

Abstract
Bioanalytical methods are widely used for quantitative estimation of drugs and their metabolites in physiological matrices. These methods could be applied to studies in areas of human clinical pharmacology and toxicology. The major bioanalytical services are method development, method validation and sample analysis (method application). Various methods such as GC, LC–MS/MS, HPLC, HPTLC, micellar electrokinetic chromatography, and UFLC have been used in laboratories for the qualitative and quantitative analysis of carbamazepine in biological samples throughout all phases of clinical research and quality control. The article incorporates various reported methods developed to help analysts in choosing crucial parameters for new method development of carbamazepine and its derivatives and also enumerates metabolites, and impurities reported so far.
Keywords: Carbamazepine, HPLC, LC–MS/MS, HPTLC, RP-UFLC, Micellar electrokinetic chromatography
1. Introduction
One of the major challenges faced by the pharmaceutical industry today is finding new ways to increase productivity, decrease costs whilst still ultimately developing new therapies that enhance human health. Bioanalytical methods are widely used for quantitative estimation of drugs and their metabolites in physiological matrices, and could be applied to studies in area of human clinical pharmacology and nonhuman pharmacology/toxicology that involves evaluation and interpretation of bioequivalence, pharmacokinetic, and toxicokinetic studies. The major bioanalytical services include method development, method validation and sample analysis (method application).
Chromatographic methods such as gas chromatography (GC), liquid chromatography–mass spectrometry (LC–MS), and high performance liquid chromatography (HPLC) are commonly used in laboratories for the qualitative and quantitative analysis of drug substances and biological samples throughout all the phases of method development of a drug in research and quality control. Further, method validation is carried out to ensure that the method developed was accurate, specific, reproducible and rugged over the specified range in which an analyte is analyzed. The present review covers the wide range of chromatographic techniques used in determination of carbamazepine (CBZ) and its congeners. It also incorporates records for simultaneous estimations performed using analytical techniques for CBZ or its congeners with other drug members, impurities and metabolites.
CBZ is an anticonvulsant and mood-stabilizing drug used primarily in the treatment of epilepsy and bipolar disorder, as well as trigeminal neuralgia in old age patients. CBZ is indicated for the treatment of partial seizures with simple or complex symptomatology (psychomotor, temporal lobe) and genaralized tonic-clonic seizures (grand mal). CBZ is also used as a diuretic and anticholinergic. CBZ is a first-choice anticonvulsant because of its relatively low psychological toxicity and the rarity of serious adverse effects. CBZ was discovered in 1953 by chemist Walter Schindler at J.R. Geigy AG in Basel, Switzerland. The drug was then synthesized in 1960 by chemist Schindler. Later, its anti-epileptic properties were discovered. In 1971, Drs. Takezaki and Hanaoka first used CBZ to control mania in patients' refractory to antipsychotics. CBZ was first marketed as a drug to treat trigeminal neuralgia in 1962. It has been used as an anticonvulsant and antiepileptic in the UK since 1965, and has been approved in the US since 1974. It is official in most of the pharmacopoeias. It is the drug of choice for many combination therapies and used in treatment of geriatric patients with multiple disease states. Method development for such combination product formulations is still a challenge.
2. Mechanism of action
CBZ acts postsynaptically by limiting the ability of neurons to sustain high frequency repetitive firing of action potentials through enhancement of sodium channel inactivation. In addition to altering neuronal excitability, it may act presynaptically to block the release of neurotransmitter by blocking the presynaptic sodium channels and the firing of action potentials, which in turn decreases synaptic transmission. Pain relief is believed to be associated with blockade of synaptic transmission in the trigeminal nucleus and seizure control with reduction of post-tetanic potentiation of synaptic transmission in the spinal cord [1].
CBZ has a narrow therapeutic index and the relationship between dose and plasma concentrations of CBZ may be unpredictable because of differences in genetics, age, gender, absorption, autoinduction and disease state between individuals. Also, the presence of numerous clinically significant drug interactions supports the need of using therapeutic monitoring of CBZ as an essential tool in designing a safe and effective therapeutic regimen for patients with epilepsy [2], [3].
CBZ (5H-dibenzo[b, f]azepine-5-carboxamide) is insoluble in water, soluble in alcohol, acetonitrile and acetone. CBZ is available in market with the brand names Carbamazepen, Carbatrol, Carbazepine, Carbelan and Epitol. Although CBZ is poorly soluble in aqueous media, it has a high oral bioavailability in humans [4].
Metabolism occurs primarily in the liver via the cytochrome P-450 oxidase system, producing carbamazepine-l0, 11-epoxide (CBZ-EP) which is as active and may reach a level up to half that of CBZ. This is almost entirely converted to carbamazepine-trans-10, 11-dihydrodiol (CBZ–Di OH) by epoxide hydrolase before excretion in the urine (Fig. 1). CBZ along with its metabolite was required to be routinely measured, since there were patients for whom high concentrations of this metabolite could be responsible for otherwise unexplained toxicity. These toxicities may be due to degradation products, metabolites present as impurities in the formulation, such impurities can be studied by forced degradation of CBZ under acid, base, oxidation, heat and photolytic conditions. Degradation was observed in CBZ samples under stress conditions like acid hydrolysis, photolysis and thermal exposure. Mild degradation was observed for alkaline hydrolysis and exposure to oxidation by hydrogen peroxide [5], [6].
Fig. 1.

Metabolites of carbamazepine.
Identification and determination of unknown organic impurities is the key to the production of high quality drug substances. ICH guidelines indicate that impurities at or above 0.1% in the drug substance require identification [7]. Fig. 2 gives the list of pharmacopoeial impurities in CBZ reported in EP and BP.
Fig. 2.

Pharmacopoeial impurities in carbamazepine.
Various chromatographic methods such as GC, LC, LC–MS, HPLC, high performance thin layer chromatography (HPTLC), ultra-force liquid chromatography (UFLC) and micellar electrokinetic chromatography (MEKC) were used in laboratories for the qualitative and quantitative analysis of CBZ.
3. GC
GC is a common type of chromatography used in analytical chemistry for separating and analyzing compounds that can be vaporized without decomposition. In GC, the mobile phase is a carrier gas, usually an inert gas such as helium or an unreactive gas such as nitrogen. The stationary phase is a microscopic layer of liquid or polymer on an inert solid support, inside a piece of glass or metal tubing called a column. The gaseous compounds being analyzed interact with the walls of the column, which is coated with a stationary phase. This causes each compound to elute at a different time, known as the retention time of the compound [8], [9], [10], [11], [12].
A simple, accurate and sensitive microextraction by packed sorbent-gas chromatography–mass spectrometry method has been developed by Rani et al. [13] for the simultaneous quantification of four antiepileptic drugs such as oxcarbazepine (OXCBZ), CBZ, phenytoin, and alprazolam in human plasma and urine as a tool for drug monitoring. Caffeine was used as internal standards for the electron ionization mode. An original pretreatment procedure on biological samples, based on microextraction in packed syringe using C18 as packing material gave high extraction yields in the range of 69.92%–99.38% with satisfactory precision of RSD<4.7% and good selectivity. Linearity was found in the range of 0.1–500 ng/mL for these drugs with limits of detection (LOD) between 0.0018 and 0.0036 ng/mL. In validation, the method was successfully applied to some plasma samples from patients undergoing therapy with one or more of these drugs. The present method was applied for the analysis of these drugs in the real urine and plasma samples of the epileptic patients.
Speed et al. [14] described a rapid method for simultaneously determining the anticonvulsant drugs such as CBZ, phenobarbitone, phenytoin, primidone and valproic acid. Calibration gives reliable quantitation from therapeutic to higher concentrations. Deuterated internal standards were extracted using Bond Elut Certify columns. Butyl derivatives were formed using n-iodobutane under mild conditions and were extracted into ethyl acetate. Recoveries were found to be greater than 50%. Sample preparation time was less than 2 h, and the GC run time was less than 20 min per injection. At least two ion pairs formed by electron impact ionization were monitored for each drug. Intraday CV׳s was less than 6.28% and interday CV׳s less than 14.1%. Linearity was observed from subtherapeutic to high fatal levels for all drugs.
4. LC
A number of LC methods with UV detection for the determination of CBZ and its metabolite in drug products and human plasma have been described [15], [16], [17]. LC–MS methods have also been reported for the determination of CBZ [18], [19] and its metabolites [20], [21] in biological fluids. Although they provide improved sensitivity and specificity compared with other analytical methods, MS procedures were more expensive than HPLC-UV.
Breton et al. [20] described a specific and sensitive liquid chromatography–electrospray ionization mass spectrometry method for the simultaneous determination of CBZ, OXCBZ and eight of their metabolites such as CBZ-10,11-epoxide (CBZ-EP), 10,11-dihydro-10,11-trans-dihydroxy-carbamazepine (DiOH-CBZ), 10-hydroxy-10,11-dihydro-CBZ (10-OH-CBZ), 2-hydroxycarbamazepine (2-OH-CBZ), 3-hydroxycarbamazepine (3-OH-CBZ), iminostilbene (IM), acridone (AO) and acridine (AI) in human plasma. Separation of the analytes was achieved within 50 min using a Zorbax eclipse XD8 C8 analytical column. The mobile phase consisted of a mixture of acetonitrile–formate buffer (2 mM, pH 3). Detection was performed using a quadrupole mass spectrometer fitted with an electrospray ion source. Mass spectrometric data were acquired in single ion recording mode at m/z 237 for CBZ, m/z 180 for CBZ-EP and AI, m/z 236 for OXCBZ, m/z 237 for 10-OH-CBZ, m/z 253 for 2-OH-CBZ, 3-OH-CBZ and DiOH-CBZ, m/z 196 for AO and m/z 194 for IM. The extraction recovery averaged 90% for CBZ, 80% for OXCBZ and was 80%–105% for the metabolites. The lower limit of quantitation (LLOQ) was 0.5 mg/L for CBZ, 0.4 mg/L for OXCBZ and ranged from 0.02 to 0.3 mg/L for the metabolites. Precision ranged from 2% to 13% and accuracy was between 86% and 112%.
