-dihydroxyvitamin D3 in humans are transcriptionally inactive in vitro. J Biol Chem. 1989;264(34):20230–20234. [PubMed] [Google Scholar]
- 86.Brooks MH, Bell NH, Love L, Stern PH, Orfei E, Queener SF, Hamstra AJ, DeLuca HF. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298(18):996–999. doi: 10.1056/NEJM197805042981804. [DOI] [PubMed] [Google Scholar]
- 87.Eil C, Liberman UA, Rosen JF, Marx SJ. A cellular defect in hereditary vitamin-D-dependent rickets type II: defective nuclear uptake of 1,25-dihydroxyvitamin D in cultured skin fibroblasts. N Engl J Med. 1981;304(26):1588–1591. doi: 10.1056/NEJM198106253042608. [DOI] [PubMed] [Google Scholar]
- 88.Pike J, Dokoh S, Haussler M, Liberman U, Marx S, Eil C. Vitamin D3--resistant fibroblasts have immunoassayable 1,25-dihydroxyvitamin D3 receptors. Science. 1984;224(4651):879–881. doi: 10.1126/science.6326262. [DOI] [PubMed] [Google Scholar]
- 89.Sone T, Marx S, Liberman U, Pike J. A unique point mutation in the human vitamin D receptor chromosomal gene confers hereditary resistance to 1,25-dihydroxyvitamin D3. Mol Endocrinol. 1990;4(4):623–631. doi: 10.1210/mend-4-4-623. [DOI] [PubMed] [Google Scholar]
- 90.Lin N, Malloy P, Sakati N, al-Ashwal A, Feldman D. A novel mutation in the deoxyribonucleic acid-binding domain of the vitamin D receptor causes hereditary 1,25-dihydroxyvitamin D-resistant rickets. J Clin Endocrinol Metab. 1996;81(7):2564–2569. doi: 10.1210/jcem.81.7.8675579. [DOI] [PubMed] [Google Scholar]
- 91.Malloy PJ, Tasic V, Taha D, Tütüncüler F, Ying GS, Yin LK, Wang J, Feldman D. Vitamin D receptor mutations in patients with hereditary 1,25-dihydroxyvitamin D-resistant rickets. Mol Genet Metab. 2014;111(1):33–40. doi: 10.1016/j.ymgme.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Christakos S, Gabrielides C, Rhoten WB. Vitamin D-dependent calcium binding proteins: chemistry, distribution, functional considerations, and molecular biology. Endocr Rev. 1989;10(1):3–26. doi: 10.1210/edrv-10-1-3. [DOI] [PubMed] [Google Scholar]
- 93.Gill RK, Christakos S. Identification of sequence elements in mouse calbindin-D28k gene that confer 1,25-dihydroxyvitamin D3- and butyrate-inducible responses. Proc Natl Acad Sci U S A. 1993;90(7):2984–2988. doi: 10.1073/pnas.90.7.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Price PA, Baukol SA. 1,25-Dihydroxyvitamin D3 increases synthesis of the vitamin K-dependent bone protein by osteosarcoma cells. J Biol Chem. 1980;255(24):11660–11663. [PubMed] [Google Scholar]
- 95.Lian JB, Stein GS, Stewart C, Puchacz E, Mackowiak S, Aronow M, Von Deck M, Shalhoub V. Osteocalcin: characterization and regulated expression of the rat gene. Connect Tissue Res. 1989;21(1–4):61–68. doi: 10.3109/03008208909049996. discussion 69. [DOI] [PubMed] [Google Scholar]
- 96.Prince CW, Butler WT. 1,25-Dihydroxyvitamin D3 regulates the biosynthesis of osteopontin, a bone-derived cell attachment protein, in clonal osteoblast-like osteosarcoma cells. Coll Relat Res. 1987;7(4):305–313. doi: 10.1016/s0174-173x(87)80036-5. [DOI] [PubMed] [Google Scholar]
- 97.Haussler MR, Chandler JS, Pike JW, Brumbaugh PF, Speer DP, Pitt MJ. Physiological importance of vitamin D metabolism. Prog Biochem Pharmacol. 1980;17:134–142. [PubMed] [Google Scholar]
- 98.Kerner SA, Scott RA, Pike JW. Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc Natl Acad Sci U S A. 1989;86(12):4455–4459. doi: 10.1073/pnas.86.12.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ozono K, Liao J, Kerner SA, Scott RA, Pike JW. The vitamin D-responsive element in the human osteocalcin gene. Association with a nuclear proto-oncogene enhancer. J Biol Chem. 1990;265(35):21881–21888. [PubMed] [Google Scholar]
- 100.Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25-dihydroxyvitamin D3 receptor and 1,25-dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (SPP-1 or osteopontin) gene expression. Proc Natl Acad Sci U S A. 1990;87(24):9995–9999. doi: 10.1073/pnas.87.24.9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ohyama Y, Ozono K, Uchida M, Shinki T, Kato S, Suda T, Yamamoto O, Noshiro M, Kato Y. Identification of a vitamin D-responsive element in the 5′-flanking region of the rat 25-hydroxyvitamin D3 24-hydroxylase gene. J Biol Chem. 1994;269(14):10545–10550. [PubMed] [Google Scholar]
