

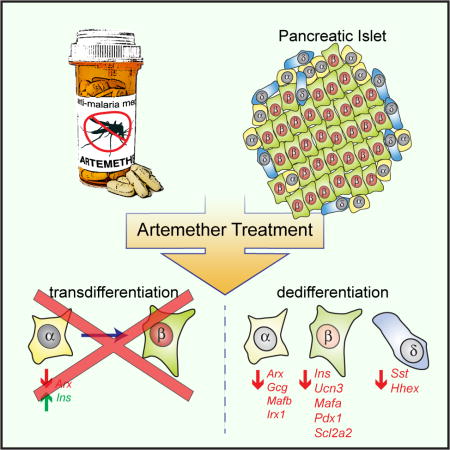
Summary
Pancreatic alpha cells retain considerable plasticity and can – under the right circumstances – transdifferentiate into functionally mature beta cells. In search of a targetable mechanistic basis, a recent paper suggested that the widely used antimalarial drug artemether suppresses the alpha cell transcription factor Arx to promote transdifferentiation into beta cells. However, key initial experiments in this paper were carried out in islet cell lines and most subsequent validation experiments implied transdifferentiation without direct demonstration of alpha to beta cell conversion. Indeed, we find no evidence that artemether promotes transdifferentiation of primary alpha cells into beta cells. Moreover, artemether reduces Ins2 expression in primary beta cells >100-fold, suppresses glucose uptake, and abrogates beta cell calcium responses and insulin secretion in response to glucose. Our observations suggest that artemether induces general islet endocrine cell dedifferentiation and call into question the utility of artemisinins to promote alpha to beta cell transdifferentiation in treating diabetes.
Keywords: artemether, artemisinins, transdifferentiation, dedifferentiation, maturation, diabetes, GCaMP6, beta cell, alpha cell, pancreatic islet, neogenesis
eTOC Blurb
The antimalaria drug artemether has been recently shown to promote transdifferentiation of alpha cells into beta cells. Van der Meulen et al. now report loss of beta cell gene expression, glucose uptake, calcium responses and insulin secretion following stimulation in intact islets treated with a high dose of artemether.
Introduction
Type 1 and Type 2 diabetes are fundamentally different diseases, yet both are associated with a deficiency in functional beta cells. This has generated intense interest in any strategy that could regenerate functional beta cells towards a cure for diabetes. Self-replication has long been viewed as the principal mechanism of beta cell maintenance and regeneration. However, beta cell replication declines rapidly with age and no methods to promote clinically meaningful restoration of beta cell mass via proliferation have been achieved to date. Recent years have seen a surge of interest into the innate plasticity of islet endocrine cells. Following near-complete ablation of beta cells, functional beta cell mass partially recovers by transdifferentiation from alpha or delta cells, depending on the age at which beta cell ablation occurred (Chera et al., 2014; Thorel et al., 2010). Conversely, beta cells in both types of diabetes dedifferentiate into a non-functional state that no longer contributes to the functional beta cell pool, but may escape death by autoimmune destruction or exhaustion (Rui et al., 2017; Talchai et al., 2012). These observations have changed the perspective on pancreatic islets that were traditionally considered to consist of terminally differentiated cells. It is now apparent that islet endocrine cells maintain considerable plasticity and can change fate from one cell type to another. In further support of such plasticity, we demonstrated that the periphery of healthy, adult mouse islets constitutes a privileged ‘neogenic niche’ that supports the conversion of alpha cells into functionally mature beta cells (van der Meulen et al., 2017).
The notion that beta cells can be generated via transdifferentiation of alpha cells has spurred considerable interest into the underlying mechanisms that establish and control islet cell fate that could therefore be targeted to turn alpha cells into beta cells. Understanding these mechanisms to target the transdifferentiation of alpha to beta cells would serve a dual purpose by not only regenerating beta cell mass, but simultaneously reducing the number of alpha cells, which are complicit in diabetic hyperglycemia via excess glucagon secretion (Unger and Cherrington, 2012). The successive waves of transcription factors that establish alpha and beta cell identity during embryonic development are well-described (reviewed by Shih et al., 2013). From these studies, the transcription factors Arx and Pax4 have emerged as critical determinants of alpha and beta cell identity, respectively. Whole body Arx loss-of-function leads to a lack of alpha cells that is accompanied by an increase in delta and beta cells, while Pax4-deficient mice present with the opposite phenotype: a gain of alpha cells at the expense of beta and delta cells (Collombat et al., 2003; Sosa-Pineda et al., 1997). This has led to a model where Arx and Pax4 drive opposing transcriptional programs towards alpha and beta cell identity. Indeed, overexpression of Pax4 (Collombat et al., 2009) or deletion of Arx in alpha cells (Chakravarthy et al., 2017; Courtney et al., 2013; Wilcox et al., 2013) both result in alpha to beta cell transdifferentiation. This has made Arx a central target in efforts to generate beta cells from alpha cells.
