

Abstract
Adipocytes differentiated from preadipocytes provide a valuable model for the study of human adipocyte metabolism. We describe methods for isolation of human stromal vascular cells, expansion of preadipocytes, differentiation into mature adipocytes, and in vitro metabolic interrogation of adipocytes.
Keywords: Adipocyte, Preadipocyte, Stromal vascular cell fraction, Adipose tissue, Differentiation, Lipogenesis, Lipolysis, Glucose uptake
1 Introduction
Obesity is a public health crisis. Adipose tissue metabolic dysfunction is a central feature of obesity and underlies the pathogenesis of metabolic disease. An understanding of adipocyte metabolism is therefore of critical importance to metabolic disease research. Murine 3T3L1 cells provide a valuable adipocyte model, but significant differences exist between murine and human adipose tissue and systemic metabolic disease phenotypes. Human adipocytes differentiated in vitro from preadipocytes retain depot- and patient- specific metabolic phenotypes in culture and provide a tractable model for studying human adipocyte cellular metabolism [1–5].
While contributing to the vast majority of total adipose tissue mass, mature adipocytes comprise only half of total adipose tissue cell number, the remainder of which consists of a diverse stromal vascular cell fraction (SVF). Forty to sixty percent of the SVF is CD45+ leukocytes including macrophages, T-cells, B-cells, and other immune cells. Of the remaining CD45− SVF population, over 80 % are CD34+CD31−, a phenotype that defines preadipocytes (a.k.a. adipocyte stem cells), a mesenchymal cell population that gives rise to mature adipocytes. Endothelial cells, fibroblasts, and other cell types comprise the remainder of the CD45−population; endothelial cells are typically described as CD45−CD34+CD31+, and comprise 1–5 % of the CD45− SVF population (Fig. 1). Other markers define specific subpopulations of human preadipocytes with variable adipogenic capacity. CD140a+ preadipocytes, for example, are a particularly adipogenic subset of the CD34+ CD31− population [6, 7]. As of this writing, the functional significance of specific human preadipocyte subpopulations is poorly defined and an active area of research.
Fig.1.
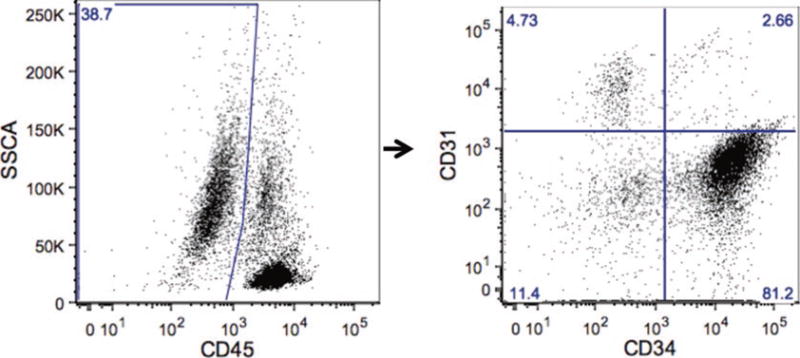
Flow cytometry of human SVF: Representative flow cytometry scatter plots of visceral human adipose tissue SVF demonstrating gating strategy for preadipocytes (CD45−CD31−CD34+); a large forward scatter-side scatter gate is used to encompass all viable cells (not shown), followed by gating on CD45− cells (left), followed by gating on CD31−CD34+ cells (right, right lower quadrant)
Preadipocytes may be isolated or enriched from SVF using a variety of methods depending on the intended downstream application. Flow cytometry sorting of CD45−CD34+CD31+ preadipocytes provides a high purity cell preparation for microarray and next-generation sequencing studies; antibody-coated magnetic bead sorting also provides a relatively high purity population. Enrichment of preadipocytes by plastic adherence of SVF provides a cell population of sufficient purity for many applications and is efficient and economical. Nonadherent CD45+ hematopoietic cells are removed after 1–2 days of adherence, and CD45− CD34+CD31− cells subsequently selectively proliferate in culture and are receptive to adipogenic differentiation.
Adipose tissue may be divided into white and brown phenotypes, although increasing data suggest functional overlap, with pluripotent stem cells giving rise to white, brown, and beige/brite (brown-in-white) adipocytes. While white and brown adipose tissues are for the most part anatomically distinct, stem cells with brown/ beige potential have been identified within white anatomic depots [8], reinforcing functional overlap between canonical adipose tissue depots. In humans, the existence of brown adipose tissue beyond the neonatal period was confirmed using positron emission tomography only in the last decade [9]. As a result, most published literature prior to this point studies white adipose tissue.
White adipose tissue in humans and mice resides in anatomically and functionally distinct depots that may be broadly categorized into visceral and subcutaneous adipose tissue compartments (VAT, SAT). Multiple sub-depots within each compartment have distinct phenotypes and functions. Adipose tissue depot anatomy differs significantly in mice and humans. The epididymal fat pad comprises the majority of VAT in mice, is often termed eWAT (epididymal white adipose tissue), and is the best-studied murine VAT depot, although mice also harbor mesenteric and retroperitoneal VAT. Murine SAT is generally derived from the inguinal and flank areas (iWAT, inguinal white adipose tissue). As in humans, murine VAT/eWAT, when compared to SAT/iWAT, is more strongly associated with systemic metabolic disease [10–12]. VAT in humans includes omental, mesenteric, and retroperitoneal depots. The greater omentum tissue consists of an apron of adipose tissue attached to the transverse colon and greater curvature of the stomach, and is the most easily accessible and best studied human VAT sub-depot (Fig. 2). Human SAT may be divided into truncal (abdominal wall) and extremity compartments, within which exist deep and superficial sub-depots.