- 102.Ohyama Y, Ozono K, Uchida M, Yoshimura M, Shinki T, Suda T, Yamamoto O. Functional Assessment of Two Vitamin D-responsive Elements in the Rat 25-Hydroxyvitamin D3 24-Hydroxylase Gene. J Biol Chem. 1996;271(48):30381–30385. doi: 10.1074/jbc.271.48.30381. [DOI] [PubMed] [Google Scholar]
- 103.Zierold C, Darwish HM, DeLuca HF. Identification of a vitamin D-response element in the rat calcidiol (25-hydroxyvitamin D3) 24-hydroxylase gene. Proc Natl Acad Sci U S A. 1994;91(3):900–902. doi: 10.1073/pnas.91.3.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zierold C, Darwish H, DeLuca H. Two vitamin D response elements function in the rat 1,25-dihydroxyvitamin D 24-hydroxylase promoter. J Biol Chem. 1995;270(4):1675–1678. doi: 10.1074/jbc.270.4.1675. [DOI] [PubMed] [Google Scholar]
- 105.Carlberg C. Molecular basis of the selective activity of vitamin D analogues. J Cell Biochem. 2003;88(2):274–281. doi: 10.1002/jcb.10337. [DOI] [PubMed] [Google Scholar]
- 106.Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, Ichiyama K, Yoshimura A, Steinman L, Christakos S, Youssef S. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol. 2011;31(17):3653–3669. doi: 10.1128/MCB.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM. Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1992;89(17):8097–8101. doi: 10.1073/pnas.89.17.8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liao J, Ozono K, Sone T, McDonnell D, Pike J. Vitamin D receptor interaction with specific DNA requires a nuclear protein and 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1990;87(24):9751–9755. doi: 10.1073/pnas.87.24.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sone T, Ozono K, Pike JW. A 55-kilodalton accessory factor facilitates vitamin D receptor DNA binding. Mol Endocrinol. 1991;5(11):1578–1586. doi: 10.1210/mend-5-11-1578. [DOI] [PubMed] [Google Scholar]
- 110.Sone T, Kerner S, Pike JW. Vitamin D receptor interaction with specific DNA. Association as a 1,25-dihydroxyvitamin D3-modulated heterodimer. J Biol Chem. 1991;266(34):23296–23305. [PubMed] [Google Scholar]
- 111.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 112.Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature. 1992;355(6359):446–449. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thompson PD, Remus LS, Hsieh JC, Jurutka PW, Whitfield GK, Galligan MA, Encinas Dominguez C, Haussler CA, Haussler MR. Distinct retinoid X receptor activation function-2 residues mediate transactivation in homodimeric and vitamin D receptor heterodimeric contexts. J Mol Endocrinol. 2001;27(2):211–227. doi: 10.1677/jme.0.0270211. [DOI] [PubMed] [Google Scholar]
- 114.Pathrose P, Barmina O, Chang CY, McDonnell DP, Shevde NK, Pike JW. Inhibition of 1,25-dihydroxyvitamin D3-dependent transcription by synthetic LXXLL peptide antagonists that target the activation domains of the vitamin D and retinoid X receptors. J Bone Miner Res. 2002;17(12):2196–2205. doi: 10.1359/jbmr.2002.17.12.2196. [DOI] [PubMed] [Google Scholar]
- 115.Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. EMBO J. 2012;31(2):291–300. doi: 10.1038/emboj.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carlson M, Laurent BC. The SNF/SWI family of global transcriptional activators. Curr Opin Cell Biol. 1994;6(3):396–402. doi: 10.1016/0955-0674(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 117.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25(1):45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 118.Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D(3) receptor: a network of coactivator interactions. Gene. 2000;246(1–2):9–21. doi: 10.1016/s0378-1119(00)00052-4. [DOI] [PubMed] [Google Scholar]
- 119.Lewis BA, Reinberg D. The mediator coactivator complex: functional and physical roles in transcriptional regulation. J Cell Sci. 2003;116(Pt 18):3667–3675. doi: 10.1242/jcs.00734. [DOI] [PubMed] [Google Scholar]