Sener et al. [21] described analytical method for determination of CBZ and its metabolite CBZ-EP using ESI–LC–MS (ion trap). The compounds were separated on a C18 (150 mm ×2.1 mm i.d., 3 µm particles) column and were isocratically eluted in the mobile phase consisting of water–acetonitrile–acetic acid (74.5:25:0.5, v/v/v) using the flow rate of 0.4 mL/min. OXCBZ was used as an internal standard. The retention time for CBZ-EP, OXCBZ and CBZ was 5.6, 6.8, and 12.8 min, respectively. Signals of the compounds were monitored under the multi-reaction monitoring mode of ESI–LC–MS (ion trap) for the quantification. Selected ions of CBZ-EP, OXCBZ and CBZ in the multi-reaction monitoring were m/z 253>210, m/z 253>180 and m/z 237>194. The method was validated over the concentration range of 5.0–500.0 ng/mL.
Liquid chromatography/tandem mass spectrometry (LC–MS/MS) is a hyphenated technique, combining the separation power of HPLC with the detection power of mass spectrometry. LC–MS/MS is highly selective and sensitive and includes high speed, low detection limits, the ability to generate structural information, the requirement of minimal sample treatment and the possibility to cover a wide range of analytes that differ in their polarities. LC–MS/MS and liquid chromatography with quadrupole-time of flight mass spectrometry (LC-Q-TOF MS) have recently been reported for the determination of CBZ in aquatic environment [22].
A sensitive method for the determination of CBZ and CBZ-EP in plasma was described by Van Rooyen et al. [23] using high-performance liquid chromatographic separation with tandem mass spectrometry. Samples were separated on a Phenomenex Luna C18 150 mm×2 mm, 5 µm column with a mobile phase consisting of acetonitrile, methanol and formic acid (0.1%) (10:70:20, v/v/v). Detection was performed by a Micromass Quattro Ultima mass spectrometer in the multi-reaction monitoring mode using electrospray ionization (ESI+). The transition of the protonated molecular ion was found for CBZ at m/z 237.05 and CBZ-EP at m/z 253.09 to the predominant ions of m/z 194.09 and 180.04, respectively. The mean recovery was 95% for CBZ and 101% for CBZ-EP, with a lower limit of quantification of 0.722 ng/mL for CBZ and 5.15 ng/mL for CBZ-EP.
LC–MS studies were performed to get molecular weight, establish mass fragmentation profile and identify an unknown impurity in CBZ active pharmaceutical ingredient. Unknown impurity was isolated using semi-preparative HPLC. An LC–MS compatible reverse phase isocratic method was developed and tandem mass spectrometry was performed using electrospray ionization source and ion trap mass analyzer [24].
A rapid tandem mass spectrometric (MS/MS) method for the quantification of OXCBZ in human plasma using imipramine as an internal standard has been developed and validated [25]. Plackett–Burman design was applied for screening of chromatographic and mass spectrometric factors. Factorial design was applied for optimization of essential factors for the robustness study such as the percentage of acetonitrile in mobile phase, flow rate, autosampler temperature, column oven temperature, declustering potential, collision energy and exit potential. Chromatographic separation was achieved isocratically on C18 reversed phase column within 3.0 min using a mobile phase of acetonitrile–10 mM ammonium formate (90:10, v/v) at a flow rate of 0.3 mL/min. Quantitation was achieved by using multiple reaction monitoring (MRM) scan at MRM transitions m/z 253>208 and m/z 281>86 for OXCBZ and imipramine, respectively. Calibration curves were linear over the concentration range of 0.2–16 µg/mL (r>0.999) with a limit of quantification of 0.2 µg/mL. Analytical recoveries of OXCBZ from spiked human plasma were in the range of 74.9%–76.3%. Stability studies showed that OXCBZ was stable for at least 3 months in both actual and spiked human plasma samples when frozen at or below −20 °C. The mean (SD) recoveries for actual samples (from the first determination) were 104%±8% (n=6) for OXCBZ. OXCBZ was stable for at least 6 h at room temperature in spiked human plasma samples; the mean recoveries from the nominal concentration were 95%–106%. OXCBZ in working solutions was found to be stable for at least two weeks at 2–8 °C. The mean recoveries (n=3) from the nominal concentrations were 88%–101% for OXCBZ, at 8.0 µg/mL and OXCBZ was also found to be stable in working solutions for at least 6 h at room temperature in darkness. The mean recoveries (n=3) from the nominal concentration of OXCBZ at LQC and HQC concentrations were at 0.2 and 16.0 µg/mL respectively. Extracts at concentrations of LQC and HQC were found to be stable on the autosampler at 10 °C for at least 12 h. Arithmetic mean recovery values after three freeze–thaw cycles were between 95% and 105% of the nominal value for LQC and HQC, respectively.
The MS and MS/MS studies [24] were performed on Thermo LCQAdvantage (Thermo Electron, San Jose, CA, USA) using electrospray ionization source and ion trap mass spectrometer. The source voltage was maintained at 3.0 kV and the capillary temperature was at 250 °C. Nitrogen was used as both sheath and auxiliary gas. The mass to charge ratio was scanned across the range of m/z 50–1000. MS/MS studies were carried out by keeping normalized collision energy at 25% and an isolation width of 1 amu. The HPLC consisted of Waters alliance 2690 separation module equipped with a 2487 UV detector and a column oven. A C18 column (Inertsil ODS-3, 250 mm×4.6 mm i.d., 5 µm particles) was used for chromatographic separation. The mobile phase consisted of a mixture of water–methanol–trifluoroacetic acid (30:70:0.05, v/v/v). The flow rate was maintained at 1.0 mL/min. Mass spectral data showed protonated molecular ion peaks at m/z 237, m/z 196 and m/z 501 for CBZ, impurity-A and impurity-B, respectively. On the basis of RRT and mass spectral data, impurity-A having molecular ion peak at m/z 196 was identified as iminodibenzyl. The mass spectral data obtained for impurity-B at RT 95.8 min did not match with any of the known impurities. Impurity-B was inferred to be unknown based on the HPLC and LC–MS spectral data. The proposed structure was further confirmed by using NMR, FT-IR and elemental analyzer studies and the molecular formula of unknown impurity was deduced as C30H20N4O2S and the corresponding structure was characterized as tetrabenzo[b,f,b׳f׳]azepino[4′,5′:4,5]thieno[2,3-d]azepine-3,9-dicarboxamide (Fig. 3).
Fig. 3.
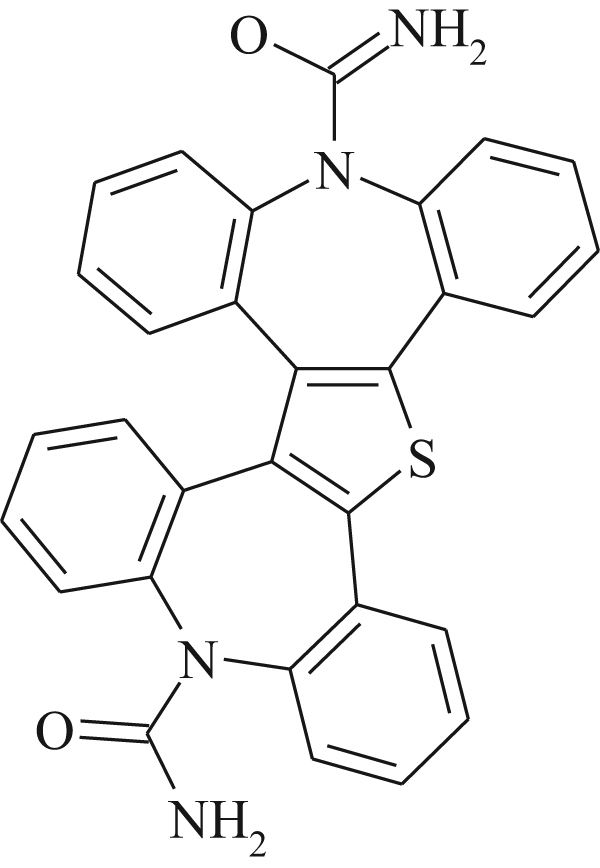
Structure of impurity tetrabenzo[b,f,b׳f׳]azepino[4′,5′:4,5]thieno[2,3-d]azepine-3,9-dicarboxamide.
LC–MS method has been proven to be powerful for the characterization of the metabolites of CBZ in patient urine [26]. CBZ-EP (9.6–15.0 µg/mL), trans-10, 11-dihydrodiol-CBZ (273.0–400.0 µg/mL) and CBZ (2.4–3.8 µg/mL) were measured in human urine by HPLC.
An LC–MS/MS method has been developed for the simultaneous determination of CBZ and its main metabolite CBZ-EP in rat plasma [27]. The method consisted of a liquid–liquid extraction procedure and electrospray LC–MS/MS analysis for the determination of CBZ and CBZ-EP in rat plasma with an isotope labeled internal standard. The LC–MS/MS system was equipped with a BASi PM-80 pump (BASi, West Lafayette, IN, USA) coupled to a Finnigan LCQ Deca ion trap mass spectrometer (ThermoQuest, San Jose, CA, USA) equipped with an ESI source. The chromatographic separation was achieved within 5 min using a C8 (150 mm×2.1 mm, 5 µm) column (Supelco, Bellefonte, PA, USA) with a mobile phase composed of water/acetonitrile/acetic acid (69.5:30:0.5, v/v/v) at a flow rate of 0.4 mL/min. Samples were injected by an autosampler (Sample Sentinel, BASi), which was set at 10 °C and fitted with a 20 µL loop. The mass spectrometer was operated in ESI positive ion mode. Nitrogen gas was used as both the sheath and auxiliary gas at a pressure of 80 and 20 units, respectively. The spray and capillary voltage were set at 5.0 kV and 22 V, respectively. The capillary temperature was set at 275 °C. D10-carbamazepine was used as the internal standard for all compounds. Analytes were determined by electrospray ionization tandem mass spectrometry in the positive ion mode using selected reaction monitoring (SRM). CBZ was monitored by scanning m/z 237>194, CBZ-EP by m/z 253>210 and d10-carbamazepine by m/z 247>204. The LLOQ was 5 ng/mL for each analyte, based on 0.1 mL aliquots of rat plasma. The extraction recovery of analytes from rat plasma was over 87%. Intra-day and inter-day assay coefficients of variations were in the range of 2.6%–9.5% and 4.0%–9.6%, respectively. Linearity was observed over the range of 5–2000 ng/mL. This method was used for pharmacokinetic studies of CBZ and CBZ-EP in response to two different blood sampling techniques (i.e. manual sampling via a jugular catheter versus automated sampling by using Culex automated blood sampler) in the rat. Several differences between the two sampling techniques suggest that the method of blood collection affects the evaluation of pharmacokinetic data. Automated blood sampling offers several advantages, such as an easy and accurate blood drawing and low animal stress.