Fig. 2.
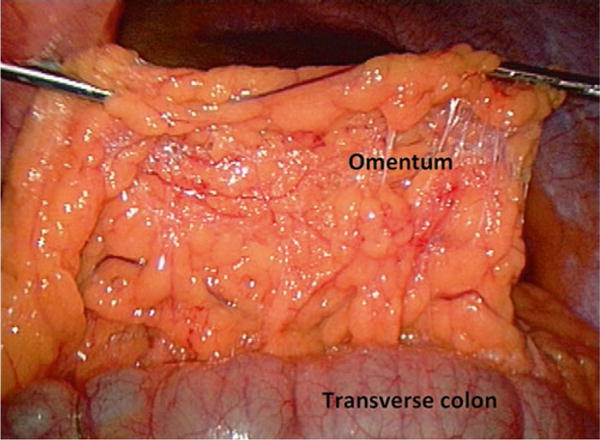
The human omentum: Intraoperative photograph of the human omentum reflected ventrally off the transverse colon; the view is from a caudad position looking cephalad, with ventral and dorsal positions at top and bottom of photo respectively; the stomach (not seen) is cephalad, behind the omentum in the photograph
Technical and anatomic considerations affect surgical adipose tissue biopsy in humans. Omental adipose tissue is easily accessible during most intra-abdominal operations and may be biopsied without significant risk or clinical sequelae using cautery or harmonic scalpel dissection. Mesenteric adipose tissue is intimately associated with the small intestine, and thus biopsy is usually performed only in the context of clinically indicated small bowel resection. Retroperitoneal VAT is intimately associated with the kidneys and other retroperitoneal structures; retroperitoneal adipose tissue biopsy is therefore only possible in the context of clinically indicated resection of the kidney or other retroperitoneal organs. SAT biopsy is usually performed at the site of the skin incision for most operations, the anatomic location of which varies depending on the specific operation performed. We obtain omental VAT and abdominal wall SAT from patients undergoing bariatric surgery and other abdominal operations via laparoscopy and laparotomy. Biopsy of these tissues via laparotomy is relatively straightforward for the surgeon. Omental VAT collection via laparoscopy is technically more challenging and time-consuming, and requires intraoperative extraction from the abdomen through a laparoscopic trocar site with a laparoscopic specimen retrieval bag, removing tissue from the bag intraoperatively through the trocar site using ringed forceps. We endeavor to collect VAT and SAT from the same anatomic sites in all patients whenever possible. VAT is collected from the apex/caudad end of the omentum. Since the majority of samples are collected from laparoscopic bariatric operations, we standardize SAT collection to the upper abdominal wall at the site of a specific trocar incision placed in every operation. We collect 20–80 g of VAT from the greater omentum, and 1–6 g of SAT from the skin incision. SAT samples larger than 6 g from a laparotomy incision, or larger than 4 g from a laparoscopy trocar site, are associated with higher rates of hematoma, seroma, and infection, and are contraindicated. Biopsy size, especially SAT, should be tailored to the amount of adipose tissue present, at the discretion of the operating surgeon. Each laboratory should work closely with surgeon investigators/collaborators to refine and standardize surgical technique.
Tissue is transported to the laboratory in a sterile plastic bag on ice and samples aliquoted and stored for various applications using a codified standard operating protocol. Here, we provide protocols for isolation of SVF from human adipose tissue, expansion of preadipocytes from SVF, and differentiation of preadipocytes into mature adipocytes, but whole tissue may be frozen, formalin-fixed, or cultured as live explants, and leukocyte and non-leukocyte cell subpopulations may be isolated from SVF using flow cytometry or antibody-coated magnetic bead sorting for multiple phenotypic and functional analyses. Our protocols have been optimized for omental VAT and abdominal wall SAT, but have also been successfully used for retroperitoneal and mesenteric tissues. Further optimization for different sub-depots may improve results and should be considered by individual laboratories.
2 Materials
2.1 Equipment
Laminar flow biosafety cabinet.
Temperature-controlled orbital shaker.
Temperature-controlled desktop centrifuge.
Tissue culture incubator, 37 °C, 5 % CO2.
Freezer, −80 °C.
Liquid nitrogen storage unit.
Microplate spectrophotometer.
Scintillation counter.
Sterile scissors, forceps.
Hemocytometer.
2.2 Disposables
Tissue culture materials, including 10 cm culture plates, T150 cell culture flasks, 24-well plates, 50 mL conical tubes.
100 μM nylon cell strainers.
Plastic weigh boats.
Internally threaded cryogenic vials.
1.5 mL microcentrifuge tubes.
Scintillation vials, scintillation fluid.
2.3 Isolation of SVF from Human Adipose Tissue
20× collagenase stock solution: Type II Collagenase (Gibco Inc.) 40 mg/mL in 1× PBS, 2 % BSA; prepare immediately prior to use.
