

Summary
We report the case of a 55-year-old woman who presented to the emergency department having woken from sleep with right sided thigh swelling. Pelvic radiographs revealed bilateral atypical subtrochanteric femoral fractures (ASFFs). In the two years leading up to this admission, the patient had experienced gradually increasing pain and weakness in her legs which had resulted in a decrease in her mobility from fully mobile to bed-bound. During this time a neurologist had organised a magnetic-resonance imaging (MRI) scan of the brain and spine which was normal. There was no history of bisphosphonate (BP) use. Historical and admission blood tests revealed a persistently low serum alkaline phosphatase (ALP), with all other results within normal limits. The patient was treated with intramedullary nailing of both femurs and histological analysis of bone reamings were characteristic of hypophosphatasia (HPP). The patient was independently mobilising with a walking frame on discharge. Subsequent genetic testing revealed bi-allelic pathogenic variants in the TNSALP gene: c.526G>A, p.(Ala176Thr) and c.1171C>T, p.(Arg391Cys).
HPP is an inborn error in metabolism caused by mutation in the gene coding for tissue non-specific alkaline phosphatase (TNSALP), resulting in a decrease in serum ALP concentrations. The age at which it presents which can vary from childhood to middle age, with symptoms ranging from perinatal death to late-onset osteomalacia. In those patients who survive to adulthood, there is a predisposition to fractures, including ASFFs. Treatment with asfotase alfa (a bone-targeted, recombinant human TNSALP) has been approved for perinatal, infantile and paediatric-onset hypophosphatasia.
This case emphasises the importance of viewing persistent low ALP as a ‘red flag’ in patients presenting with musculoskeletal symptoms. Timely diagnosis and treatment of HPP can reduce the risk of serious complications, such as those experienced by this patient.
Keywords: hypophosphatasia, genetics, bone turnover, atypical fracture
Introduction
Hypophosphatasia (HPP) is an inborn error in metabolism that is caused by mutation in the gene coding for tissue nonspecific alkaline phosphatase (TNSALP), resulting in a decrease in serum ALP concentrations (1, 2). The age at which it presents which can vary from early childhood to middle age (3), with symptoms ranging from perinatal death to late-onset osteomalacia. The loss-of-function mutation in HPP results in the accumulation of TNSALP substrates outside of the cell, including inorganic pyrophosphate which inhibits bone mineralisation (4). In those patients who survive to adulthood, there is a subsequent predisposition to fractures, including atypical subtrochanteric femoral fractures (ASFFs) (5). These are defined as any fracture meeting four out of five of the major criteria described by the American Society for Bone and Mineral Research (ASBMR) task force in 2013 (Table 1) (7). They are most commonly associated with the use of bisphosphonates, which are thought to have adverse effects on bone biology in a small proportion of patients (8, 9). Outcomes following these fractures are generally poor with 25% patients surviving less than two years and greater than half of all patients failing to return to their previous level of functioning in this time (10).
Table 1.
Atypical femoral fracture: major and minor features.
| Major features |
|---|
|
| Minor features |
|
We discuss a 55-year-old lady who woke who noticed swelling and deforming in her right thigh when waking from sleep. Radiographs revealed bilateral ASFFs, which together with low serum ALP led to a diagnosis of adult-onset HPP which was later confirmed from bone histology and genetic testing.
Case report
A 55-year-old Caucasian woman presented to the emergency department in August 2016 having woken that morning with painless right sided thigh swelling in the context of increasing pain and weakness in the lower limbs over the preceding two years. This began with an awareness of a worsening ache in the right leg and by one year prior to admission she was unable to walk more than a short distances. Six months prior to admission she required the use of a single crutch due to a feeling of unsteadiness and following this fell from standing onto her right side. Two further falls from standing occurred six weeks prior to admission with no injury sustained. The patient’s mobility continued to decline and the patient was bed bound in the two weeks prior to admission.
In March 2016 she had been investigated by a neurologist for her symptoms however following normal magnetic resonance imaging (MRI) scans of the brain and spinal cord and normal cerebrospinal fluid (CSF) analysis (including electrophoresis) no cause for the patient’s symptoms could be identified. She had subsequently been referred to a physiotherapy service. Other medical problems included well-controlled bronchiectasis, bilateral carpal tunnel syndrome, chronic kidney disease stage 1 following pre-eclampsia, frequent cystitis, trigeminal neuralgia and juvenile tooth loss. There was no history of osteoporosis or rickets and menopause had been at the age of 53. There was no history of bisphosphonate therapy.