A simple, accurate, and sensitive LC–MS/MS method has been developed by Kima et al. [28] for the simultaneous quantification of CBZ in human plasma as a rapid tool for drug monitoring using d10-phenytoin and d6-valproic acid as internal standards for the positive- and negative-ionization modes, respectively. Plasma samples were precipitated by the addition of acetonitrile and supernatants were analyzed on a C18 reverse-phase column using an isocratic elution. The mobile phase consisted of 5 mM ammonium formate buffer (pH 7.8) and 90% acetonitrile containing 5 mM ammonium formate (60:40, v/v). The flow rate was 0.2 mL/min. The HPLC system was coupled to a Qtrap 4000 triple–quadrupole mass spectrometer (AB Sciex, Foster City, CA, USA) equipped with an electrospray ionization (ESI) source. Detection was carried out in SRM mode. The calibration curves were linear over a 50-fold concentration range, with correlation coefficients (r2) greater than 0.997 for all antiepileptic drugs. The intra- and inter-day precision was less than 12%, and the accuracy was between 85.9% and 114.5%. The intra-day coefficients of variation were between 4.0% and 8.2%, and accuracy ranged from 85.2% to 105%. The inter-day coefficients of variation were between 6.7% and 12.0%, and the accuracy was between 90.6% and 107%. The absolute recovery of antiepileptic drugs was between 94.6% and 105%. Stability was assessed by analyzing three replicate plasma samples after three different manipulations: (i) short-term storage (6 h at room temperature); (ii) three freeze–thaw cycles and (iii) post-treatment storage (24 h at room temperature). The concentrations obtained were compared with the nominal values of the QC samples. The stabilities of the stock solutions of analytes after three weeks at 4 °C and after 4 months at 80 °C were evaluated by comparison with freshly prepared solutions of the same concentrations.
Loureiro et al. [29] developed and validated a sensitive and specific enantioselective LC–MS/MS method, for the simultaneous quantification of eslicarbazepine acetate (ESL), eslicarbazepine (S-Lic), OXCBZ and R-licarbazepine (R-Lic) in human plasma. Analytes were extracted from human plasma using solid phase extraction, and the chromatographic separation was achieved using a mobile phase of 80% n-hexane and 20% ethanol/isopropyl alcohol (66.7/33.3, v/v). A Daicel CHIRALCEL OD-H column (5 μm, 50 mm×4.6 mm) was used with a flow rate of 0.8 mL/min, and a run time of 8 min. ESL, S-Lic, R-Lic, OXCBZ and the internal standard, 10,11-dihydrocarbamazepine, were quantified by positive ion electrospray ionization mass spectrometry. This method was fully validated, demonstrating acceptable accuracy, precision, linearity, and specificity in accordance with FDA regulations for the validation of bioanalytical methods. Linearity was proven over the range of 50.0–1000.0 ng/mL for ESL and OXCBZ and over the range of 50.0–25,000.0 ng/mL for S-Lic and R-Lic. The intra- and inter-day coefficient of variation in plasma was less than 9.7% for ESL, 6.0% for OXCBZ, 7.7% for S-Lic and 12.6% for R-Lic. The accuracy was between 98.7% and 107.2% for all the compounds quantified. The LLOQ was 50.0 ng/mL for ESL, S-Lic, OXCBZ and R-Lic in human plasma. The short-term stability in plasma, freeze–thaw stability in plasma, frozen long-term stability in plasma, autosampler stability and stock solution stability all met acceptance criteria.
A quantitative method was described for solid-phase extraction (SPE) followed by LC–MS/MS for the simultaneous analysis of CBZ and its five metabolites, 10,11-dihydro-10,11-epoxycarbamazepine, 10,11-dihydro-10,11-dihydroxycarbamazepine, 2-hydroxycarbamazepine, 3-hydroxycarbamazepine and 10,11-dihydro-10-hydroxycarbamazepine [30]. An SPE procedure was used to concentrate target compounds from aqueous samples collected from sewage treatment plant (STP) wastewater and surface water. Extracts were analyzed using electrospray LC–MS/MS with time-scheduled SRM. The recoveries of the analytes were 83.6%–102.2% from untreated sewage (influent), 90.6%–103.5% from treated sewage (effluent) and 95.7%–102.9% from surface water samples. The instrumental detection limits were 0.8–4.8 pg for the analytes. Matrix effects were investigated for the analytes in HPLC-grade water, surface water, and STP influent and effluent. Ion suppression was increased for analytes in the order of surface water to STP effluent to STP influent, but no ion suppression was observed for analytes in HPLC-grade water. The developed method was validated by analysis of environmental aqueous samples: STP influent and effluent and surface water. CBZ and all five metabolites were detected in STP influent and effluent samples. Only CBZ and 10,11-dihydro-10,11-dihydroxycarbamazepine were detected in the surface water sample. Notably, 10,11-dihydro-10,11-dihydroxycarbamazepine was detected at the concentration approximately three times higher than that of the parent drug, CBZ, in all of the aqueous samples.
5. HPLC
HPLC is a chromatographic technique used to separate the components in a mixture, to identify each component, and to quantify each component. In general, the method involves a liquid sample being passed over a solid adsorbent material packed into a column using a flow of liquid solvent. Each analyte in the sample interacts slightly differently with the adsorbent material, thus retarding the flow of the analytes. If the interaction is weak, the analytes flow off the column in a short amount of time and if the interaction is strong, then the elution time is long.
HPLC methods for the determination of CBZ related impurities were reported in USP, EP, BP and IP. A number of HPLC methods for simultaneous determination of CBZ and its metabolites in plasma have been published, using pretreatment techniques such as liquid–liquid extraction [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], SPE [46], [47], [48], [49], [50], [51], [52], deproteinization [53], [54], stir bar-sorptive extraction [55], fluorescence polarization immunoassay [56], [57], chemiluminescence [58], spectrophotometry [59], spectrofluorimetry method [60], FT-Raman spectroscopy [61], and planar chromatography [62]. However, with some of these techniques, the obtained extraction yields were not satisfactory: most of them were time-consuming, and some required expensive instrument, rendering them not appropriate for routine drug monitoring [63].
In bioanalytcal methods, the determination of CBZ in biological fluids usually involves an isolation procedure prior to the chromatographic step. The most frequently used procedures are liquid–liquid extraction [64], [65], [66], [67], [68], [69], protein precipitation [70], [71] and off-line SPE with disposable extraction disk cartridge [72] or Bond-Elut cartridge [73]. These methods are labor intensive and time consuming and they also require a large volume of sample, as well as addition of an internal standard. Therefore, it is advantageous to inject the sample directly into a liquid chromatographic system without an off-line procedure. Different approaches have been used for direct-injection determination of CBZ in biological fluids by HPLC. The methods include micellar chromatography [74], [75], restricted access media columns such as Biotrap amine and acid precolumn [76], [77] and column switching techniques employing conventional reversed-phase precolumn [78].
5.1. API and metabolites
HPLC was performed by using reversed-phase 1.5 µm monosized non-porous silicon dioxide microspheres column instead of regular columns containing spherical porous C18 material and was studied to achieve decrease in run time and specificity [41]. Determination of CBZ and its active metabolite, CBZ-EP, in human plasma or serum was performed to demonstrate the utility of these columns. The sample was prepared in autosampler vials by protein precipitation with acetonitrile, followed by a quick centrifugation. Without any change to a conventional HPLC system, CBZ and CBZ-EP were well separated in less than 2.5 min using a Kovasil MS C14 column. No interference was observed with endogenous compounds and with nine antiepileptic drugs commonly prescribed as co-medication, and their metabolites. Due to the very low specific surface area of the packing, the required organic modifier volume per chromatographic run was decreased by a factor of 25. The developed method was found to be well suited for the determination of CBZ and CBZ-EP in clinical trials.
A simple, rapid, sensitive, and reproducible HPLC method for simultaneous determination of CBZ and two metabolites, carbamazepine-diol and CBZ-EP, in human plasma has been developed and validated by Matar et al. [79]. The procedure involved extraction of the antiepileptic drugs such as ethosuximide, primidone, lamotrigine, phenobarbital, phenytoin, and CBZ and two metabolites such as carbamazepine-diol and CBZ-EP from human plasma (100 µL) with ether using 9-hydroxymethyl-10-carbamyl acridan as an internal standard. The extract was evaporated and reconstituted with mobile phase and then injected onto the chromatograph. The drugs and the internal standard were eluted from a Supelcosil LC-18 stainless steel column at ambient temperature with a mobile phase consisting of a 0.01 M phosphate buffer/methanol/acetonitrile (65:18:17, v/v/v) adjusted to a pH of 7.5 with phosphoric acid and a flow rate of 1 mL/min. The effluent was monitored at 220 nm. Quantitation was achieved by using peak area ratio of each drug to the internal standard. The intraassay and interassay coefficients of variation (CV) ranged from 2.43% to 6.25% and from 3.02% to 5.85%, respectively. The absolute extraction and relative analytical recoveries for the drugs ranged from 70.7% to 104.4% and from 88.3% to 106.1%, respectively. Stability tests showed that the drugs were stable in plasma for at least 4 weeks when stored at −20 °C. The method was applied clinically for monitoring the antiepileptic drugs in epileptic patients.