RBC lysis buffer: 10× stock solution: 82.9 g ammonium chloride + 10.0 g potassium bicarbonate + 0.37 g EDTA, QS to 1 liter with sterile H2O; dilute to 1× with sterile H2O prior to use.
Trypan blue.
2.4 Expansion and Storage of Preadipocytes
Expansion medium: DMEM/F12 (50:50), 15 % FBS, 1 antibiotic-antimycotic.
Freezing medium: DMEM/F12 (50:50), 15 % FBS, 10 DMSO, 1 % antibiotic-antimycotic.
Trypsin solution: 0.25 % Trypsin-EDTA.
2.5 In Vitro Differentiation of Human Adipocytes
Differentiation medium: Serum-free DMEM/F12 (50:50), 2.5 mM glutamine, 15 mM HEPES, 1 % antibiotic-antimycotic, 10 mg/ml transferrin, 33 μM biotin, 0.5 μM human insulin, 17 μM pantothenate, 0.1 μM dexamethasone, 2 nM T3, 540 μM IBMX, 1 μM ciglitazone.
Maintenance medium: Serum-free DMEM/F12 (50:50), 2.5 mM glutamine, 15 mM HEPES, 1 % antibiotic-antimycotic, 10 mg/ml transferrin, 33 μM biotin, 0.5 μM human insulin.
2.6 Oil Red-O Staining
Oil Red-O working solution: Add 30 mL of Oil Red-O stock solution (Sigma Inc.) to 20 mL of H2O and filter solution through 15 cm Grade 201 Whatman-Reeve Angel filter paper.
Formalin, 4 %.
Isopropanol, 60 %.
2.7 Lipolysis Assay
Maintenance medium without insulin: Serum-free DMEM/ F12 (50:50), 2.5 mM glutamine, 15 mM HEPES, 1 % antibiotic-antimycotic, 10 mg/ml transferrin, 33 μM biotin.
Triglyceride Determination Kit (Sigma Inc.).
Isoproterenol hydrochloride.
2.8 Labeled Acetate Lipogenesis Assay
Serum Starvation Medium: DMEM:F12 50:50, 1 % antibiotic-antimycotic.
Lipogenesis Medium: Serum Starvation Medium + 100 nM insulin, 10 μM sodium acetate, 0.5uCi 3H-acetate.
Human insulin stock solution, 1 μM; dilute to 100 nM working solution in 1× PBS.
Sodium acetate 10 mM stock solution.
3H–acetate (Perkin Elmer Inc.).
HCl stock solution, 1 N; dilute to 0.1 N HCl with H2O on the day of procedure to obtain working solution.
Chloroform:methanol 2:1 v/v.
3 Methods
3.1 Isolation of SVF from Human Adipose Tissue
Transport human adipose tissue to laboratory in a sterile plastic bag on ice. All tissue processing is done in a laminar flow biosafety cabinet under sterile tissue culture conditions. Weigh tissue and then transfer to a sterile plastic tray. Remove and discard blood clots, vessels, and cauterized tissue with sterile scissors and forceps. Mince remaining tissue into 2–3 mm (~5 mg) pieces with scissors and forceps. Mincing should be performed rapidly, for not more than 15–20 min, to prevent decreased cell viability. Use multiple personnel to mince tissue in parallel for large samples, to keep mincing time to a minimum (see Note 1a).
Aliquot 2 mL (2 g) of minced adipose tissue into a 50 mL conical tube with a transfer pipette; use multiple tubes for larger samples. To each tube, add 17 mL of 1×PBS + 2 % BSA. At this point, if any remaining visible pieces of tissue >2 mm are present, then further mince tissue in buffer in the tube with scissors until tissue fragments are <2 mm.
Add 1 mL of 20× collagenase stock solution to the 19 mL tissue suspension to a total volume of 20 mL and a final working (1×) collagenase concentration of 2 mg/mL. Agitate samples on an orbital shaker for 30–60 min, 130 rpm, 37 °C, removing tubes every 5 min to agitate by hand more vigorously for 5–10 s. The resulting digestate should be cloudy, homogenous, and almost fully liquefied with some strands of gelatinous, fibrous tissue aggregates (see Notes 1b–d).
While tissue is digesting, pre-wet 100 μM nylon cell strainers with 1× PBS and place on top of fresh 50 mL conical tubes (one for each digestate tube). After digestion is complete, apply the tissue digestate from a single 50 mL tube onto a single strainer and allow it to gravity drip into the 50 mL tube; use 10 mL of 1×PBS to rinse the initial digestion 50 mL tube, removing any residual digestate, and apply to the strainer to rinse, letting it gravity drip into tube. Repeat for all digestate tubes. Rinse each strainer with an additional 10 ml 1× PBS. The strainer will remove strands of gelatinous, fibrous tissue aggregates. Discard the strainer with these fibrous tissue aggregates and retain the filtrate (see Note 1e).
Centrifuge the filtrate for 10 min, 250 rcf, 4 °C; the sample will layer into three fractions from bottom to top: an SVF pellet, an aqueous supernatant, and a lipid/adipocyte layer (Fig. 3). Pipette off the lipid/adipocyte and aqueous layers with a transfer pipette, being careful not to disturb the SVF pellet, leaving only the SVF pellet with 1–2 mL of overlying aqueous phase. Combine SVF pellets from three to five conical tubes into one fresh 50 mL conical tube.