- 120.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 121.Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, Yang H, Wang T, Lee AY, Swanson SA, Zhang J, Zhu Y, Kim A, Nery JR, Urich MA, Kuan S, Yen CA, Klugman S, Yu P, Suknuntha K, Propson NE, Chen H, Edsall LE, Wagner U, Li Y, Ye Z, Kulkarni A, Xuan Z, Chung WY, Chi NC, Antosiewicz-Bourget JE, Slukvin I, Stewart R, Zhang MQ, Wang W, Thomson JA, Ecker JR, Ren B. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153(5):1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, Zhang X, Coyne M, Fostel JL, Holmes L, Meldrim J, Guttman M, Epstein C, Park H, Kohlbacher O, Rinn J, Gnirke A, Lander ES, Bernstein BE, Meissner A. Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell. 2013;153(5):1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463(7280):474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McInerney EM, Rose DW, Flynn SE, Westin S, Mullen TM, Krones A, Inostroza J, Torchia J, Nolte RT, Assa-Munt N, Milburn MV, Glass CK, Rosenfeld MG. Determinants of coactivator LXXLL motif specificity in nuclear receptor transcriptional activation. Genes Dev. 1998;12(21):3357–3368. doi: 10.1101/gad.12.21.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13(24):3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 1998;395(6698):199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- 127.Zella LA, Meyer MB, Nerenz RD, Lee SM, Martowicz ML, Pike JW. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol Endocrinol. 2010;24(1):128–147. doi: 10.1210/me.2009-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meyer MB, Zella LA, Nerenz RD, Pike JW. Characterizing early events associated with the activation of target genes by 1,25-dihydroxyvitamin D3 in mouse kidney and intestine in vivo. J Biol Chem. 2007;282(31):22344–22352. doi: 10.1074/jbc.M703475200. [DOI] [PubMed] [Google Scholar]
- 129.Martowicz ML, Meyer MB, Pike JW. The mouse RANKL gene locus is defined by a broad pattern of histone H4 acetylation and regulated through distinct distal enhancers. J Cell Biochem. 2011;112(8):2030–2045. doi: 10.1002/jcb.23123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, Hardison RC, Dunham I, Kellis M, Noble WS. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41(2):827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28(8):817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, Dunham I, Elnitski LL, Farnham PJ, Feingold EA, Gerstein M, Giddings MC, Gilbert DM, Gingeras TR, Green ED, Guigo R, Hubbard T, Kent J, Lieb JD, Myers RM, Pazin MJ, Ren B, Stamatoyannopoulos JA, Weng Z, White KP, Hardison RC. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111(17):6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stamatoyannopoulos JA. What does our genome encode? Genome Res. 2012;22(9):1602–1611. doi: 10.1101/gr.146506.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015;12(7):1184–1195. doi: 10.1016/j.celrep.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 138.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hille F, Charpentier E. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci. 2016;371(1707) doi: 10.1098/rstb.2015.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Singh A, Chakraborty D, Maiti S. CRISPR/Cas9: a historical and chemical biology perspective of targeted genome engineering. Chem Soc Rev. 2016 doi: 10.1039/c6cs00197a. [DOI] [PubMed]
- 142.Kim S, Shevde N, Pike J. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J Bone Miner Res. 2005;20(2):305–317. doi: 10.1359/JBMR.041112. [DOI] [PubMed] [Google Scholar]
- 143.Meyer MB, Goetsch PD, Pike JW. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J Steroid Biochem Mol Biol. 2010;121(1–2):136–141. doi: 10.1016/j.jsbmb.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Meyer MB, Benkusky NA, Lee CH, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Biol Chem. 2014;289(28):19539–19554. doi: 10.1074/jbc.M114.578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Meyer MB, Benkusky NA, Sen B, Rubin J, Pike JW. Epigenetic Plasticity Drives Adipogenic and Osteogenic Differentiation of Marrow-derived Mesenchymal Stem Cells. J Biol Chem. 2016;291(34):17829–17847. doi: 10.1074/jbc.M116.736538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.St John HC, Bishop KA, Meyer MB, Benkusky NA, Leng N, Kendziorski C, Bonewald LF, Pike JW. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014;28(7):1150–1165. doi: 10.1210/me.2014-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, Handunnetthi L, Handel AE, Disanto G, Orton SM, Watson CT, Morahan JM, Giovannoni G, Ponting CP, Ebers GC, Knight JC. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20(10):1352–1360. doi: 10.1101/gr.107920.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Heikkinen S, Väisänen S, Pehkonen P, Seuter S, Benes V, Carlberg C. Nuclear hormone 1α,25-dihydroxyvitamin D3 elicits a genome-wide shift in the locations of VDR chromatin occupancy. Nucleic Acids Res. 2011;39(21):9181–9193. doi: 10.1093/nar/gkr654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Meyer MB, Goetsch PD, Pike JW. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: impact on c-FOS and c-MYC gene expression. Mol Endocrinol. 2012;26(1):37–51. doi: 10.1210/me.2011-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12(4):283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee SM, Meyer MB, Benkusky NA, O’Brien CA, Pike JW. Mechanisms of Enhancer-mediated Hormonal Control of Vitamin D Receptor Gene Expression in Target Cells. J Biol Chem. 2015;290(51):30573–30586. doi: 10.1074/jbc.M115.693614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol. 2006;20(6):1447–1461. doi: 10.1210/me.2006-0031. [DOI] [PubMed] [Google Scholar]
- 153.Deng W, Blobel GA. Do chromatin loops provide epigenetic gene expression states? Curr Opin Genet Dev. 2010;20(5):548–554. doi: 10.1016/j.gde.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149(6):1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Deng W, Blobel GA. Manipulating nuclear architecture. Curr Opin Genet Dev. 2014;25:1–7. doi: 10.1016/j.gde.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Deng W, Blobel GA. Detecting Long-Range Enhancer-Promoter Interactions by Quantitative Chromosome Conformation Capture. Methods Mol Biol. 2017;1468:51–62. doi: 10.1007/978-1-4939-4035-6_6. [DOI] [PubMed] [Google Scholar]
- 157.Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet. 2016;48(5):488–496. doi: 10.1038/ng.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Meyer MB, Benkusky NA, Pike JW. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J Biol Chem. 2014;289(23):16016–16031. doi: 10.1074/jbc.M114.552216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Meyer MB, Benkusky NA, Pike JW. Selective Distal Enhancer Control of the Mmp13 Gene Identified through Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) Genomic Deletions. J Biol Chem. 2015;290(17):11093–11107. doi: 10.1074/jbc.M115.648394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Meyer MB, Benkusky NA, Onal M, Pike JW. Selective regulation of Mmp13 by 1,25(OH)2D3, PTH, and Osterix through distal enhancers. J Steroid Biochem Mol Biol. 2015 doi: 10.1016/j.jsbmb.2015.1009.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Meyer MB, Pike JW. Corepressors (NCoR and SMRT) as well as coactivators are recruited to positively regulated 1α,25-dihydroxyvitamin D3-responsive genes. J Steroid Biochem Mol Biol. 2013;136:120–124. doi: 10.1016/j.jsbmb.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, Lau SL, Atkins AR, Barish GD, Gunton JE, Liddle C, Downes M, Evans RM. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Seth-Vollenweider T, Joshi S, Dhawan P, Sif S, Christakos S. Novel mechanism of negative regulation of 1,25-dihydroxyvitamin D3-induced 25-hydroxyvitamin D3 24-hydroxylase (Cyp24a1) Transcription: epigenetic modification involving cross-talk between protein-arginine methyltransferase 5 and the SWI/SNF complex. J Biol Chem. 2014;289(49):33958–33970. doi: 10.1074/jbc.M114.583302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Meyer MB, Benkusky NA, Pike JW. Profiling histone modifications by chromatin immunoprecipitation coupled to deep sequencing in skeletal cells. Methods Mol Biol. 2015;1226:61–70. doi: 10.1007/978-1-4939-1619-1_6. [DOI] [PubMed] [Google Scholar]
- 165.Pike JW, Meyer MB, St John HC, Benkusky NA. Epigenetic histone modifications and master regulators as determinants of context dependent nuclear receptor activity in bone cells. Bone. 2015 doi: 10.1016/j.bone.2015.03.012. [DOI] [PMC free article] [PubMed]