A method by using on-line coupling of SPE has been developed based on a restricted-access support with high performance reverse phase chromatography for the analysis of CBZ and CBZ-EP in human plasma samples [80]. The liquid chromatographic system consisted of a Waters Alliance HT HPLC system (Milford, MA, USA) connected to a Waters 996 photodiode array (PDA) detector. For the column-switching purposes, a column switching six-port valve (Waters) controlled by the work station and an additional Knauer 64 pump (Berlin, Germany) to deliver the analysis eluent were used. The following columns used in this study are ODS-Hypersil, (125 mm×4 mm i.d., 5 µm) (Hewlett-Packard) as analytical column and a LiChrocart 25-4 LiChrospher RP-18 ADS (25 mm×4 mm i.d., 25 µm particle diameter; Merck, Darmstadt, Germany) as extraction precolumn. A precolumn packed with 25 µm C18 alkyl-diol-silica support was used for plasma injection using column-switching technique. The analyte was enriched on the precolumn by a 5 µm phosphate buffer (pH 7) with 2% of methanol solution at a flow-rate of 0.8 mL/min, while proteins and endogenous hydrophilic substances in plasma were washed off to waste. The enriched analytes were then back-flushed onto the analytical C18 column, separated by a mixture of 10 mM phosphate buffer (pH 7) and acetonitrile solution (70:30, v/v) at a flow-rate of 1.0 mL/min and detected by the ultraviolet absorbance set at 212 and 285 nm and without transfer loss. Linear calibration graphs were obtained for sample injection volume of 50 µL containing 0.2–4.0 µg/mL of CBZ and 0.1–5.0 µg/mL of CBZ-EP, and 20 µL containing 5.0–20.0 µg/mL of CBZ; in either case the r-value was>0.9963. Recoveries from spiked plasma samples were quantitative for both the analytes and the coefficient of variation was below 3.83%. The lowest sample concentration that can be quantified with acceptable accuracy and precision was 0.2 µg/mL of CBZ and 0.1 µg/mL of CBZ-EP when a sample volume of 50 µL was injected. Concentrations of 0.08 and 0.05 µg/mL of CBZ and CBZ-EP were considered the limit of detection for a signal-to-noise ratio of 3. The developed column-switching method was successfully applied to the determination of CBZ and CBZ-EP in plasma samples.
Ribarska et al. [81] optimized and validated a simple and reliable SPE method followed by a reversed-phase (RP)-HPLC for the simultaneous determination of plasma levels of CBZ and CBZ-EP, in order to assure the implementation of the method for therapeutic monitoring. Nitrazapam was used as an internal standard. The extraction of the analytes from the plasma samples was performed by means of an SPE procedure. Separation was performed on a reversed-phase column Zorbax Extend C18 (150 mm×4.6 mm, 5 μm) using isocratic elution with acetonitrile and water (35:65, v/v) as a mobile phase, at a flow rate of 1 mL/min. The temperature was 30 °C, the volume of injection was 20 μL, and UV detection was set at 220 nm. Within-run assay precision ranged from 0.6% to 1.2% for CBZ and from 0.7% to 2.0% for CBZ-EP, while within-run assay accuracy ranged from 98.4% to 101.3% and 97.0% to 100.6% for CBZ and CBZ-EP, respectively. The extraction yields were in the range of 98.5%–99.6% for CBZ and 99.1%–99.7% for CBZ-EP, while the recovery value for nitrazapam was 98.9%. The extraction yield values were more than 98% for all the analytes, measured at four concentration levels of the linear concentration range. The method has shown excellent selectivity, sensitivity, linearity, precision and accuracy. Stability studies indicate that stock solutions and plasma samples were stabile under different storage conditions at least during the observed period. Stability tests were performed on three replicates of low and high QC samples after 24 h at room temperature (short term stability), after three freeze–thaw cycles, autosampler stability for 12 h, and after 90 days on samples stored at −20 °C (long term stability). Stability tests were also performed on stock solutions of analytes after 24 h at room temperature and after 3 months at 2–8 °C. The method was successfully applied for determination of CBZ and CBZ-EP in plasma of epileptic patients treated with CBZ as monotherapy and in polytherapy.
SPE-HPLC method has been developed and validated for rapid analysis of CBZ and its two metabolites, CBZ-EP and carbamazepine trans-diol, in human plasma [82]. The analysis was performed using C18 Bakerbond-BDC analytical column (250 mm×4.6 mm i.d., 5 μm). The optimal conditions for the separation were established with the mobile phase acetonitrile–10 mM phosphate buffer, pH 7.0 (30:70, v/v) at the flow rate of 1.5 mL/min, temperature 35 °C, and UV detection at 210 nm. Total run time was about 8 min. Phenobarbital was used as an internal standard. SPE procedure for extraction of the analyte from plasma sample was developed using Oasis HLB cartridges and subsequently eluate was injected into the HPLC system for analysis. Afterwards, SPE-HPLC method was subjected to validation. Linearity was obtained over the concentration range of 0.2–25 μg/mL for CBZ, CBZ-EP and carbamazepine trans-diol with correlation coefficients higher than 0.995. The method showed good intra-day and inter-day precision with relative standard deviation below 7.96%, while accuracy ranged from 92.09% to 108.5% for all the analytes. Absolute recovery was found to be from 87.39% to 104.04%. The method was successfully applied to analysis of plasma samples of epileptic patients in monotherapy and polytherapy.
5.2. Combination product
A simple and sensitive method has been developed by Bhatti et al. [83] and validated for the simultaneous determination of PHT, CBZ and CBZ-EP in human plasma by HPLC with 10,11-dihydrocarbamazepine as the internal standard. Acetonitrile was added to plasma samples containing PHT, CBZ and CBZ-EP to precipitate the plasma proteins. After centrifugation, the acetonitrile supernatant was transferred to a clean tube and evaporated under nitrogen gas. The dried sample extract was reconstituted in 0.4 mL of mobile phase and injected for analysis by HPLC. Separation was achieved on a Spherisorb ODS2 analytical column with a mobile phase of acetonitrile:methanol:potassium phosphate buffer (18:18:70, v/v/v). Detection was at 210 nm using an ultraviolet detector. The mean retention time of CBZ-EP, PHT and CBZ was 5.8, 9.9 and 11.8 min, respectively. Peak height ratios were fit to a least squares linear regression algorithm with a 1/(concentration)2 weighting. The method produces acceptable linearity, precision and accuracy to a minimum concentration of 0.05 µg/mL in human plasma. The method showed no observable matrix interferences.
Szabo et al. [84] described a method by which the heavily deuterated 2H10 analogs of carbamazepine (2H10 CBZ) and phenytoin (2H10 PHT) were chromatographically separated by HPLC from unlabeled CBZ and PHT. All compounds were quantitated against an internal standard, 10,11-dihydrocarbamazepine, and measured using conventional UV detection rather than mass spectrometry. Baseline resolution of extracted serum containing 2H10 CBZ, CBZ, 2H10 PHT, PHT and the internal standard achieved on a heated (55 °C) 25 cm×4.6 mm BioAnalytical Systems Phase II 5 μm ODS column with an isocratic mobile phase consisting of water–acetonitrile–tetrahydrofuran (80:16:4, v/v/v) at 1.2 mL/min. Eluting compounds were monitored at a UV wavelength of 214 nm. Calculated resolution of 2H10 CBZ from CBZ and of 2H10 PHT from PHT was 1.3. Serum standard curves were linear (R=0.999) over a range of 0.5–14 µg/mL for 2H10 CBZ, 0.5–20 µg/mL for CBZ, 0.5–20 µg/mL for 2H10 PHT, and 0.5–30 µg/mL for PHT. Within-day percent relative standard deviations (precision) were less than 6% in all cases.
An isocratic simple rapid assay has been developed and validated for the determination of CBZ in both solution form and rabbit plasma [85] using propylparaben as an internal standard. The assay was performed using a µ-Bondapak C18 (150 mm×4.6 mm) with a mobile phase consisting of methanol and water (50:50, v/v), at a flow rate of 1 mL/min and UV detection at 285 nm. The method was found to be specific for CBZ, and no interfering peaks were observed with an overall analytical run time of 15 min. Inter-day and intra-day accuracies reported as percent recovery were found to be 98.37%–100.45% and 97.53%–103.58%, respectively. Inter-day precision (reproducibility) was found to be 0.53%–2.75% RSD, while intra-day precision (repeatability) was found to be 1.06%–3.7% RSD for the samples studied. The calibration curve was found to be linear with a correlation coefficient (R2) of 0.999 over a concentration range of 0.5–40 µg/mL. The limit of quantitation was the lowest concentration. The method is simple and rapid and does not require any preliminary treatment of the sample.
According to literature survey, HPLC–MS methods were published for quantitative analysis of CBZ, its metabolites and other medications in human plasma. The HPLC–MS techniques are not widely used because this expensive equipment is not available in most clinical laboratories. Prior to HPLC–MS analysis, human plasma samples were subjected to protein precipitation, liquid–liquid extraction, stir bar-sorptive extraction or SPE. Unfortunately, the methods are time-consuming on account of eluate evaporation and subsequently reconstitution in comparison with the proposed SPE-HPLC method [86], [87], [88], [89], [90].