A plain-film anterior-posterior (AP) radiograph of the pelvis was organised which revealed a shortened, laterally angulated atypical subtrochanteric femoral fracture (ASFF) on the right side with endosteal thickening visible in the subtrochanteric region of the left femur (Figure 1 a, b). A subsequent radiograph of the left femur revealed an incomplete ASFF involving the lateral cortex only (Figure 1 c).
Figure 1 a–c.

Pelvic radiograph showing bilateral atypical subtrochanteric femoral fractures.
Admission blood tests revealed a low alkaline phosphatase (ALP) of 5 U/L (44–147 NI), with remaining blood tests all within normal limits.
The patient was consented for intramedullary fixation of the right sided ASFF and taken to the operating room (OR) the following day. The procedure went as planned, with reamings taken during the procedure sent for histological analysis. The patient made a good post-operative recovery and underwent intramedullary nailing of the contralateral femur four days later. Reamings showed deposited nuclear/cellular material at the osteoid - bone interface in keeping with hypophosphatasia (Figure 2 a, b).
Figure 2 a, b.

Post operative radiographs of both femora following intramedullary nailing.
The patient continued to recover and on discharge seventeen days after admission was mobilising with a walking frame. A skeletal survey was performed at orthopaedic follow-up which revealed further fractures in the right tibia, right 4th and 5th metatarsals and sclerosis of the left 5th metatarsal (Figures 3, 4 a, b). The tibial fracture was treated with intramedullary nailing, with the remaining fractures treated non-operatively and monitored with serial radiographs which showed gradual improvement to follow-up in June 2017 (Figure 5).
Figure 3.
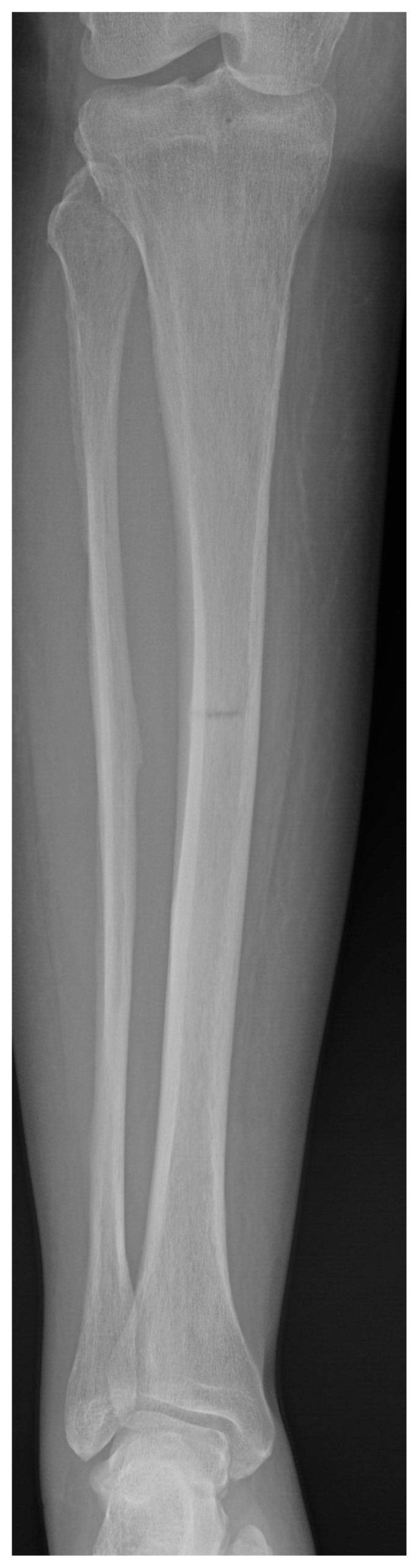
Radiograph showing incomplete right tibial fracture.
Figure 4 a, b.

Radiographs showing right 4th and 5th metatarsal fractures and sclerosis of the left 5th metatarsal.
Figure 5.

Post-operative radiograph of the right tibia following intramedullary nailing.