Add 20 mL 1× RBC lysis solution to the SVF pellet; vortex to mix, and incubate 5 min, 25 °C. Dilute RBC lysate with 30 mL of 1× PBS to tube and invert tube to mix. Centrifuge 10 min, 250rcf, 4 °C. Remove supernatant from cell pellet with a pipette.
Resuspend SVF cell pellet in either in 1×PBS or culture medium depending on intended downstream application (e.g., PBS for flow cytometry; medium for culture/differentiation) (see Note 1f). Remove a 10 μL aliquot and mix with 10 μL trypan blue in a microcentrifuge tube. Count viable SVF cells using a hemocytometer or automated cell counter.
Fig. 3.

Collagenase-digested human visceral adipose tissue: Collagenase-digested human visceral adipose tissue after gradient centrifugation demonstrating lipid and aqueous layers and the SVF cell pellet
3.2 Expansion and Storage of Preadipocytes
Seed 5 × 105 fresh SVF cells into a 10 cm cell culture dish (~1 × 104 cells/cm2) (see Note 1g). Add 10 mL expansion medium. Incubate for 24 h. Place in an incubator, change medium every 2–3 days until 90 % + confluent, usually 2–3 days. Non-preadipocytes (leukocytes, endothelial cells) will not adhere and will be removed with subsequent medium changes, leaving preadipocytes as an adherent cell population.
At confluence, remove medium from cell culture dish. Rinse with 5 mL PBS and then remove PBS. Add 2.5 mL of trypsin solution and incubate for 5–10 min at 37 °C, periodically gently swirling and tapping the culture dish to detach cells. When 90 % of cells have detached, add 10 mL expansion medium to cell culture dish, and swirl dish to mix. Transfer the 12.5 mL cell suspension to a T150 flask and add an additional 12.5 mL of expansion medium to a total volume of 25 mL. Place in an incubator, change medium every 2–3 days until 90 % confluent, usually 2–3 days.
Remove medium from T150 flask. Rinse with 10 mL PBS and then remove PBS. Add 5 mL trypsin solution to flask and remove cells as above. When 90 % of cells have detached, add 35 mL of expansion medium to flask and swirl flask to mix. Divide the 50 mL cell suspension into eight new T150 flasks (5 mL per flask). Add an additional 20 mL of expansion medium to each flask to a total volume of 25 mL. Place in an incubator, change medium every 3 days until 95 % confluent, usually 2–5 days. Time to confluence is variable and patient-specific.
Remove medium from flasks. Rinse each flask with 10 mL 1× PBS then remove PBS. Add 5 mL trypsin solution to each flask and remove cells as above. When 90 % of cells have detached, add 5 mL expansion medium to each flask and swirl to mix. Combine cell suspensions from all flasks into two 50 mL conical tubes and centrifuge for 10 min, 250 rcf, 4 °C. Remove medium, combine cell pellets by resuspending in 10 mL expansion medium. Count cells and repeat centrifugation step. Remove medium and resuspend cell pellet in freezing medium at 2 × 106 cells/mL.
Aliquot 1 mL of cell suspension into 2 mL screw-capped tubes. Transfer to −80° freezer for 24 hrs, then transfer tubes to liquid nitrogen storage unit for long-term storage (see Note 2h).
3.3 In Vitro Differentiation of Human Adipocytes
Thaw a frozen vial of PA containing two million cells by gently swirling cryovial in a 37 °C water bath; resuspend in 20 mL of expansion medium, in a single T150 flask, culture at 37 °C for 2–3 days until 90 % confluence.
Trypsinize, centrifuge, and resuspend PA in 10 mL of expansion medium; count cells using a hemocytometer and trypan blue exclusion. Seed cells at a density of 60,000 cells per well in a 24-well tissue culture plate (3 × 104 cells/cm2) in 0.5 mL of expansion medium.
Remove expansion medium when cells are 90 % confluent usually at day 2–3; wash with 1× PBS and add differentiation medium. Replenish with fresh differentiation medium every 2–3 days.
Remove differentiation medium after 3–7 days; wash with 1× PBS and add maintenance medium (see Note 3i). Replenish with fresh maintenance medium every 2–3 days. Cells will begin to accumulate visible lipid droplets by day 10. By day 14, cells are lipid-laden and ready for experimental use (see Notes 3j–l, Fig. 4).
Fig. 4.