5.3. Formulation
An RP-HPLC method for the quantitative determination of CBZ in pure forms and in pharmaceutical preparations was developed and validated [91]. Good separation was achieved using Bondolone C18 column (150 mm×3.9 mm, 5 µm). RP-HPLC was carried out using a mobile phase of acetonitrile-Milli-Q grade water (30:70,v/v). The retention time was 8.2 min at a flow-rate of 1 mL/min. The injection volume was 10 µL and the peaks were detected at 220 nm. Linearity was obeyed in the range of 0.25–25 µg/mL. The correlation coefficient (r) obtained for regression equation was found to be 0.9995. Accuracy of the method was checked for six days at three concentration levels of 2.5, 7.5 and 15 mg/mL in six replicates. The precision of the RP-HPLC method was demonstrated by the relative standard derivation (RSD%) of lower than 5.88% for intra-day and 6.68% for inter-day. The limit of quantitation (LOQ) value of the RP-HPLC method was determined as 0.07 µg/mL. The LOD for CBZ determination was approximately 0.05 µg/mL. Recovery was found to be 98%–101%. The stability measurements were carried out for CBZ in solution at 4 °C and −20 °C using three sets of 2.5, 7.5 and 15 mg/mL concentrations. Developed method was successfully applied for assay of CBZ in pharmaceutical preparations.
Sensitive RP-HPLC and second derivative spectrophotometric methods for determination of CBZ in tablets have been developed [92]. In the HPLC method CBZ was separated using Phenomenex C18 column and acetonitrile:water (75:25, v/v) as mobile phase system. The speed of the mobile phase flowing was 1 mL/min and the detection was actualized at 285 nm. Enalapril was used as an internal standard. For the second derivative spectrophotometric method CBZ was determined by applying the techniques of the peak to peak amplitude. The assay was linear in the concentration range of 0.2–2.0 µg/mL for HPLC and 4.0–10.0 for second derivative spectrophotometric method. The detection limits of CBZ were 0.055 and 1.25 µg/mL for HPLC and second derivative spectrophotometric method, respectively. The recovery (%RSD) in HPLC and derivative spectrophotometric methods was (99.22±0.25)% and (99.05±0.25)%, respectively.
6. HPTLC
HPTLC is an enhanced form of thin layer chromatography (TLC). A number of enhancements can be made to the basic method of TLC to automate the different steps, to increase the resolution achieved, and to allow more accurate quantitative measurements [93]. The major advantage of HPTLC is its ability to analyze several samples simultaneously using a small quantity of mobile phase. This reduces time and cost of analysis. In addition, it minimizes exposure risks and significantly reduces disposal problems of toxic organic effluents, thereby reducing possibilities of environment pollution. HPTLC also facilitates repeated detection of chromatogram with the same or different parameters. The ability of HPTLC to analyze many samples in parallel has the advantage over other techniques in that the separation of 10 or 20 samples only takes the same amount of time as the separation of one sample. Furthermore, in case of HPTLC, there are no restrictions on the choice of solvents and mobile phases; drug and lipophilic excipients can be dissolved in a suitable solvent that would evaporate during spotting on TLC plate, leaving behind analyte as a thin band. Therefore, for such methods, extraction procedure is not always required and could be developed for analyzing drug without any interference from excipients [94].
A simple and rapid HPTLC method was developed and validated for quantitative determination of CBZ [95]. CBZ was chromatographed on silica gel 60 F254 TLC plate using ethyl acetate–toluene–methanol (5.0: 4.0: 1.0, v/v/v) as the mobile phase. CBZ was quantified by densitometric analysis at 285 nm. The method was found to give compact spots for the drug (Rf=0.47±0.01). The linear regression analysis data for the calibration plots showed good linear relationship with r2 of 0.9995 in the concentration range of 100–600 ng/spot. The minimum detectable amount was found to be 16.7 ng/spot, whereas LOQ was found to be 50.44 ng/spot. Statistical analysis of the data showed that the method is precise, accurate, reproducible and selective for the analysis of CBZ. The method was successfully employed for the estimation of equilibrium solubility, quantification of CBZ as a bulk drug, in commercially available preparation and in-house developed mucoadhesive microemulsion formulations and solution.
An instrumental planar chromatography method for quantitative analysis of CBZ in saliva [96] and in human serum [62] was developed. Chromatography was carried out on silica gel F254 HPTLC plates, previously washed in methanol and activated at 130 °C during 20 min. For the chromatographic development, ethyl acetate/toluene/methanol (5:4:1, v/v/v) was used. The length of development was 5 cm occurring in a period of 10 min. Horizontal development chambers were used. The method was linear between 0.5 and 15.0 ng/spot, with a regression coefficient of 0.999. The intra-assay variation (repeatability) was between 5.1% and 7.4%, and the inter-assay (reproducibility) was between 5.6% and 7.4%. The detection limit was 0.18 ng, and the quantification limit was 0.54 ng. The method proved accurate, with a recovery percentage of 109.8%, and it was selective for the active principle tested. It is a good method for the quantitative determination of CBZ in saliva.
7. UFLC
UFLC offers high-speed and separation even at normal pressure levels. By maximizing the performance of the column and the entire system, UFLC minimizes the analysis time and offers fast LC analysis.
A novel, accurate and precise RP-UFLC method for determination of OXCBZ has been developed and validated by Panda et al. [97]. Separation was achieved on an Enable C18G column (250 mm ×4.6 mm i.d., 5 μm) using acetonitrile: 10 mM tetra butyl ammonium hydrogen sulfate (60:40, v/v) as mobile phase at a flow rate of 1.0 mL/min and photodiode array detection at 254 nm. The retention time for OXCBZ was found to be 3.896 min. The run time was 6 min. Linearity was observed in the concentration range of 0.1–250 μg/mL with a regression coefficient of 0.999. The average recovery was in the range of 100.38%–105.5%. Forced degradation was performed by using HCl, NaOH, H2O2, thermal and UV radiation. The method was used successfully for the determination of OXCBZ in tablet dosage form.
8. MEKC
MEKC is a chromatography technique used in analytical chemistry. It is a modification of capillary electrophoresis (CE), where the samples are separated by differential partitioning between micelles (pseudo-stationary phase) and a surrounding aqueous buffer solution (mobile phase). Capillary electrophoresis has also been reported for the separation of CBZ and its metabolites [98] and monitoring the concentrations of CBZ and its metabolites in plasma [74], [99].
MEKC is based on the addition to the buffer solution of a micellar “pseudostationary” phase, which interacts with the analytes according to partitioning mechanisms, just like in a chromatographic method. The “pseudostationary” phase is composed of a surfactant added to the buffer solution in a concentration above its critical micellar concentration (CMC) [100].
A reliable MEKC method for the determination of CBZ and its two main metabolites, 10-hydroxycarbamazepine and 10,11-trans-dihydroxy-10,11-dihydroxycarbamazepine, in human plasma has been developed by Pucci et al. [101]. The separation and determination of the analytes was done using a system consisting of 60 mM sodium dodecylsulfate in phosphate buffer (30 mM, pH 8.0), to which 20% (v/v) methanol was added. Melatonine was used as an internal standard. Separation was carried out in an uncoated fused-silica capillary with a separation voltage of 25 kV and currents typically less than 40 µA. Spectrophotometric detection was at 205 nm. Isolation of OXCBZ and its metabolites from plasma was accomplished by an SPE procedure. The mean extraction yield of the analytes from plasma was higher than 94%. The linear correlation coefficients were better than 0.994 for all analytes. LOD was 0.05 µg/mL while LOQ was 0.15 µg/mL. The intraday repeatability parameter for the spiked blank plasma samples was lower than 1.9% RSD and the interday intermediate precision was lower than 2.1% RSD. The results obtained by analyzing real plasma samples from epileptic patients were found to be satisfactory in terms of precision, accuracy and detectability.
A rapid and feasible method for the analysis of CBZ and its five metabolites (10,11-dihydro-10,11-epoxycarbamazepine, 10,11-dihydro-10,11-dihydroxycarbamazepine, 10,11-dihydro-10-hydroxycarbamazepine, 2-hydroxycarbamazepine and 3-hydroxycarbamazepine) in human plasma has been developed by Raggi et al. [102]. Separation of the analytes was based on MEKC, in untreated fused-silica capillary (48.5/40.0 cm length, i.d. 50 µm) with phosphate buffer (30 mM, pH 8.0) as background electrolyte, containing 50 mM sodium dodecylsulfate and methanol (15%, v/v) as organic modifier. Clean up of human plasma samples was carried out by means of an SPE procedure, which gave a high extraction yield for all six carbamazepines (>88%). The mean extraction yield was 96.8% for CBZ and 93.7% for the metabolites. The overall precision of the method gave a mean RSD of about 1.8%. LOQ for all the analytes was ≤0.30 µg/mL while LOD was ≤0.12 µg/mL.
9. Overview
Bioanalytical and analytical methods require time-consuming and laborious extraction procedures or relatively large sample volumes (approx. 1 mL) as well as lengthy chromatographic run times, limiting their throughput capacity and sensitivity. The present review incorporates outline of various methods and techniques used in quantifying CBZ and its congeners using reports of analytical studies. Numbers of analytical techniques have been used with variables such as instruments, type of stationary phases, analytical columns and mobile phases. Numbers of combination of solvent used suits the analytical methods and tools. The review would help analytical chemists in knowing the key solvents and their combinations for their available set of instruments in the analytical laboratory. Analytical chemists can have the knowledge of advantage of one technique over another with the due comparisons mentioned in the published records. Apart from single drug profile records, the review also incorporates the records of comparative studies of one or more members of the same class of compounds. The following official methods for identification and qualitative and quantitative methods of estimations are prerequisites, but for multiple components studying the previous analytical records guide the proper selection of methods, instruments and solvents. The effective combination of parameters should minimize the cost of the analysis and reduce the time required for producing a reliable analytical method. The methods are also useful for determining parameters for in-process evaluation during the manufacturing of API.
Footnotes
Peer review under responsibility of Xi׳an Jiaotong University.