- 166.Deplancke B, Alpern D, Gardeux V. The Genetics of Transcription Factor DNA Binding Variation. Cell. 2016;166(3):538–554. doi: 10.1016/j.cell.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 167.Pike JW, Lee SM, Meyer MB. Regulation of gene expression by 1,25-dihydroxyvitamin D3 in bone cells: exploiting new approaches and defining new mechanisms. Bonekey Rep. 2014;3:482. doi: 10.1038/bonekey.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M, He M, Liddle C, Downes M, Evans RM. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci U S A. 2015;112(51):15713–15718. doi: 10.1073/pnas.1522163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ong CT, Corces VG. Modulation of CTCF insulator function by transcription of a noncoding RNA. Dev Cell. 2008;15(4):489–490. doi: 10.1016/j.devcel.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Denker A, de Laat W. A Long-Distance Chromatin Affair. Cell. 2015;162(5):942–943. doi: 10.1016/j.cell.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 171.Denker A, de Laat W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev. 2016;30(12):1357–1382. doi: 10.1101/gad.281964.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Galli C, Zella LA, Fretz JA, Fu Q, Pike JW, Weinstein RS, Manolagas SC, O’Brien CA. Targeted deletion of a distant transcriptional enhancer of the receptor activator of nuclear factor-kappaB ligand gene reduces bone remodeling and increases bone mass. Endocrinology. 2008;149(1):146–153. doi: 10.1210/en.2007-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie M, Martin T. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20(3):345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 174.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 175.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 176.Kitazawa R, Kitazawa S. Vitamin D(3) augments osteoclastogenesis via vitamin D-responsive element of mouse RANKL gene promoter. Biochem Biophys Res Commun. 2002;290(2):650–655. doi: 10.1006/bbrc.2001.6251. [DOI] [PubMed] [Google Scholar]
- 177.Kitazawa S, Kajimoto K, Kondo T, Kitazawa R. Vitamin D3 supports osteoclastogenesis via functional vitamin D response element of human RANKL gene promoter. J Cell Biochem. 2003;89(4):771–777. doi: 10.1002/jcb.10567. [DOI] [PubMed] [Google Scholar]
- 178.Bishop KA, Coy HM, Nerenz RD, Meyer MB, Pike JW. Mouse Rankl expression is regulated in T cells by c-Fos through a cluster of distal regulatory enhancers designated the T cell control region. J Biol Chem. 2011;286(23):20880–20891. doi: 10.1074/jbc.M111.231548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bishop KA, Wang X, Coy HM, Meyer MB, Gumperz JE, Pike JW. Transcriptional regulation of the human TNFSF11 gene in T cells via a cell type-selective set of distal enhancers. J Cell Biochem. 2015;116(2):320–330. doi: 10.1002/jcb.24974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Onal M, St John HC, Danielson AL, Pike JW. Deletion of the Distal Tnfsf11 RL-D2 Enhancer That Contributes to PTH-Mediated RANKL Expression in Osteoblast Lineage Cells Results in a High Bone Mass Phenotype in Mice. J Bone Miner Res. 2016;31(2):416–429. doi: 10.1002/jbmr.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Onal M, St John HC, Danielson AL, Markert JW, Riley EM, Pike JW. Unique Distal Enhancers Linked to the Mouse Tnfsf11 Gene Direct Tissue-Specific and Inflammation-Induced Expression of RANKL. Endocrinology. 2016;157(2):482–496. doi: 10.1210/en.2015-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Selvamurugan N, Jefcoat SC, Kwok S, Kowalewski R, Tamasi JA, Partridge NC. Overexpression of Runx2 directed by the matrix metalloproteinase-13 promoter containing the AP-1 and Runx/RD/Cbfa sites alters bone remodeling in vivo. J Cell Biochem. 2006;99(2):545–557. doi: 10.1002/jcb.20878. [DOI] [PubMed] [Google Scholar]
- 183.Shimizu E, Selvamurugan N, Westendorf JJ, Partridge NC. Parathyroid hormone regulates histone deacetylases in osteoblasts. Ann N Y Acad Sci. 2007;1116:349–353. doi: 10.1196/annals.1402.037. [DOI] [PubMed] [Google Scholar]
- 184.Shimizu E, Nakatani T, He Z, Partridge NC. Parathyroid hormone regulates histone deacetylase (HDAC) 4 through protein kinase A-mediated phosphorylation and dephosphorylation in osteoblastic cells. J Biol Chem. 2014;289(31):21340–21350. doi: 10.1074/jbc.M114.550699. [DOI] [PMC free article] [PubMed] [Google Scholar]