The notion that inhibition of Arx suffices to promote beta cell identity was the premise for one recent study that screened 280 clinically approved drugs and discovered that the anti-malaria drug artemether suppressed Arx function and promoted the conversion of alpha into beta cells (Li et al., 2017). However, many of the key observations in this paper were obtained in experiments that used αTC-1 alpha or Min6 beta cell lines. This is potentially problematic, as islet tumor cell lines lose properties of the primary cell type they model, while gaining traits associated with other primary islet cells (Huising et al., 2011; Oie et al., 1983). Particularly for studies that address the mechanisms underlying the establishment and maintenance of mature islet cell fate and identity, primary tissues are preferable over cell lines. Because the ramifications of this study – the repurposing of an approved drug to promote restoration of beta cells in T1D – are significant, we conducted the pertinent experiments at the basis of the conclusion that artemether drives transdifferentiation of alpha to beta cells on primary islets. Following treatment with artemether, we also observed significant inhibition of Arx expression, as previously reported in human islets (Li et al., 2017). However, we observed no alpha to beta cell transdifferentiation upon artemether treatment of intact mouse primary islets. Moreover, the application of artemether at the dose and duration originally used (Li et al., 2017) reduces Ins2 expression by 100-fold, downregulates Slc2a2 mRNA (which encodes the beta cell Glut2 glucose transporter), inhibits glucose uptake, and abolishes glucose-induced Ca2+ responses and insulin secretion. Our observations that artemether 1) does not turn primary alpha cells into beta cells and 2) severely affects beta cell identity and function cast doubt on the original suggestion that artemisinins could turn alpha cells into functional beta cells.
Results and discussion
The main finding behind the idea that artemisinins could drive transdifferentiation of alpha to beta cells was the observation that artemether suppressed glucagon protein content or otherwise antagonized the effects of Arx (Li et al., 2017). However, these observations were largely made in αTC-1 alpha or Min6 beta cell lines. Furthermore, artemether was suggested to promote restoration of beta cell mass following beta cell ablation in zebrafish or rat and increase beta cell function in human islets, but none of these experiments offered direct evidence that alpha to beta transdifferentiation contributed to the observed effect. The direct evidence that was offered for alpha to beta transdifferentiation – based on lineage tracing using Gcg-Cre – consisted of an experiment using isolated islets of Gcg-Cre x Rosa26-stop-RFP reporter mice that were dissociated after treatment (Li et al., 2017). Slightly over 1% of all cells in control cultures was an RFP+ alpha lineage positive cell that stained for insulin, in line with the frequency of alpha to beta transdifferentiation that accumulates at a specialized neogenic niche at the periphery of adult islets (van der Meulen et al., 2017). This fraction of beta cells with an alpha cell lineage marker increased to 4% following 24 hr of culture with artemether (Li et al., 2017), although the absolute number of beta cells and beta cells with an alpha cell lineage mark in this experiment was not reported. As the local micro-environment within the islet is important in establishing and maintaining islet cell fate (van der Meulen et al., 2017), we treated intact C57BL/6 wild type mouse islets with 10 μM artemether for 72 hr as described by (Li et al., 2017) and assessed whole islet gene expression by quantitative PCR (qPCR). We observed loss of Arx mRNA expression in artemether-treated islets compared to untreated control islets cultured in parallel (Figure 1A), consistent with the original observations on human islets (Li et al., 2017). Moreover, expression of other alpha cell genes, including Gcg, Mafb, and Irx1 was also downregulated, suggesting a general loss of alpha cell identity (Figure 1A).
Figure 1. Artemether does not promote the transdifferentiation of alpha to beta cells but instead suppresses overall islet cell identity.