Follow-up with a clinical geneticist was arranged for October 2016 and mutation analysis revealed bi-allelic pathogenic variants in TNSALP gene. Subsequent testing of the patient’s two children revealed that each had inherited only of the two one variant alleles.
The patient did not meet the criteria for treatment with asfotase alfa (a bone-targeted, recombinant human TNSALP) which is only currently approved for use in perinatal, infantile and paediatric-onset HPP [although these treatments are not available on the National Health Service (NHS) in the UK]. The patient was mobilising full weight bearing with crutches for support.
Materials and methods
Serum biochemistry
Informed verbal consent was obtained from the patient for blood sampling in the emergency department in August 2016 using the Sarstedt Monovette blood collection system (Sarstedt AG & Co, Numbrecht, Germany). Analysis was performed using the Siemens Dimension RxL Integrated Chemistry System (Siemens Healthcare, Erlangen, Germany).
Radiographs
Informed verbal consent was obtained from the patient at admission and post-operatively for radiographs of the pelvis and both femurs. Images were analysed using the Centricity Enterprise Web version 3.0 Picture Archiving and Communication System (General Electric, Boston, Massachusetts, USA).
Bone histological analysis
Reaming of the medullary canal was performed intraoperatively in keeping with the standard protocol for intramedullary fixation of the femur using an intramedullary nail. Samples obtained from the reamer were deposited into 70% ethanol for transport to the laboratory, where they were embedded in 5ppm hydroquinone hydrophilic acrylic resin prior to sectioning. Samples were treated with toluidine blue and von Kossa stains prior to light microscopy.
Mutation analysis
Next Generation Sequencing (NGS) of the coding region of the TNSALP gene was performed using the Illumina TruSight One sequencing panel (Illumina, San Diego, California, USA). Variants detected by this method were confirmed using Sanger sequencing.
Results
Serum biochemistry results on admission revealed a low ALP activity of 5 U/L (44–147 NI). Inspection of historical blood test results revealed an ALP of 9 U/L (44–147 NI) from samples taken by the patient’s general practitioner six month’s previously. Remaining liver function tests, full blood count, urea & electrolytes and coagulation were within normal limits on admission. Secondary screening blood tests for osteoporosis, including vitamin D, folate, calcium, vitamin B12, thyroid function and erythrocyte sedimentation rate (ESR) were all within normal limits.
Histological analysis of bone samples using Von Kossa stain revealed mineralised bone with wide osteoid seams and scalloping of cement lines. Further analysis with toluidine blue stain revealed deposited nuclear/cellular material at the osteoid - bone interface in keeping with hypophosphatasia.
Mutation analysis revealed two heterozygous single nucleotide polymorphisms in the TNSALP gene: c.526G>A, p.(Ala176Thr) and c.1171C>T, p.(Arg391Cys).
Discussion
Hypophosphatasia (HPP) is an inborn error in metabolism that caused by loss-of-function mutations in the gene coding for tissue non-specific alkaline phosphatase (TNSALP) located on chromosome 1p36.1–34 (1, 2, 11). Over 300 mutations have been identified to date which result in a variable loss of function in the enzyme and a consequent decrease in serum ALP concentrations (1, 2, 6). TNSALP substrates including inorganic phosphate accumulate in the extracellular space, inhibiting bone mineralisation (4). The age at which it presents which can vary from early childhood to middle age, with symptoms ranging from perinatal death to late-onset osteomalacia (3). Adult-onset HPP can be a consequence of both autosomal dominant and autosomal recessive inheritance of TNSALP mutations, and has characteristically variable penetrance and severity (12).
The single nucleotide polymorphisms [SNPs] identified in this patient’s TNSALP gene have been previously identified as causing HPP. The SNP c.526G>A, p.(Ala176Thr) is known to result in approximately 70% loss of enzyme function and is associated with mild hypophosphatasia (12). The SNP c.1171C>T, p.(Arg391Cys) has been previously reported in cases of childhood-onset HPP and is known to result in approximately 90% loss of enzyme activity (13–15).