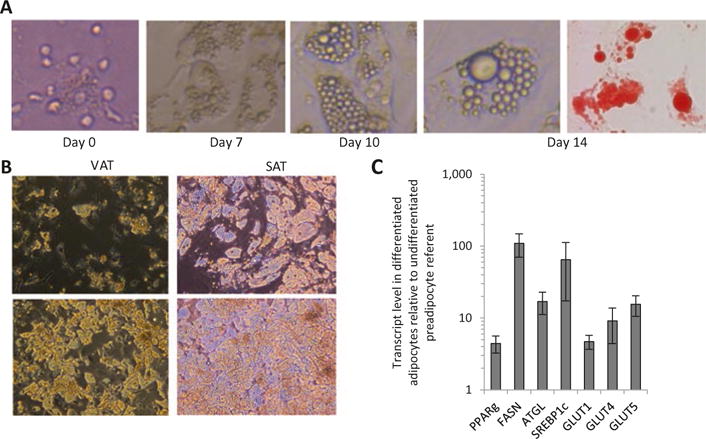
Differentiated human adipocytes: (a) Human visceral adipocytes at various stages of differentiation; far right- Oil Red-O stained adipocytes. (b). Examples of VAT and SAT adipocytes from two different patients (top, bottom) at day 14 of differentiation; note greater degree of differentiation in SAT adipocytes, and moderate and good differentiation efficiencies (top, bottom respectively) between patients. (c). QRTPCR for adipogenic genes in RNA from VAT adipocytes after 14 days of differentiation; ordinate: fold difference in transcript levels in mature adipocytes compared with undifferentiated SVF referent; n = 5 samples
3.4 Oil Red-O Staining
Aspirate medium from cells that have been differentiated in a 24-well plate and wash once with 500 μL 1× PBS.
Add 200 μL 4 % formalin, fix for 15 min.
Aspirate formalin, and wash samples twice with 1× PBS
Add 200 μL of 60 % isopropanol, incubate for 5 min.
Aspirate 60 % isopropanol, and add 200 μL Oil Red-O working solution (see Notes 4m and 4n).
Stain with Oil Red-O working solution for 15 min (see Note 4o).
Aspirate Oil Red-O solution from wells (try to aspirate as much as possible in this step to avoid formation of precipitate).
Wash with 500 μL 1× PBS three times.
Image cells with light microscopy.
Aspirate PBS from well and allow to dry completely.
Add 200 μL 100 % isopropanol to well; place plate on rocker to disperse 60 % isopropanol for 15 min.
Aliquot 100 μL of solubilized Oil Red-O in isopropanol from each well in a well of a 96-well plate and read absorbance at 525 nM on a microplate spectrophotometer.
3.5 Lipolysis Assay
Culture cells as desired. For measurement of lipolysis, we seed 60,000 preadipocytes/well in a 24-well plate in 0.5 mL medium and differentiate into mature adipocytes as outlined above, then once cells are fully differentiated, proceed with lipolysis assay.
Remove maintenance medium and wash cells twice with warm 1× PBS; add 0.5 mL maintenance medium (without insulin) with or without 3 μM isoproterenol, then culture adipocytes for 6–72 h, then collect culture supernatants.
Pipet 2 μL of each supernatant into a 96-well plate. Reserve at least two wells to allow for one blank and one standard. The blank used is distilled H2O; the glycerol standard solution is provided in the Triglyceride Determination Kit (see Notes 4p–r).
Add 270 μL of free glycerol reagent from the Triglyceride Determination Kit to each well, pipetting to mix. Incubate plate at 37 °C for 5 min.
Read plate at 540 nm on a microplate spectrophotometer.
Calculate the concentration of glycerol: (absorbance of sample − absorbance of blank) /(absorbance of standard − absorbance of blank) × 2.5 mg/mL = concentration of sample.
3.6 Labeled Acetate Lipogenesis Assay
Culture cells as desired. For measurement of basal lipogenesis, we seed 60,000 preadipocytes/well in a 24-well plate in 0.5 mL medium and differentiate into mature adipocytes as outlined above, then once cells are fully differentiated, culture for 72 h in maintenance medium and proceed with lipogenesis assay.
Remove maintenance medium and wash cells twice with warm 1× PBS; add 0.5 mL serum starvation medium supplemented with 100 nM insulin; incubate cells for 24 h at 37 °C.
Remove serum starvation medium; add 0.5 mL Lipogenesis medium; incubate at 37 °C overnight.
Wash cells twice with 1× PBS. Add 120 μL 0.1 N HCl to each well and pipette vigorously to lyse cells. Reserve 10 μL of lysate for protein assay (assess via Bradford assay); transfer 100 μL of remaining lysate to a 1.5 mL microcentrifuge tube.
Add 500 μL of 2:1 chloroform:methanol (v/v). Vortex briefly and incubate at room temperature for 5 min. Add 250 μL H2O, vortex, and incubate at room temperature for an additional 5 min. Centrifuge samples for 10 min at 3000 × g, 25 °C.
Carefully transfer lower lipid/organic phase to a scintillation vial containing 2 mL liquid scintillation fluid; measure 3H activity on scintillation counter. Normalize cpm to protein concentration as determined by protein assay (see Note 4s).
3.7 Glucose Uptake Assay
Culture cells per desired experimental measurement in 24-well plates. For measurement of glucose uptake, we seed 60,000 preadipocytes/well in a 24-well plate in 0.5 mL medium and differentiate into mature adipocytes as outlined above, then once cells are fully differentiated, proceed with glucose uptake assay.
Remove medium and wash cells once with 1× PBS; add 0.5 mL/well serum starvation medium and incubate at 37 °C for 12 h.
Remove medium and wash cells twice with 1× PBS; add 0.5 mL/well 1 % BSA in PBS and incubate at 37 °C for 2 h.