References
- 1.Liu L., Zheng T., Morris M.J. The mechanism of carbamazepine aggravation of absence seizures. J. Pharmacol. Exp. Ther. 2006;319:790–798. doi: 10.1124/jpet.106.104968. [DOI] [PubMed] [Google Scholar]
- 2.Silanpaa M., Haataja L., Tomson T. In: The Treatment of Epilepsy. third ed. Shorvon S., Perucca E., Engel J., editors. Blackwell Publishing Ltd.; Oxford: 2009. pp. 459–474. [Google Scholar]
- 3.Patsalos P.N., Berry D.J., Bourgeois B.F.D. Antiepileptic drugs – best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on the therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia. 2008;49:1239–1276. doi: 10.1111/j.1528-1167.2008.01561.x. [DOI] [PubMed] [Google Scholar]
- 4.Jung H., Milan R.C., Girard M.E. Bioequivalence study of carbamazepine tablets: in vitro/in vivo correlation. Int. J. Pharm. 1997;152:37–44. [Google Scholar]
- 5.Srinivasa R.K., Belorkar N. Development and validation of a specific stability indicating liquid chromatographic method for carbamazepine in bulk and pharmaceutical dosage forms. J. Adv. Pharm. Res. 2010;1:36–47. [Google Scholar]
- 6.Vogna D., Marotta R., Andreozzi R. Kinetic and chemical assessment of the UV/H2O2 treatment of antiepileptic drug carbamazepine. Chemosphere. 2004;54:497–505. doi: 10.1016/S0045-6535(03)00757-4. [DOI] [PubMed] [Google Scholar]
- 7.ICH Guidelines, Impurities in New Drug Substances Q3A (R2), October 25, 2006.
- 8.Ranise A., Benassi E., Besio G. Rapid gas chromatography method for the determination of carbamazepine and unrearranged carbamazepine-10,11-epoxide in human plasma. J. Chromatogr. 1981;222:120–124. doi: 10.1016/s0378-4347(00)81040-3. [DOI] [PubMed] [Google Scholar]
- 9.Cocks D.A., Dyer T.F., Edgar K. Simple and rapid gas-liquid chromatographic method for estimating carbamazepine in serum. J. Chromatogr. 1981;222:496–500. doi: 10.1016/s0378-4347(00)84154-7. [DOI] [PubMed] [Google Scholar]
- 10.Riva R., Albani F., Baruzzi A. Rapid quantitation of flurazepam and its major metabolite, N-desalkylflurazepam, in human plasma by gas–liquid chromatography with electron-capture detection. J. Chromatogr. 1981;222:491–495. doi: 10.1016/s0378-4347(00)84153-5. [DOI] [PubMed] [Google Scholar]
- 11.Kumps A., Mardens Y. Improved gas–liquid chromatographic method for the simultaneous determination of phenobarbital, phenytoin, carbamazepine and primidone in biological fluids. J. Chromatogr. 1980;182:116–120. doi: 10.1016/s0378-4347(00)81659-x. [DOI] [PubMed] [Google Scholar]
- 12.Chen K., Bashi H.K. Comparative analysis of antiepileptic drugs by gas chromatography using capillary or packed columns and by fluorescence polarization immunoassay. J. Anal. Toxicol. 1991;15:82–85. doi: 10.1093/jat/15.2.82. [DOI] [PubMed] [Google Scholar]
- 13.Rani S., Malik A.K. A novel microextraction by packed sorbent-gas chromatography procedure for the simultaneous analysis of antiepileptic drugs in human plasma and urine. J. Sep. Sci. 2012;35:2970–2977. doi: 10.1002/jssc.201200439. [DOI] [PubMed] [Google Scholar]
- 14.Speed D.J., Dickson S.J., Cairns E.R. Analysis of six anticonvulsant drugs using solid-phase extraction, deuterated internal standards, and gas chromatography–mass spectrometry. J. Anal. Toxicol. 2000;24:685–690. doi: 10.1093/jat/24.8.685. [DOI] [PubMed] [Google Scholar]
- 15.Cyr T.D., Matsui F., Sears R.W. Liquid chromatographic methods for assay of carbamazepine, 10,11-dihydrocarbamazepine, and related compounds in carbamazepine drug substance and tablets. J. Assoc. Off. Anal. Chem. 1987;70:836–840. [PubMed] [Google Scholar]
- 16.Rouan M.C., Campestrini J., Le Clanche V. Automated microanalysis of carbamazepine and its epoxide and trans-diol metabolites in plasma by column liquid chromatography. J. Chromatogr. 1992;573:65–68. doi: 10.1016/0378-4347(92)80475-6. [DOI] [PubMed] [Google Scholar]
- 17.He J., Shibukawa A., Nakagawa T. Direct injection analysis of carbamazepine and its active 10,11-epoxide metabolite in plasma by use of a semipermeable surface (SPS) silica column in LC. J. Pharm. Biomed. Anal. 1992;10:289–294. doi: 10.1016/0731-7085(92)80041-k. [DOI] [PubMed] [Google Scholar]
- 18.Heinig K., Henion J. Fast liquid chromatographic-mass spectrometric determination of pharmaceutical compounds. J. Chromatogr. B. 1999;732:445–458. doi: 10.1016/s0378-4347(99)00313-8. [DOI] [PubMed] [Google Scholar]
- 19.Abdel-Hamid M.E. Comparative LC–MS and HPLC analyses of selected antiepileptics and beta-blocking drugs. Farmaco. 2000;55:136–145. doi: 10.1016/s0014-827x(00)00006-9. [DOI] [PubMed] [Google Scholar]
- 20.Breton H., Cociglio M., Bressolle F. Liquid chromatography–electrospray mass spectrometry determination of carbamazepine, oxcarbazepine and eight of their metabolites in human plasma. J. Chromatogr. B. 2005;828:80–90. doi: 10.1016/j.jchromb.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 21.Sener E., Korkmaz O.T., Yeniceli D. Determination of CBZ and its main metabolite CBZ-10,11-epoxide in rat brain microdialysate and blood using ESI-LC-MS (ion trap) Chromatographia. 2007;66:S31–S36. [Google Scholar]
- 22.Stolker A.A.M., Niesing W., Hogendoorn E.A. Liquid chromatography with triple-quadrupole or quadrupole-time of flight mass spectrometry for screening and confirmation of residues of pharmaceuticals in water. Anal. Bioanal. Chem. 2004;378:955–963. doi: 10.1007/s00216-003-2253-y. [DOI] [PubMed] [Google Scholar]
- 23.Van Rooyen G.F., Badenhorst D., Swart K.J. Determination of carbamazepine and carbamazepine 10,11-epoxide in human plasma by tandem liquid chromatography–mass spectrometry with electrospray ionization, J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002;25:1–7. doi: 10.1016/s1570-0232(01)00590-6. [DOI] [PubMed] [Google Scholar]
- 24.Thomas S., Chandra S., Mathela T. Identification and structural elucidation of unknown impurity in carbamazepine active pharmaceutical ingredient by liquid chromatography–tandem mass spectrometry and semipreparative chromatographic isolation. J. Pharmaceut. Biomed. Anal. 2011;56:423–428. doi: 10.1016/j.jpba.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 25.Shrinubabu G., Ratnam B., Rao A. Development and validation of LC–MS/MS method for the quantification of oxcarbazepine in human plasma using an experimental design. Chem. Pharm. Bull. 2008;56:28–33. doi: 10.1248/cpb.56.28. [DOI] [PubMed] [Google Scholar]
- 26.Maggs J.L., Pirmohamed M., Kitteringham N.R. Characterization of the metabolites of carbamazepine in patient urine by liquid chromatography/mass spectrometry. Drug Metab. Dispos. 1997;25:275–280. [PubMed] [Google Scholar]
- 27.Zhu Y., Chiang H., Wulster M. Liquid chromatography/tandem mass spectrometry for the determination of carbamazepine and its metabolite in rat plasma utilizing an automated blood sampling system. J. Pharm. Biomed. Anal. 2005;38:119–125. doi: 10.1016/j.jpba.2004.11.058. [DOI] [PubMed] [Google Scholar]
- 28.Kima K., Kyung A., Sung E. Simple and accurate quantitative analysis of ten antiepileptic drugs in human. J. Pharm. Biomed. Anal. 2011;56:771–777. doi: 10.1016/j.jpba.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Loureiro A., Fernandes C., Wright L. Development and validation of an enantioselective liquid chromatography/tandem mass spectrometry method for the separation and quantification of eslicarbazepine acetate, eslicarbazepine, R-licarbazepine and oxcarbazepine in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011;879:2611–2618. doi: 10.1016/j.jchromb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 30.Miao X.S., Metcalfe C.D. Determination of carbamazepine and its metabolites in aqueous samples using liquid chromatography–electrospray tandem mass spectrometry. Anal. Chem. 2003;75:3731–3738. doi: 10.1021/ac030082k. [DOI] [PubMed] [Google Scholar]
- 31.Chelberg R.D., Gunawan S., Treiman D.M. Simultaneous high-performance liquid-chromatographic determination of carbamazepine and its principal metabolites in human plasma and urine. Ther. Drug Monit. 1988;10:188–193. doi: 10.1097/00007691-198802000-00013. [DOI] [PubMed] [Google Scholar]
- 32.Oh E., Ban E., Woo J.S. Analysis of carbamazepine and its active metabolite carbamazepine-10,11-epoxide, in human plasma using HPLC. Anal. Bioanal. Chem. 2006;386:1931–1936. doi: 10.1007/s00216-006-0724-7. [DOI] [PubMed] [Google Scholar]
- 33.Moreno J., Belmont A., Jaimes O. Pharmacokinetic study of carbamazepine and its carbamazepine 10,11-epoxide metabolite in a group of female epileptic patients under chronic treatment. Arch. Med. Res. 2004;35:168–171. doi: 10.1016/j.arcmed.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 34.Pienimaki P., Fuchs S., Isojarvi J. Improved detection and determination of carbamazepine and oxcarbazepine and their metabolites by high-performance liquid chromatography. J. Chromatogr. 1995;B 673:97–105. doi: 10.1016/0378-4347(95)00246-f. [DOI] [PubMed] [Google Scholar]