This patient presented with bilateral atypical subtrochanteric femoral fractures (ASFFs). This fracture type was given an official definition in 2009 (later updated in 2013) by the American Society for Bone and Mineral Research (ASBMR) as any fracture meeting four of five major features (Table 1) (9). There have been several cases of bilateral ASFFs documented in the literature, however the vast majority of these are associated with bisphosphonate use, which is thought to contribute to the accumulation of bone microdamage and cause dysregulation of bone mineralisation, bone turnover, collagen cross-linking and bone vascularity in some patients (8, 16–18). Whilst this case represents an unusual injury pattern for a patient with HPP, approximately 18% of the adult-onset form of disease is diagnosed following a fracture, with 23% of all patients having sustained a femoral fracture at some point in their life (19).
Prior to presentation at our institution, this patient had been extensively investigated for musculoskeletal symptoms. Included in this diagnostic workup were three sets of blood tests in the six months leading up to her admission which revealed persistently low serum ALP of between 9–11 U/L. This emphasises the importance of viewing persistent low ALP as a ‘red flag’ in patients presenting with musculoskeletal symptoms. Timely diagnosis and treatment of HPP can reduce the risk of serious complications, such as those experienced by this patient.
There is currently no approved treatment available for adult-onset HPP, though there is some evidence to support the use of teraparatide in this cohort (4, 20, 21). The use of bisphosphonates in this disease is not advised, as this treatment can exacerbate the already dysfunctional bone turnover in HPP (22, 23). Treatment with asfotase alfa (a bone-targeted, recombinant human TNSALP) has been shown to improve survival in perinatal and infantile HPP and this drug has been approved for the treatment of these variants of the disease [though is not currently available on the UK National Health Service (NHS) (24)].
Acknowledgements
We thank Dr. Mardhavi Vindlacheruvu, Dr. Kenneth Poole, Dr. Soo-Mi Park, Mr. Ross Coomber, Mr. Philip Johnston and Mr. Alan Norrish for their expert input in the care of this patient.
Authors’ roles: DS and JEL admitted the patient from the emergency department and arranged initial investigations. AC was the supervising clinician. JEL, DS and JB performed the literature search and compiled the manuscript. AC edited the manuscript.
Footnotes
Disclosures
The Authors report no conflicts of interest. There are no grant supporters for this manuscript.
References
- 1.Whyte MP. Atypical femoral fractures, bisphosphonates, and adult hypophosphatasia. J Bone Miner Res. 2009;24(6):1132–4. doi: 10.1359/jbmr.081253. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ML. Hypophosphatasia: an overview of the disease and its treatment. Osteoporos Int. 2015;26(12):2743–57. doi: 10.1007/s00198-015-3272-1. [DOI] [PubMed] [Google Scholar]
- 3.Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54(1):21–7. doi: 10.1016/j.bone.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 4.Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016;12(4):233–46. doi: 10.1038/nrendo.2016.14. [DOI] [PubMed] [Google Scholar]
- 5.McKiernan FE, Berg RL, Fuehrer J. Clinical and radiographic findings in adults with persistent hypophosphatasemia. J Bone Miner Res. 2014;29(7):1651–60. doi: 10.1002/jbmr.2178. [DOI] [PubMed] [Google Scholar]
- 6.Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366(10):904–13. doi: 10.1056/NEJMoa1106173. [DOI] [PubMed] [Google Scholar]
- 7.Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster DW, Ebeling PR. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29(1):1–23. doi: 10.1002/jbmr.1998. [DOI] [PubMed] [Google Scholar]
- 8.Kharwadkar N, Mayne B, Lawrence JE, Khanduja V. Bisphosphonates and atypical subtrochanteric fractures of the femur. Bone Joint Res. 2017 Mar;6(3):144–153. doi: 10.1302/2046-3758.63.BJR-2016-0125.R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster DW, Ebeling PR, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O’Keefe R, Papapoulos S, Howe TS, van der Meulen MC, Weinstein RS, Whyte MP. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014 Jan;29(1):1–23. doi: 10.1002/jbmr.1998. [DOI] [PubMed] [Google Scholar]