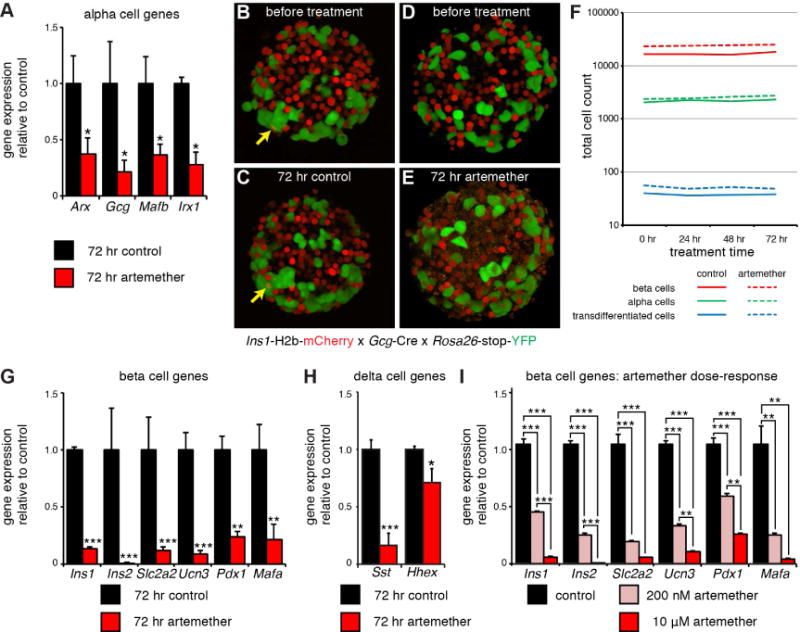
(A) Real time quantitative PCR analysis of Arx, Gcg, Mafb, and Irx gene expression in artemether treated islets (n=4 replicates). *p<0.05.
(B) 3D reconstruction of a representative image of an islet from an Ins1-H2B-mCherry x Gcg-Cre x Rosa26-stop-YFP triple transgenic reporter mouse at the onset of the experiment. Arrow indicates an mCherry/YFP co-positive cell that represents a spontaneous alpha to beta transdifferentiated cell.
(C) Islet in (B), re-imaged after 72 hr of incubation. Arrow indicates the same alpha to beta transdifferentiated cell that was present prior to the 72 hr culture window.
(D) 3D reconstruction of a representative image of another islet from the same mouse as in (B) prior to artemether treatment.
(E) Islet in (D), re-imaged after 72 hr of culture in the presence of 10 μM artemether. No alpha to beta transdifferentiated cells are present, note the appearance of a ‘speckled’ background across the entire islet. See also Movie S1.
(F) Quantification of the total number of alpha, beta and alpha to beta transdifferentiated cells. The same islets were imaged repeatedly in 3D at 24 hr intervals. n = 5 mice, 8–17 islets per animal for each treatment.
(G) Real time quantitative PCR analysis of the expression of a panel of beta cell genes in artemether treated islets (n=4 replicates). *p<0.05, **p<0.01, ***p<0.001.
(H) Real time quantitative PCR analysis of the expression of a panel of delta cell genes in artemether treated islets (n=4 replicates). *p<0.05, **p<0.01, ***p<0.001.
(I) Real time quantitative PCR analysis of a panel of beta cell genes in islets treated for 72 hr with 200 nM or 10 μM artemether (n=4 replicates). *p<0.05, **p<0.01, ***p<0.001.
Artemether inhibits Arx but fails to promote transdifferentiation of primary alpha cells
To determine whether this inhibition of Arx initiated a fate switch of alpha to beta cells in intact islets, we turned to a lineage labelling strategy combining the Ins1-H2b-mCherry beta cell reporter (Benner et al., 2014) with alpha lineage labelling using Gcg-Cre (Herrera, 2000) and Rosa26-stop-YFP, similar to what was done in the original paper. In triple transgenic offspring of these mice all current beta cells express nuclear mCherry, while the alpha lineage will be indelibly labelled with YFP, as previously reported (van der Meulen et al., 2017). Any alpha to beta transdifferentiation events induced by artemether in these islets should therefore present as cells that retain YFP and acquire a red nucleus following artemether exposure. These mice are thus well-suited to study transdifferentiation from primary alpha to beta cells within intact islets. We imaged islets of five different mice (3 female, 2 male) in 3D over the course of 72 hr treatment with 10 μM artemether. DMSO-treated controls cultured in parallel were included. The fact that the islets attach to the glass coverslip enabled us to re-image the same islets at multiple time points. This allowed us to track the fate of a large number of alpha cells to detect any that transdifferentiated into beta cells in response to artemether. We observed alpha to beta transdifferentiated cells at the islet periphery prior to treatment (Figure 1B), in agreement with our recent description of a ‘neogenic niche’ in the periphery of mouse islets (van der Meulen et al., 2017). However, of the 2344 conventional alpha cells we observed prior to artemether treatment (n = 5 mice, 8–17 islets per treatment for each animal), not a single alpha cell acquired a red nucleus that would indicate induction of Ins1 expression and thus alpha to beta cell transdifferentiation during the course of 72 hr treatment (Figure 1C–F; movies S1). We verified on the islets we imaged of two mice (both female) that Arx was inhibited at the conclusion of the experiment (Supplemental Figure 1).