- 35.Miller R.B., Vranderick M. A validated HPLC method for the determination of carbamazepine and carbamazepine-10,11-epoxide in human plasma. J. Liq. Chromatogr. 1993;16:1249–1261. [Google Scholar]
- 36.Wad N.J. Simultaneous determination of eleven antiepileptic compounds in serum by high-performance liquid chromatography. J. Chromatogr. 1984;305:127–133. doi: 10.1016/s0378-4347(00)83320-4. [DOI] [PubMed] [Google Scholar]
- 37.Elizabeth G.S., Darla R.L., Mohamed A.V. Simultaneous determination of lamotrigine, zonisamide, and carbamazepine in human plasma by high-performance liquid chromatography. Biomed. Chromatogr. 2007;21:225–228. doi: 10.1002/bmc.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owen A., Tettey J.N., Morgan P. LC determination of carbamazepine in murine brain. J. Pharm. Biomed. Anal. 2001;26:573–577. doi: 10.1016/s0731-7085(01)00477-0. [DOI] [PubMed] [Google Scholar]
- 39.Scheyer R.D., During M.J., Cramer J.A. Simultaneous HPLC analysis of carbamazepine and carbamazepine epoxide in human brain microdialysate. J. Liq. Chromatogr. 1994;17:1567–1576. [Google Scholar]
- 40.Chan K. Simultaneous determination of carbamazepine and its epoxide metabolite in plasma and urine by high-performance liquid chromatography. J. Chromatogr. 1985;342:341–347. doi: 10.1016/s0378-4347(00)84525-9. [DOI] [PubMed] [Google Scholar]
- 41.Chollet D., Castella E., Combe P. High-speed liquid chromatographic method for the monitoring of carbamazepine and its active metabolite, carbamazepine- 10,11-epoxide, in human plasma. J. Chromatogr. B. 1996;683:237–243. doi: 10.1016/0378-4347(96)00116-8. [DOI] [PubMed] [Google Scholar]
- 42.Reith D.M., Cannell G.R. An HPLC assay for carbamazepine phase I metabolites and their glucuronides in urine. J. Liq. Chromatogr. Relat. Technol. 1999;22:1907–1918. [Google Scholar]
- 43.Dumortier G., Pons D., Zerrouk A. Concomitant HPLC method for determination of lamotrigine, carbamazepine, and 10,11-carbamazepine epoxide in plasma using dual UV 240–220 nm wavelength detection. J. Liq. Chromatogr. Relat. Technol. 2001;24:3171–3180. [Google Scholar]
- 44.MacKichan J.J. Simultaneous liquid chromatographic analysis for carbamazepine and carbamazepine 10,11-epoxide in plasma and saliva by use of double internal standardization. J. Chromatogr. 1980;180:373–383. doi: 10.1016/s0378-4347(00)81140-8. [DOI] [PubMed] [Google Scholar]
- 45.Raggi M.A., Casamenti G., Mandrioli R. A rapid LC method for the identification and determination of CNS drugs in pharmaceutical formulations. J. Pharm. Biomed. Anal. 2000;23:161–167. doi: 10.1016/s0731-7085(00)00265-x. [DOI] [PubMed] [Google Scholar]
- 46.Vermeij T.A., Edelbroek P.M. Robust isocratic HPLC method for simultaneous determination of seven antiepileptic drugs including lamotrigine, oxcarbamazepine and zonisamide in serum after SPE. J. Chromatogr. B. 2007;857:40–46. doi: 10.1016/j.jchromb.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 47.Franceschi L., Furlanut M. A simple method to monitor plasma concentrations of oxcarbazepine, carbamazepine, their main metabolites and lamotrigine in epileptic patients. Pharmacol. Res. 2005;51:297–302. doi: 10.1016/j.phrs.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 48.Bugamelli F., Sabbioni C., Mandrioli R. Simultaneous analysis of six antiepileptic drugs and two selected metabolites in human plasma by liquid chromatography after solid-phase extraction. Anal. Chim. Acta. 2002;472:1–10. [Google Scholar]
- 49.Fortuna A., Sousa J., Alves G. Development and validation of an HPLC-UV method for the simultaneous quantification of carbamazepine, oxcarbazepine, eslicarbazepine acetate and their main metabolites in human plasma. Anal. Bioanal. Chem. 2010;397:1605–1615. doi: 10.1007/s00216-010-3673-0. [DOI] [PubMed] [Google Scholar]
- 50.Mandrioli R., Ghedini N., Albani F. Liquid chromatographic determination of oxcarbazepine and its metabolites in plasma of epileptic patients after solid-phase extraction. J. Chromatogr. B. 2003;783:253–263. doi: 10.1016/s1570-0232(02)00664-5. [DOI] [PubMed] [Google Scholar]
- 51.Hartley R., Lucock M., Cookman J.R. High-performance liquid-chromatographic determination of carbamazepine and carbamazepine-10,11-epoxide in plasma and saliva following solid phase sample extraction. J. Chromatogr. 1986;380:347–356. doi: 10.1016/s0378-4347(00)83663-4. [DOI] [PubMed] [Google Scholar]
- 52.Mandrioli R., Albani F., Casamenti G. Simultaneous HPLC determination of carbamazepine and five of its metabolites in plasma of epileptic patients. J. Chromatogr. B. 2001;762:109–116. doi: 10.1016/s0378-4347(01)00328-0. [DOI] [PubMed] [Google Scholar]
- 53.Leite C.E., Petersen G.O., Lunardelli A. A HPLC method for the determination of carbamazepine and carbamazepine 10,11-epoxide and its comparison with chemiluminescent immunoassay. Clin. Chem. Lab. Med. 2009;47:458–463. doi: 10.1515/CCLM.2009.105. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida T., Imai K., Motohashi S. Simultaneous determination of zonisamide, carbamazepine and carbamazepine-10,11-epoxide in infant serum by HPLC. J. Pharm. Biomed. Anal. 2006;16:1386–1390. doi: 10.1016/j.jpba.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 55.Queiroz R.H.C., Bertucci C., Malfara W.R. Quantification of carbamazepine, carbamazepine-10,11-epoxide, phenytoin and phenobarbital in plasma samples by stir bar-sorptive extraction and liquid chromatography. J. Pharm. Biomed. Anal. 2008;48:428–434. doi: 10.1016/j.jpba.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 56.Ashy A.R., El Sayed Y.M., Islam S.I. Comparison of fluorescence polarization immunoassay and high performance liquid chromatography for the quantitative determination of phenytoin, phenobarbitone and carbamazepine in serum. J. Pharm. Pharmacol. 1986;38:572–577. doi: 10.1111/j.2042-7158.1986.tb03083.x. [DOI] [PubMed] [Google Scholar]
- 57.Sanchez A., Garcia R., Abadin J.A. Determination of free serum carbamazepine by protein precipitation with sulfosalicylic acid. Pharm. Pharmacol. Commun. 1999;5:435–438. [Google Scholar]
- 58.Lee S.H., Li M., Suh J.K. Determination of carbamazepine by chemiluminescence detection using chemically prepared tris(2,2-V-bipyridine)ruthenium(III) as oxidant. Anal. Sci. 2003;19:903–906. doi: 10.2116/analsci.19.903. [DOI] [PubMed] [Google Scholar]
- 59.Rezaei Z., Hemmateenejad B., Khabnadideh S. Simultaneous spectrophotometric determination of carbamazepine and phenytoin in serum by PLS regression and comparison Ruth HPLC. Talanta. 2005;65:21–28. doi: 10.1016/j.talanta.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 60.Huang C., He Q., Chen H. Flow injection photochemical spectrofluorimetry for the determination of carbamazepine in pharmaceutical preparations. J. Pharm. Biomed. Anal. 2002;30:59–65. doi: 10.1016/s0731-7085(02)00200-5. [DOI] [PubMed] [Google Scholar]
- 61.Auer M.E., Griesser U.J., Sawatzki J. Qualitative and quantitative study of polymorphic forms in drug formulations by near infrared FT-Raman spectroscopy. J. Mol. Struct. 2003;661–662:307–317. [Google Scholar]
- 62.Mennickent S., Fierro R., Vega M. Instrumental planar chromatographic method for determination of carbamazepine in human serum. J. Sep. Sci. 2009;32:1454–1458. doi: 10.1002/jssc.200800675. [DOI] [PubMed] [Google Scholar]
- 63.Budakova L., Brozmanova H., Grundmann M. Simultaneous determination of antiepileptic drugs and their two active metabolites by HPLC. J. Sep. Sci. 2008;31:1–8. doi: 10.1002/jssc.200700253. [DOI] [PubMed] [Google Scholar]
- 64.Szabo G.K., Browne T.R. Improved isocratic liquid-chromatographic simultaneous measurement of phenytoin, phenobarbital, primidone, carbamazepine, ethosuximide, and N-desmethylmethsuximide in serum. Clin. Chem. 1982;28:100–104. [PubMed] [Google Scholar]
- 65.Sawchuk R.J., Cartier L.L. Simultaneous liquid-chromatographic determination of carbamazepine and its epoxide metabolite in plasma. Clin. Chem. 1982;28:2127–2130. [PubMed] [Google Scholar]
- 66.Riad L.E., Sawchuk R.J. Simultaneous determination of carbamazepine and its epoxide and transdiol metabolites in plasma by microbore liquid chromatography. Clin. Chem. 1988;3:1863–1866. [PubMed] [Google Scholar]
- 67.Bonato P.S., Lanchote V.L., de Carvalho D. Measurement of carbamazepine and its main biotransformation products in plasma by HPLC. J. Anal. Toxicol. 1992;16:88–92. doi: 10.1093/jat/16.2.88. [DOI] [PubMed] [Google Scholar]
- 68.Meyler M., Kelly M.T., Smyth M.R. New method for the determination of four antiepileptic drugs in human plasma by high performance liquid chromatography. Chromatographia. 1993;36:27–32. [Google Scholar]
- 69.Romanyshyn L.A., Wichmann J.K., Kucharczyk N. Simultaneous determination of felbamate, primidone, phenobarbital, carbamazepine, two carbamazepine metabolites, phenytoin, and one phenytoin metabolite in human plasma by high-performance liquid chromatography. Ther. Drug Monit. 1994;16:90–99. doi: 10.1097/00007691-199402000-00015. [DOI] [PubMed] [Google Scholar]