Wash cells once with 1× PBS, add 0.5 mL/well 1× PBS with or without 100 nM insulin (exclude insulin for basal experimental arm), and incubate at 37 °C for 40 min.
Aspirate PBS, add 0.5 mL/well 1× PBS, 0.1 mM 2-deoxy glucose, 2 μCi/mL deoxy-D-glucose, 2- [1,2- 3H(N)], with or without 200 nM insulin (exclude insulin for basal experimental arm), and incubate at 37 °C for 40 min.
Remove medium and wash cells three times with 1× PBS; add 420 μL 1 % SDS solution, lyse cells with vigorous pipetting; incubate at 25 °C for 10 min.
Collect 10 μL from each well for Bradford protein assay. Transfer 400 μL of whole cell lysate into 2 mL scintillation fluid in a scintillation vial. Count activity on scintillation counter.
4 Notes
4.1 Isolation of SVF from human adipose tissue
-
(a)
Typical SVF yields from the above protocol are variable and patient-dependent, average yields are ~0.7 million cells per gram of tissue and range from 0.3 to 1.0 million per gram of tissue.
-
(b)
Type I[13, 14] and type II[1, 5]collagenases, as well as liberase, a combination of type I and II collagenases [13], may be used for human SVF isolation. Vendors include Sigma Inc., Worthington Inc., Gibco Inc., and Roche Inc. (Liberase). In our experience, cell yields are incrementally higher with type II collagenase.
-
(c)
The duration and concentration of collagenase digestion affect cell yield and viability. Yields increase and viability decreases with increasing digestion times and collagenase concentrations. We have experimented with 30–60 min digestion times and working (1×) collagenase concentrations of 0.5–3 mg/mL, ranges similar to other published protocols. In addition, collagenase activity varies among different lots, and some protocols calculate collagenase concentrations in units/mL rather than mg/mL. Type II collagenase (Sigma Inc.) typically ranges from 200 to 275 units/mg depending on lot. Digestion times, collagenase concentrations, and activity of specific lots of collagenase are variables each laboratory should consider during protocol optimization.
-
(d)
Collagenase digestion may eliminate specific cell surface markers on leukocytes and preadipocytes. For SVF flow cytometry analysis, variable digestion times and collagenase concentrations should be tested to confirm that specific cell surface markers of interest are not eliminated by collagenase digestion.
-
(e)
Particularly fibrous or less digested tissue may generate cell suspensions with excessive debris, elimination of which is helpful if SVF flow cytometry is planned (but not necessary for preadipocyte expansion, as debris is eliminated with medium changes during culture). Three interventions may aid in removing excessive debris from the final SVF suspension: first, an additional wash step may be added after RBC lysis; second, an additional filtration step may be added after mincing of tissue but before digestion; finally filtration of the SVF pellet retrieved after centrifugation may be performed [15]. Use of 250 μM, 150 μM, or 100 μM cell strainers is reported [13, 15, 16].
-
(f)
Tissue may be stored overnight at 4 °C prior to processing, but with reduced cell yields and viability.
4.2 Expansion and storage of preadipocytes
-
(g)
If starting adipose tissue amounts or SVF yields are low, then seed 50,000 SVF cells into a single well of a 6-well cell culture plate, then expand this into a 10 cm plate, then proceed with further expansion as outlined above into T150 flasks.
-
(h)
Frozen preadipocytes may be stored for at least 6 months, beyond which viability decreases.
4.3 In vitro differentiation of human adipocytes
-
(i)
Published protocols vary with respect to durations of culture in differentiation and maintenance media. Initial culture for 3–4 days in expansion medium containing serum is necessary to allow preadipocytes to adhere, after which differentiation is induced by changing cells to serum-free differentiation medium for 3–7 days or longer, after which culture is continued in serum-free maintenance medium to maintain ongoing differentiation. Differentiation and maintenance media contain similar base constituents, but the former contains IBMX and a thiazolidinedione. Some protocols include dexamethasone in differentiation and maintenance media [13], while we and others include dexamethasone only in differentiation medium. We observe better differentiation in some but not all samples with longer periods of culture in differentiation medium, and we typically culture cells for 7 days in differentiation medium, and up to 14 days if differentiation is poor. Others have reported similar longer periods of culture in differentiation medium [17, 18]. Recombinant human fibroblast growth factors (FGF-1, FGF-2) have been reported to enhance preadipocyte proliferation and adipocyte differentiation, but are not standard components of human adipocyte differentiation protocols as of this writing [19–24].
-
(j)
Adipocytes are usually fully differentiated within 14 days, by which time they contain multiple lipid droplets that begin to coalesce into larger droplets. Longer periods of culture will result in further development of dominant and in some cases unilocular lipid droplets, but as time passes, adipocytes become less adherent and more difficult to manipulate in downstream assays. Care must be taken not to detach cells from plates during downstream assays, especially with longer periods of differentiation. We typically use adipocytes for assays after 14 days of differentiation, but cells can be used up to 4 weeks after induction of differentiation.
-
(k)
We assess differentiation using Oil Red-O staining, QRTPCR for adipogenic gene expression, and lipolysis assay. We quantify lipogenesis using both quantification of Oil Red-O staining and incorporation of 3H–acetate into lipid. Mature adipocytes accumulate Oil-Red-O in lipid droplets, markedly increase adipogenic gene expression, and demonstrate lipolytic capacity manifested by glycerol release in response to beta-adrenergic stimulation (Fig. 4).