Artemether effectively suppresses beta cell identity
Artemether-treated islets showed an obvious pattern of speckles or fragmentation in the red channel after 72 hr, which was absent prior to treatment or in control islets at 72 hr (compare Figure 1D, E). We suspected this pattern to reflect a decline in beta cell health. Indeed, expression of Ins1 and Ins2 was downregulated >10-fold and >100-fold, respectively. Many mature beta cell markers, including Ucn3, Mafa, Pdx1, and Slc2a2 are also significantly inhibited by 72 hr of artemether treatment (Figure 1G). Moreover, two delta cell markers, somatostatin (Sst) and Hhex, a delta cell-specific transcription factor that is important for delta cell development and function (Zhang et al., 2014), were both downregulated as well (Figure 1H). These results establish that artemether does not selectively inhibit Arx, but instead causes broad inhibition of alpha, beta, and delta cell-specific transcription factors, in addition to key beta cell maturity genes. These observations suggested the possibility that the continued presence of artemether had prevented the transdifferentiation of alpha cells into beta cells following Arx downregulation (Figure 1F). Therefore, we performed a 48 hr washout after stimulating with 10 μM artemether for 24 or 72 hr, but still did not observe marked transdifferentiation of alpha cells into beta cells (Supplemental Figure 1).
Li et al. reported significant inhibition of ARX expression by artemether in human islets, but did not show the effect of artemether treatment on the expression of insulin or any other key beta cell markers in the same experiment. We therefore reanalyzed their human single islet cell RNAseq data, which revealed no differences in INS expression between control and artemether-treated beta cells. However, ARX expression between control and artemether-treated alpha cells was also not different (Supplemental Figure 2), which is internally inconsistent with the robust inhibition of ARX in human islets reported by quantitative PCR in the same paper (Li et al., 2017).
Inhibition of Ins2 by artemether occurs in excess of its normal therapeutic concentration
Our observations that artemether inhibits expression of key beta cell genes would suggest that a widely used class of anti-malaria drugs impairs beta cell function. Therefore, we compared the 10 μM dose of artemether that was chosen by Li et al. and thus adopted in our study, to a 50-fold lower dose of artemether that is representative of the plasma artemether concentration in patients on a standard Artemether-lumefantrine oral anti-malarial drug regimen (four or six doses within a 48 hr period) (Lefevre et al., 2001). While artemether applied directly at islets in vitro at both doses inhibits key beta cell genes, the effects of artemether at 200 nM are significantly attenuated (Figure 1I) and 72 hr stimulation exceeds the 48 hr exposure that is common in artemether-based malaria therapies. Therefore, we do not believe that our observations of the adverse consequences of 72 hr treatment with 10 μM artemether on isolated mouse islets in vitro should give reason for pause for the safety and efficacy of artemether for the treatment of malaria, its primary indication. Artemisinins save lives and have been safely prescribed to millions of malaria patients for years (Miller and Su, 2011).
Artemether does not induce beta cell death
To determine if the robust inhibition of beta cell genes by artemether was attributable to beta cell death, we assessed the expression of a small panel of pro-apoptosis (Bad, Bax and Txnip), and anti-apoptosis markers (Bcl2, Bclxl and Bip) (Danial, 2007; Minn et al., 2005) and observed no consistent changes (Figure 2A). We also did not detect an increase in the amount of cleaved caspase 3 (Figure 2B) or the number of Sytox Blue-positive dead beta cells (Figure 2C–H) that would indicate increased beta cell death following 72 hr artemether treatment.
Figure 2. Artemether does not induce beta cell death.