- 70.Liu H., Delgado M., Iannaccone S.T. Determination of total and free carbamazepine and the principal metabolites in serum by high-performance liquid chromatography with photodiode-array detection. Ther. Drug Monit. 1993;15:317–327. doi: 10.1097/00007691-199308000-00010. [DOI] [PubMed] [Google Scholar]
- 71.Fedorova G.A., Baram G.I., Grachev M.A. Application of micro-column HPLC to the determination of phenobarbital and carbamazepine in human blood serum. Chromatographia. 2001;53:495–497. [Google Scholar]
- 72.Lensmeyer G.L., Gidal B.E., Wiebe D.A. Optimized high-performance liquid chromatographic method for determination of lamotrigine in serum with concomitant determination of phenytoin, carbamazepine, and carbamazepine epoxide. Ther. Drug Monit. 1997;19:292–300. doi: 10.1097/00007691-199706000-00009. [DOI] [PubMed] [Google Scholar]
- 73.Kabra P.M., Nelson M.A., Morton L.J. Simultaneous very fast liquid-chromatographic analysis of ethosuximide, primidone, phenobarbital, phenytoin and carbamazepine in serum. Clin. Chem. 1983;29:473–476. [PubMed] [Google Scholar]
- 74.Härtter S., Jensen B., Hiemke C. Micellar electrokinetic capillary chromatography for therapeutic drug monitoring of carbamazepine and its main metabolites. J. Chromatogr. B Biomed. Sci. Appl. 1998;712:253–258. doi: 10.1016/s0378-4347(98)00169-8. [DOI] [PubMed] [Google Scholar]
- 75.Lancas F.M., Sozza M.A., Queiroz M.E.C. Simultaneous plasma lamotrignine analysis with carbamazepine, carbamazepine-10,11-epoxide, primidone, phenytoin, phenobarbital, and PEMA by micellar electrokinetic capillary chromatography (MECC) J. Anal. Toxicol. 2003;27:304–308. doi: 10.1093/jat/27.5.304. [DOI] [PubMed] [Google Scholar]
- 76.Nimura N., Itoh H., Kinoshita T. Diol-bonded silica gel as a restricted access packing forming a binary-layered phase for direct injection of serum for the determination of drugs. J. Chromatogr. A. 1995;689:203–210. doi: 10.1016/0021-9673(94)00896-h. [DOI] [PubMed] [Google Scholar]
- 77.Hermansson J., Grahn A. Determination of drugs by direct injection of plasma into a biocompatible extraction column based on a protein-entrapped hydrophobic phase. J. Chromatogr A. 1994;660:119–129. doi: 10.1016/0021-9673(94)85105-0. [DOI] [PubMed] [Google Scholar]
- 78.Juergens U. Pre-column switching techniques for the determination of drugs and metabolites in body fluids in research and routine analysis. Int. J. Environ. Anal. Chem. 1986;25:221–233. doi: 10.1080/03067318608077090. [DOI] [PubMed] [Google Scholar]
- 79.Matar K.M., Nicholls P.J., Tekle A. Liquid chromatographic determination of six antiepileptic drugs and two metabolites in microsamples of human plasma. Ther. Drug Monit. 1999;21:559–566. doi: 10.1097/00007691-199910000-00013. [DOI] [PubMed] [Google Scholar]
- 80.Brunetto M., Obando M., Fernández A. Column-switching high-performance liquid chromatographic analysis of carbamazepine and its principal metabolite in human plasma with direct sample injection using an alkyl-diol silica (ADS) precolumn. Talanta. 2002;58:535–542. doi: 10.1016/s0039-9140(02)00326-0. [DOI] [PubMed] [Google Scholar]
- 81.Tonic-Ribarska J., Haxhiu A., Sterjev Z. Development and validation of a bioanalytical LC-UV method with solid-phase extraction for determination of valproic acid in saliva. Acta Pharm. 2012;62:211–220. doi: 10.2478/v10007-012-0015-0. [DOI] [PubMed] [Google Scholar]
- 82.Predrag D., Ljiljana Z., Ana P. Development and validation of SPE-HPLC method for the determination of carbamazepine and its metabolites carbamazepine epoxide and carbamazepine trans-diol in plasma. J. Serb. Chem. Soc. 2012;77:1–23. [Google Scholar]
- 83.Bhatti M.M., Hanson G.D., Schultz L. Simultaneous determination of phenytoin, carbamazepine, and 10,11-carbamazepine epoxide in human plasma by high-performance liquid chromatography with ultraviolet detection. J. Pharm. Biomed. Anal. 1998;16:1233–1240. doi: 10.1016/s0731-7085(97)00265-3. [DOI] [PubMed] [Google Scholar]
- 84.Szabo G.K., Pylilo R.J., Perchalski R.J. Simultaneous separation and determination (in serum) of phenytoin and carbamazepine and their deuterated analogues by high-performance liquid chromatography-ultraviolet detection for tracer studies. J. Chromatogr. 1990;535:271–277. doi: 10.1016/s0021-9673(01)88952-4. [DOI] [PubMed] [Google Scholar]
- 85.Hammam A.M., Fars K.A., Gamal M. Development and validation of an HPLC–UV method for the quantification of carbamazepine in rabbit plasma. Saudi Pharm. J. 2012;20:29–34. doi: 10.1016/j.jsps.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Rooyen G.F., Badenhorst D., Swart K.J. Determination of carbamazepine and carbamazepine 10,11-epoxide in human plasma by tandem liquid chromatography-mass spectrometry with electrospray ionization. J. Chromatogr. B. 2002;769:1–7. doi: 10.1016/s1570-0232(01)00590-6. [DOI] [PubMed] [Google Scholar]
- 87.Ates Z., Özden T., Özilhan S. Simultaneous determination of carbamazepine and its active metabolite carbamazepine-10,11-epoxide in human plasma by UPLC. Chromatographia. 2007;66:S123. [Google Scholar]
- 88.Ma C.L., Jiao Z., Jie Y. Isocratic reversed-phase HPLC for simultaneous separation and determination of seven antiepileptic drugs and two of their active metabolites in human plasma. Chromatographia. 2007;65:267–269. [Google Scholar]
- 89.Heideloff C., Bunch D.R., Wang S. A novel HPLC method for quantification of 10 antiepileptic drugs or metabolites in serum/plasma using a monolithic column. Ther. Drug Monit. 2010;32:102–106. doi: 10.1097/FTD.0b013e3181c324c8. [DOI] [PubMed] [Google Scholar]
- 90.Subramanian M., Birnbaum A.K., Remmel R.P. High-speed simultaneous determination of nine antiepileptic drugs using liquid chromatography-mass spectrometry. Ther. Drug Monit. 2008;30:347–351. doi: 10.1097/FTD.0b013e3181678ecb. [DOI] [PubMed] [Google Scholar]
- 91.Demirkaya F., Kadioglu Y. Determination of carbamazepine using RP-HPLC method in pharmaceutical preparations. J. Pharm. Sci. 2005;30:78–82. [Google Scholar]
- 92.Tatar S. Determination of carbamazepine in pharmaceutical preparation using high performance liquid chromatography and derivative spectrophotometry. Turkish J. Pharm. Sct. 2006;3:123–139. [Google Scholar]
- 93.D.H. Shewiyoa, E. Kaaleb, P.G. Rishab, et al., HPTLC methods to assay active ingredients in pharmaceutical formulations: A review of the method development and validation steps, J. Pharm. Biomed. Anal. 66 (2012) 11–23. [DOI] [PubMed]
- 94.Patel R.B., Patel A.B., Patel M.R. Estimation of alprazolam and sertraline in pure powder and tablet formulations by high-performance liquid chromatography and high-performance thin-layer chromatography. Anal. Lett. 2009;42:1588–1602. [Google Scholar]
- 95.Patel R.B., Patel M.R., Bhatt K.K. Development and validation of HPTLC method for estimation of carbamazepine in formulations and its in vitro release study. Chromatogr. Res. Int. 2011 [Google Scholar]
- 96.Mennickent S., Vega M., Godoy C. Development and validation of a method using instrumental planner chromatography for quantitative analysis of carbamazepine in saliva. J. Chil. Chem. Soc. 2003;48:3–7. [Google Scholar]
- 97.Panda S., Ravi Kumar V., Panda R. Development and validation of a Stability-indicating RP-UFLC method for determination of oxcarbazepine in tablet dosage form. J. Adv. Pharm. Res. 2012;3:77–84. [Google Scholar]
- 98.Izzo G., Raggi M.A., Maichel B. Separation of olanzapine, carbamazepine and their main metabolites by capillary electrophoresis with pseudo-stationary phases. J. Chromatogr. B. 2001;752:47–53. doi: 10.1016/s0378-4347(00)00514-4. [DOI] [PubMed] [Google Scholar]
- 99.Thormann W., Theurillat R., Wind M. Therapeutic drug monitoring of antiepileptics by capillary electrophoresis. Characterization of assays via analysis of quality control sera containing 14 analytes. J. Chromatogr. A. 2001;924:429–437. doi: 10.1016/s0021-9673(01)00854-8. [DOI] [PubMed] [Google Scholar]
- 100.Gabriel H., Brigitta S., Aura R. Principles of micellar electrokinetic capillary chromatography applied in pharmaceutical analysis. Adv. Pharm. Bull. 2013;3:1–8. doi: 10.5681/apb.2013.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pucci V., Kenndler E., Raggi M. Quantitation of oxcarbazepine and its metabolites in human plasma by micellar electrokinetic chromatograph. Biomed. Chromatogr. 2003;17:231–238. doi: 10.1002/bmc.217. [DOI] [PubMed] [Google Scholar]
- 102.Raggi M., Pucci V., Maurizio A. Separation of carbamazepine and five metabolites, and analysis in human plasma by micellar electrokinetic capillary chromatography. J. Chromatogr. B. 2002;770:217–225. doi: 10.1016/s0378-4347(01)00524-2. [DOI] [PubMed] [Google Scholar]