- 10.Ekstrom W, Nemeth G, Samnegard E, Dalen N, Tidermark J. Quality of life after a subtrochanteric fracture: a prospective cohort study on 87 elderly patients. Injury. 2009;40:371–376. doi: 10.1016/j.injury.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg CR, Evans JA, McKendry-Smith S, Redekopp S, Haworth JC, Mulivor R, Chodirker BN. Infantile hypophosphatasia: localization within chromosome region 1p36. 1–34 and prenatal diagnosis using linked DNA markers. Am J Hum Genet. 1990 Feb;46(2):286. [PMC free article] [PubMed] [Google Scholar]
- 12.Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, De Mazancourt P, Mornet E. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet. 2009 Jun 6;10(1):51. doi: 10.1186/1471-2350-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orimo H, Girschick HJ, Goseki-Sone M, Ito M, Oda K, Shimada T. Mutational analysis and functional correlation with phenotype in German patients with childhood-type hypophosphatasia. J Bone Miner Res. 2001;16:2313–2319. doi: 10.1359/jbmr.2001.16.12.2313. [DOI] [PubMed] [Google Scholar]
- 14.Girschick HJ, Mornet E, Beer M, Warmuth-Metz M, Schneider P. Chronic multifocal non-bacterial osteomyelitis in hypophosphatasia mimicking malignancy. BMC Pediatr. 2007 Jan 23;7(1):3. doi: 10.1186/1471-2431-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Utsch B, Brun-Heath I, Staatz G, Gravou-Apostolatou C, Karle S, Jacobs U, Ludwig M, Zenker M, Dörr HG, Rascher W, Mornet E. Infantile Hypophosphatasia due to a New Compound Heterozygous TNSALP Mutation-Functional Evidence for a Hydrophobic Side-Chain? Exp Clin Endocrinol Diabetes. 2009 Jan;117(01):28–33. doi: 10.1055/s-2008-1073157. [DOI] [PubMed] [Google Scholar]
- 16.Higgins M, Morgan-John S, Badhe S. Simultaneous, bilateral, complete atypical femoral fractures after long-term alendronate use. J Orthop. 2016 Dec 31;13(4):401–3. doi: 10.1016/j.jor.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo N, Yoda T, Fujisawa J, Arai K, Sakuma M, Ninomiya H, Sano H, Endo N. Bilateral atypical femoral subtrochanteric fractures in a premenopausal patient receiving prolonged bisphosphonate therapy: evidence of severely suppressed bone turnover. Clin Cases Miner Bone Metab. 2015 Sep;12(3):273. doi: 10.11138/ccmbm/2015.12.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JK. Bilateral atypical femoral diaphyseal fractures in a patient treated with alendronate sodium. Int J Rheum Dis. 2009 Jul 1;12(2):149–54. doi: 10.1111/j.1756-185X.2009.01396.x. [DOI] [PubMed] [Google Scholar]
- 19.Berkseth KE, Tebben PJ, Drake MT, Hefferan TE, Jewison DE, Wermers RA. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013 May 31;54(1):21–7. doi: 10.1016/j.bone.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 20.Camacho PM, Mazhari AM, Wilczynski C, Kadanoff R, Mumm S, Whyte MP. Adult hypophosphatasia treated with teriparatide: report of 2 patients and review of the literature. Endocr Pract. 2016 Apr 4;22(8):941–50. doi: 10.4158/EP15890.OR. [DOI] [PubMed] [Google Scholar]
- 21.Whyte MP, Mumm S, Deal C. Adult hypophosphatasia treated with teriparatide. J Clin Endocrinol Metab. 2007 Apr 1;92(4):1203–8. doi: 10.1210/jc.2006-1902. [DOI] [PubMed] [Google Scholar]
- 22.Cundy T, Michigami T, Tachikawa K, Dray M, Collins JF, Paschalis EP, Gamsjaeger S, Roschger A, Fratzl-Zelman N, Roschger P, Klaushofer K. Reversible deterioration in hypophosphatasia caused by renal failure with bisphosphonate treatment. J Bone Miner Res. 2015 Sep 1;30(9):1726–37. doi: 10.1002/jbmr.2495. [DOI] [PubMed] [Google Scholar]
- 23.Sutton RA, Mumm S, Coburn SP, Ericson KL, Whyte MP. “Atypical femoral fractures” during bisphosphonate exposure in adult hypophosphatasia. J Bone Miner Res. 2012 May 1;27(5):987–94. doi: 10.1002/jbmr.1565. [DOI] [PubMed] [Google Scholar]
- 24.Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, Thompson DD, Bishop N, Hofmann C. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016 Jan 1;101(1):334–42. doi: 10.1210/jc.2015-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]