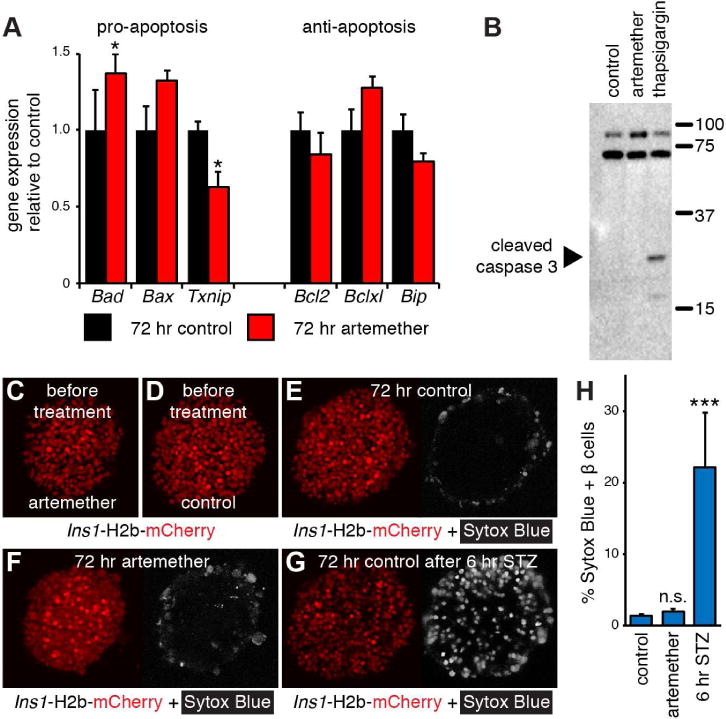
(A) Real time quantitative PCR analysis of the expression of a panel of pro- and anti-apoptotic genes in artemether treated islets (n=4 replicates). *p<0.05.
(B) Western blot analysis of the apoptosis marker cleaved caspase 3 in islets treated for 72 hr with 10 μM artemether. Thapsigargin (750 nM) was used as a positive control.
(C) 3D reconstruction of a representative Ins1-H2b-mCherry islet before artemether treatment.
(D) 3D reconstruction of a representative Ins1-H2b-mCherry control islet before culture.
(E) Islet in (D) re-imaged after 72 hr in culture. The nuclear dead cell marker Sytox Blue was added at 72 hr.
(F) Islet in (C) re-imaged after 72 hr in culture with 10 μM artemether. The nuclear dead cell marker Sytox Blue was added at 72 hr.
(G) 3D reconstruction of the same islet as in (E) after 6 hr in the presence of STZ to induce beta cell death.
(H) Quantification of the fraction of Sytox Blue positive beta cells after 72 hr in control (12 islets), and artemether-treated cultures (16 islets) compared to control islets exposed to STZ for an additional 6 hr (5 islets). ***p<0.001 compared to control.
Artemether suppresses beta cell glucose uptake
Prompted by the consistent downregulation of key beta cell genes in our experiments, we next set out to establish how 72 hr of artemether treatment would affect beta cell function. The downregulation of Slc2a2, which encodes the beta cell surface Glut2 glucose transporter, suggested impairment of beta cell glucose sensing. We therefore compared the rate of glucose uptake using the fluorescent glucose marker 6-NBDG in islets from Ins1-H2b-mCherry beta cell reporter mice (van der Meulen et al., 2017). The rate of glucose uptake was similar in freshly isolated islets and after 72 hr of culture, but was significantly suppressed in beta cells after 72 hr exposure to artemether (Figure 3, Movie S2).
Figure 3. Artemether inhibits glucose uptake by beta cells.
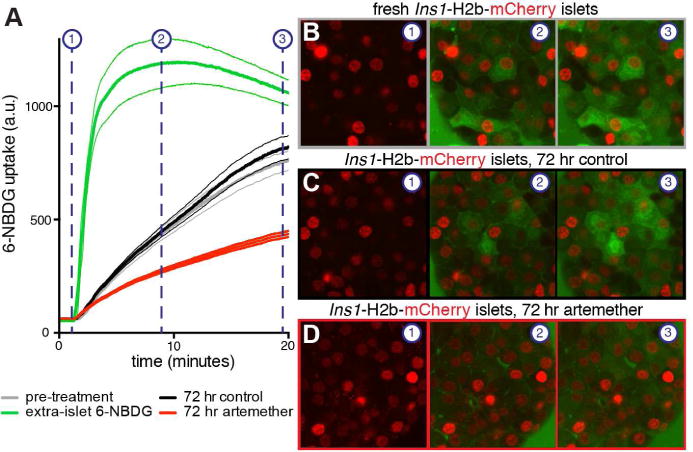
(A) Comparison of the rate of uptake of the glucose analog 6-NBDG in islets of Ins1-H2b-mCherry beta cell reporter mice in isolated islets after isolation (black), after 72 hr in culture in the presence of DMSO (gray) or artemether (red). 6-NBDG signal in the media outside of the islet is quantified as a reference (green). Averages and 95% confidence intervals are given for 19–26 individual beta cells of 2–3 islets for each treatment.