-
(l)
Significant patient-specific variability is observed in degree of differentiation as assessed by the percentage of preadipocytes that accumulate cytoplasmic lipid. Depot-specific differences in differentiation efficiency are also observed, typically 50–70 % for VAT and 90 + % for SAT.
4.4 Metabolic phenotyping
-
(m)
Oil Red-O solution will precipitate/adhere to the filter; prepare double what you intend to use that day to account for this. Stain immediately after preparation/filtration to avoid precipitation of Oil Red-O.
-
(n)
For Oil Red-O staining, dispense all reagents with a pipette against the side of the tissue culture well, as pipetting directly onto cells will disrupt the cell monolayer.
-
(o)
Oil Red-O staining longer than 15 min should be avoided, as this will lead to precipitation of Oil Red-O and falsely elevated readings.
-
(p)
The Triglyceride Determination Kit for lipolysis assay measures free glycerol via a coupled enzymatic reaction that produces a quinone imine dye, absorbance of which at 540 nm is directly proportional to glycerol concentration.
-
(q)
Free glycerol reagent for lipolysis assay comes pre-aliquoted in powdered form and should be prepared for use by adding 40 mL of H2O to one bottle of powdered reagent. Gently invert the bottle to mix the reagent. Each bottle can be stored at 4 °C for up to 60 days after it is reconstituted. This reagent should be colorless; discard bottle if a purple tint is observed. Free glycerol reagent should not be vigorously shaken or vortexed, as this will deactivate it.
-
(r)
Place the standard in the lipolysis assay at least one well away from other samples on the 96-well plate, as its strong signal may affect absorbance readings of other samples.
-
(s)
Lipogenesis may be quantified by measurement of incorporation of labeled fatty acid precursors into cellular lipids. Many assays utilize C14 labeling, but we have found good results with the above tritium-labeled acetate-based assay, which is modified from previously published protocols used in adipocytes, hepatocytes, and other cell types [25, 26]. Incorporation into lipid involves transfer of a—CH2 moiety, and while some label is lost by H-D exchange, we have found the assay to be reproducible and reliably distinguish patient-specific adipocyte lipogenic capacities.
Acknowledgments
Funding
Supported by NIH grants R01DK097449 (R.W.O.), DK090262 (C.N.L.), T32DK101357, F32DK105676 (L.A.M.).
Abbreviations
- eWAT
Inguinal white adipose tissue (iWAT)
- PA
Preadipocyte
- PBS
Phosphate buffered saline
- QRTPCR
Quantitative real-time polymerase chain reaction
- SAT
Subcutaneous adipose tissue
- SVF
Stromal-vascular cell fraction
- VAT
Visceral adipose tissue
References
- 1.O’Rourke RW, Meyer KA, Gaston G, White AE, Lumeng CN, Marks DL. Hexosamine biosynthesis is a possible mechanism underlying hypoxia’s effects on lipid metabolism in human adipocytes. PLoS One. 2013;8:e71165. doi: 10.1371/journal.pone.0071165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, Forse RA, Chinnappan D, Martin-Ruiz C, von Zglinicki T, Kirkland JL. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–2578. doi: 10.2337/db06-0540. [DOI] [PubMed] [Google Scholar]
- 3.Tchkonia T, Tchoukalova YD, Giorgadze N, Pirtskhalava T, Karagiannides I, Forse RA, Koo A, Stevenson M, Chinnappan D, Cartwright A, Jensen MD, Kirkland JL. Abundance of two human preadipocyte subtypes with distinct capacities for replication, adipogenesis, and apoptosis varies among fat depots. Am J Physiol Endocrinol Metab. 2005;288:E267–E277. doi: 10.1152/ajpendo.00265.2004. [DOI] [PubMed] [Google Scholar]
- 4.Tchkonia T, Giorgadze N, Pirtskhalava T, Tchoukalova Y, Karagiannides I, Forse RA, DePonte M, Stevenson M, Guo W, Han J, Waloga G, Lash TL, Jensen MD, Kirkland JL. Fat depot origin affects adipogenesis in primary cultured and cloned human preadipocytes. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1286–R1296. doi: 10.1152/ajpregu.00653.2001. [DOI] [PubMed] [Google Scholar]
- 5.Tchoukalova YD, Koutsari C, Votruba SB, Tchkonia T, Giorgadze N, Thomou T, Kirkland JL, Jensen MD. Sex- and depot- dependent differences in adipogenesis in normal-weight humans. Obesity (Silver Spring) 2010;18:1875–1880. doi: 10.1038/oby.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013;18:355–367. doi: 10.1016/j.cmet.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sengenès C, Lolmède K, Zakaroff-Girard A, Busse R, Bouloumié A. Preadipocytes in the human subcutaneous adipose tissue display distinct features from the adult mesenchymal and hematopoietic stem cells. J Cell Physiol. 2005;205:114–122. doi: 10.1002/jcp.20381. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, Huang K, Tu H, van Marken Lichtenbelt WD, Hoeks J, Enerbäck S, Schrauwen P, Spiegelman BM. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 10.Carey VJ, Walters EE, Colditz GA, et al. Body fat distribution and risk of non-insulin-dependent diabetes mellitus in women. Nurses’ Health Study. Am J Epidemiol. 1997;145:614–619. doi: 10.1093/oxfordjournals.aje.a009158. [DOI] [PubMed] [Google Scholar]