(B) Video stills of 6-NBDG uptake in a freshly isolated Ins1-H2b-mCherry islet. See also Movie S2.
(C) Video stills of 6-NBDG uptake in an Ins1-H2b-mCherry control islet after 72 hr of culture.
(D) Video stills of 6-NBDG uptake in an Ins1-H2b-mCherry islet after 72 hr of artemether treatment.
Artemether inhibits the normal calcium response
Artemisinins have several proposed mechanisms of action. One of those is inactivation of the Plasmodium falciparum homolog of SERCA (Eckstein-Ludwig et al., 2003), a Ca2+-ATPase that normally transports cytosolic free Ca2+ back into the endoplasmic reticulum. As calcium is required for normal insulin secretion, we sought to determine the effect of 72 hr artemether exposure to the calcium response of beta cells within primary islets using triple transgenic offspring of a cross between Ins1-H2b-mCherry x Ucn3-Cre x Rosa26-stop-GCaMP6. These islets exhibit efficient expression of the genetically encoded green fluorescent calcium indicator GCaMP6 across the mature beta cell lineage and independent of current Ucn3 expression status in addition to expression of nuclear mCherry across all beta cells (van der Meulen et al., 2017). Islets imaged 24 hr after isolation invariably showed a strong calcium response to an increase in glucose concentration from 5.5 to 16.8 mM, with a majority of islets exhibiting robust pulsatile calcium behavior (Figure 4A, Movie S3). After 72 hr in culture, islets from the same animal continue to respond robustly to glucose, although the pulsatility of the second phase has been replaced with sustained, but uncoordinated calcium activity that returns to baseline when ambient glucose is lowered back to 5.5 mM (Figure 4B). In sharp contrast, 72 hr of continuous treatment with 10 μM artemether completely abolished any detectable calcium response to glucose, leaving only a muted response to 30 mM KCl-induced depolarization (Figure 4C). Li et al. reported increased excitability of αTC-1 cells using Fura2 following 72 hr artemether treatment in response to excitation with 15 or 30 mM KCl (Li et al., 2017), but did not measure the calcium responses to physiologically relevant cues such as glucose on primary islet cells. Our observations that calcium responses of artemether-treated primary beta cells within intact islets to glucose and even KCl all but disappear illustrates the detrimental effect of prolonged stimulation with 10 μM artemether on beta cell function.
Figure 4. Artemether abrogates beta cell calcium responses and insulin secretion.
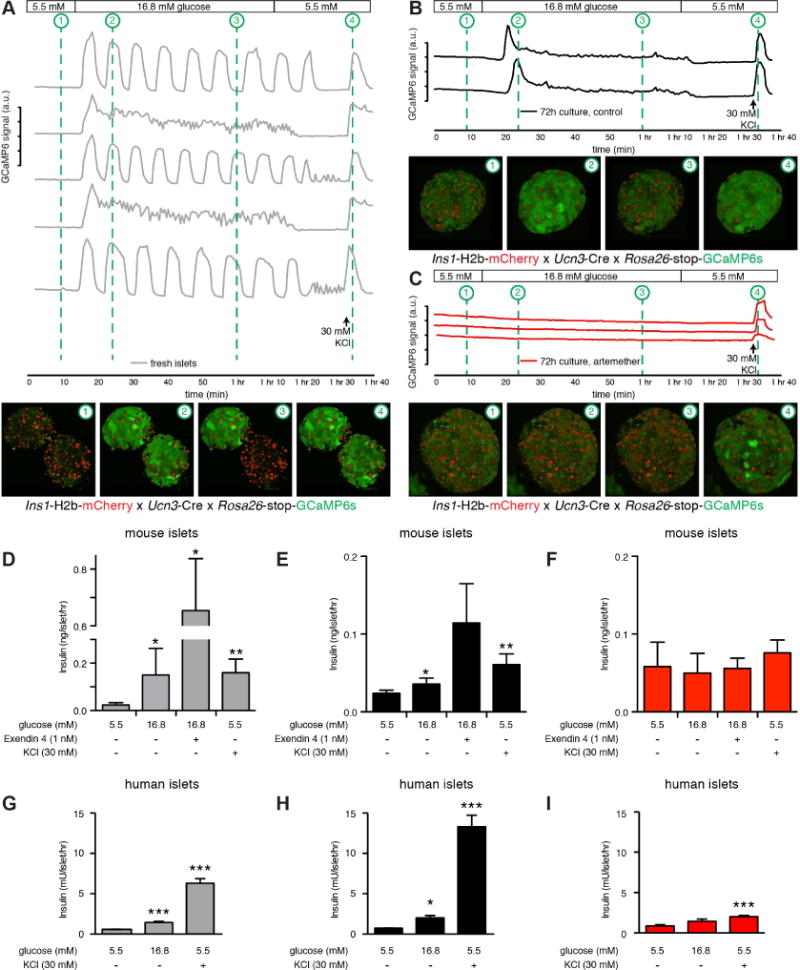
(A) The calcium responses of freshly isolated islets from Ins1-H2B-Chy x Ucn3-Cre x Rosa26-stop-GCaMP6 triple transgenic mice were imaged in 3D during a standard glucose stimulation protocol from 5.5 to 16.8 to 5.5 mM glucose, followed by a brief depolarization by 30 mM KCl to demonstrate responsiveness of the islets throughout the experiment. Each trace represents the combined response of a single islet. Thumbnails provide snapshots of the calcium response at key points during the trace for 2 of the 5 islets imaged. See also Movie S3.
(B) Islets from the same animal and subjected to the same stimulation protocol as in (A), but after 72 hr in culture. First phase secretion is intact, but the second phase is muted and pulsatility is lost.
(C) Islets from the same animal and subjected to the same stimulation protocol as in (A), but after 72 hr in culture in the presence of 10 μM artemether. The glucose response is completely abolished and only a modest response to forced depolarization with 30 mM KCl can be detected.
(D) Insulin secretion from freshly isolated mouse islets in response to glucose, glucose + Exendin 4, or 30 mM KCl (n=4 replicate treatments). *p<0.05, **p<0.01 compared to 5.5 mM glucose. (E) Insulin secretion on islets from the same batch as in (D), but after 72 hr in culture (n=4 replicate treatments). *p<0.05, **p<0.01 compared to 5.5 mM glucose.
(F) Insulin secretion on islets from the same batch as in (D), but after 72 hr of treatment with 10 μM artemether (n=4 replicate treatments). *p<0.05, **p<0.01 compared to 5.5 mM glucose.
(G) Insulin secretion from freshly isolated human islets in response to glucose or 30 mM KCl (n=8 replicate treatments). *p<0.05, **p<0.01, ***p<0.001 compared to 5.5 mM glucose.
(H) Insulin secretion on islets from the same batch as in (G), but after 72 hr in culture (n=6 replicate treatments). *p<0.05, **p<0.01, ***p<0.001 compared to 5.5 mM glucose.
(I) Insulin secretion on islets from the same batch as in (G), but after 72 hr of treatment with 10 μM artemether (n=6 replicate treatments). *p<0.05, **p<0.001, ***p<0.001 compared to 5.5 mM glucose.
Artemether abrogates insulin secretion
As calcium is required for insulin secretion, we followed these functional calcium imaging experiments with studies to directly measure insulin secretion. Fresh islets secreted significantly more insulin in response to 16.8 mM glucose, by itself or in combination with 1 nM Exendin 4, or depolarization with 30 mM KCl (Figure 4D). Islets remain responsive to these cues after 72 hr in culture (Figure 4E). However, islets from the same animals cultured in the presence of 10 μM artemether no longer responded to glucose, incretins or even depolarization by 30 mM KCl with increased insulin release (Figure 4F). These findings conflict with the reported effect of artemether on human islets, where 72 hr artemether treatment of human islets was reported to significantly increase glucose-stimulated insulin secretion (Li et al., 2017), although it was not explained how a relatively small contribution of transdifferentiating alpha cells to the existing beta cell mass promoted such a significant increase in normalized insulin secretion. We therefore repeated the artemether treatment on human islets and observed robust insulin secretion in response to 16.8 mM glucose and KCl shortly after receipt of the islets (Figure 4G) and following 72 hr of culture (Figure 4H). However, insulin secretion after 72 hr of culture in the presence of artemether completely abrogated normal insulin secretion (Figure 4I). These results are fully in line with the body of observations we reported here on mouse islets, but do not reproduce the stimulation of insulin secretion in response to artemether treatment reported previously (Li et al., 2017).