- 11.Kissebah AH, Vydelingum N, Murray R, Evans DJ, Hartz AJ, Kalkhoff RK, et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab. 1982;54:254–260. doi: 10.1210/jcem-54-2-254. [DOI] [PubMed] [Google Scholar]
- 12.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7:410–420. doi: 10.1016/j.cmet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee MJ, Fried SK. Optimal protocol for the differentiation and metabolic analysis of human adipose stromal cells. Methods Enzymol. 2014;538:49–65. doi: 10.1016/B978-0-12-800280-3.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lystedt E, Westergren H, Brynhildsen J, Lindh-Astrand L, Gustavsson J, Nystrom FH, et al. Subcutaneous adipocytes from obese hyperinsulinemic women with polycystic ovary syndrome exhibit normal insulin sensitivity but reduced maximal insulin responsiveness. Eur J Endocrinol. 2005;153:831–835. doi: 10.1530/eje.1.02027. [DOI] [PubMed] [Google Scholar]
- 15.Hutley LJ, Herington AC, Shurety W, Cheung C, Vesey DA, Cameron DP, Prins JB. Human adipose tissue endothelial cells promote preadipocyte proliferation. Am J Physiol Endocrinol Metab. 2001;281:E1037–E1044. doi: 10.1152/ajpendo.2001.281.5.E1037. [DOI] [PubMed] [Google Scholar]
- 16.Hauner H, Röhrig K, Petruschke T. Effects of epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) on humanadipocyte development and function. Eur J Clin Invest. 1995;25(2):90–96. doi: 10.1111/j.1365-2362.1995.tb01532.x. [DOI] [PubMed] [Google Scholar]
- 17.Dicker A, Ryden M, Naslund E, Muehlen IE, Wiren M, Lafontan M, et al. Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia. 2004;47:420–428. doi: 10.1007/s00125-003-1324-0. [DOI] [PubMed] [Google Scholar]
- 18.Yu G, ZE F, Wu X, Hebert T, Halvorsen YD, Buehrer BM, et al. Adipogenic differentiation of adipose-derived stem cells. Methods Mol Biol. 2011;702:193–200. doi: 10.1007/978-1-61737-960-4_14. [DOI] [PubMed] [Google Scholar]
- 19.Hutley LJ, Shurety W, Newell F, McGeary R, Pelton N, Grant J, Herington A, Cameron D, Whitehead J, Prins J. Fibroblast growth factor 1: a key regulator of human adipogenesis. Diabetes. 2004;53:3097–3106. doi: 10.2337/diabetes.53.12.3097. [DOI] [PubMed] [Google Scholar]
- 20.Kakudo N, Shimotsuma A, Kusumoto K. Fibroblast growth factor-2 stimulates adipogenic differentiation of human adipose- derived stem cells. Biochem Biophys Res Commun. 2007;359:239–244. doi: 10.1016/j.bbrc.2007.05.070. [DOI] [PubMed] [Google Scholar]
- 21.Newell FS, Su H, Tornqvist H, Whitehead JP, Prins JB, Hutley LJ. Characterization of the transcriptional and functional effects of fibroblast growth factor-1 on human preadipocyte differentiation. FASEB J. 2006;20:2615–2617. doi: 10.1096/fj.05-5710fje. [DOI] [PubMed] [Google Scholar]
- 22.Skurk T, Ecklebe S, Hauner H. A novel technique to propagate primary human preadipocytes without loss of differentiation capacity. Obesity (Silver Spring) 2007;15:2925–2931. doi: 10.1038/oby.2007.349. [DOI] [PubMed] [Google Scholar]
- 23.Skurk T, Hauner H. Primary culture of human adipocyte precursor cells: Expansion and differentiation. Methods Mol Biol. 2012;806:215–226. doi: 10.1007/978-1-61779-367-7_15. [DOI] [PubMed] [Google Scholar]
- 24.Widberg CH, Newell FS, Bachmann AW, Ramnoruth SN, Spelta MC, Whitehead JP, Hutley LJ, Prins JB. Fibroblast growth factor receptor 1 is a key regulator of early adipogenic events in human preadipocytes. Am J Physiol Endocrinol Metab. 2009;296:E121–E131. doi: 10.1152/ajpendo.90602.2008. [DOI] [PubMed] [Google Scholar]
- 25.Akie TE, Cooper MP. Determination of fatty acid oxidation and lipogenesis in mouse primary hepatocytes. J Vis Exp. 2015;102:e52982. doi: 10.3791/52982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez-Diaz S, Johnson LA, DeKroon RM, Moreno-Navarrete JM, Alzate O, Fernandez- Real JM, Maeda N, Arbones-Mainar JM. Polymerase I and transcript release factor (PTRF) regulates adipocyte differentiation and determines adipose tissue expandability. FASEB J. 2014;28:3769–3779. doi: 10.1096/fj.14-251165. [DOI] [PMC free article] [PubMed] [Google Scholar]