The mechanism of artemether action
Li et al. proposed a model where GABA (from neighboring beta cells) inhibits glucagon secretion from alpha cells via the artemether-induced upregulation of alpha cell GABA receptors (Li et al., 2017). Reduced extracellular glucagon concentration then would induce the loss of alpha cell identity – presumably via glucagon receptors on alpha cells. However, we did not detect expression of the glucagon receptor (GCGR) or most GABA receptors in these single alpha cell libraries (Supplemental figure 2), which are key parts of their model. Furthermore, it is well-known that alpha cells in Gcgr null mice undergo impressive hyperplasia with only limited evidence of insulin/glucagon co-positive cells (Solloway et al., 2015). This does not fit a model where loss of glucagon-mediated autocrine signaling causes alpha cells to transdifferentiate into beta cells. Li et al. also reported that expression of ABCC8 (the sulfonylurea receptor subunit of the KATP channel) is undetectable in most single alpha cells, but is induced in alpha cells upon artemether treatment. Instead, upon reanalysis of their data we detect robust expression of ABCC8 in all single alpha cells irrespective of artemether treatment (Supplemental figure 2). This is in keeping with the fact that KATP channel subunit expression by alpha cells is in fact well established and supported by transcriptomic analysis (Benner et al., 2014) and direct functional measurements (MacDonald et al., 2007; Zhang et al., 2013). While the mechanistic basis for its actions are unresolved, the actions of artemether at high doses could prove to be a useful tool for the experimental induction of beta cell dedifferentiation in vitro.
Summary and conclusion
The anti-malarial drug artemether was recently reported to inhibit the alpha cell-specific transcription factor Arx to promote alpha to beta transdifferentiation (Li et al., 2017). However, this study relied heavily on experiments conducted on immortalized islet cell lines and was limited in its ability to directly demonstrate transdifferentiation in primary islet cells. As we have a vested interest in transdifferentiation of islet cells and have developed state-of-the art tools to follow this process over time (van der Meulen et al., 2017), we set out to reproduce the reported effects of artemether on alpha to beta cell transdifferentiation within intact, live islets. While we were able to reproduce the original observation that artemether treatment reduces Arx mRNA expression along with the expression of other alpha cell markers, we observed no evidence that this promotes alpha to beta transdifferentiation. This is at odds with the notion that Arx deletion in adult alpha cells leads to the formation of mature beta cells from alpha cells (Chakravarthy et al., 2017; Courtney et al., 2013), although others have also reported that ablation of Arx in adult alpha cells was insufficient to promote their transdifferentiation into beta cells (Wilcox et al., 2013). The lack of alpha to beta cell conversion in our experiments may be attributed to the fact that artemether inhibited Arx mRNA, but may not have fully ablated its expression. Moreover, 72 hr artemether treatment on primary islets caused sustained loss of identity across all islet endocrine cell types including the dedifferentiation of existing beta cells as evidenced by severe impairments in glucose uptake, calcium responses and insulin secretion. While artemether at high doses impacts islet cell function in vitro, we find no evidence to corroborate the key conclusion (Li et al., 2017) that this drug upregulates GABA receptor expression on alpha cells to inhibit glucagon secretion and thereby promote their conversion into beta cells. As sustained systemic GABA administration was reported in a separate paper to drive robust beta cell neogenesis by promoting alpha to beta cell conversions (Ben-Othman et al., 2017), it will be important to elucidate the mechanisms that explain GABA’s potent beta cell neogenic actions.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Huising (mhuising@ucdavis.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
For in vitro stimulation of islets with artemether for quantitative PCR, commercial C57BL6/NHsd mice were obtained from Envigo (Indianapolis, IN). A number of transgenic mouse lines were employed. The Rosa26-stop-eYFP reporter mouse (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J) (Srinivas et al., 2001) was used to label cells for lineage tracing. Calcium levels in the cell were visualized using the Rosa26-stop-GCaMP6 mouse line (B6;129S6-Gt(ROSA)26Sortm96(CAG-GCaMP6s)Hze/J) (Madisen et al., 2015). A Ucn3 BAC transgenic reporter mouse based on BAC clone RP23-332L13, which contains the Ucn3 gene flanked by more than 197 kb of genomic context was used: the Ucn3-Cre line (B6.FVB(Cg)-Tg(Ucn3-cre)KF43Gsat/Mmucd (van der Meulen et al., 2017). For alpha cell lineage labeling we employed a Gcg-Cre mouse line (Herrera, 2000). To label beta cells we used the B6.Cg-Tg(Ins1-HIST1H2BB/mCherry)5091Mhsg/J mouse line (Benner et al., 2014). All transgenic lines are maintained by back crossing to commercially obtained C57BL6/NHsd (Envigo). Isolated islets of mice between 3 and 13 months of age were used. Male islets were used for most experiments, with the following exceptions. The Western blot in Figure 2B was conducted on islets pooled from 8 animals of mixed sex. The 3D imaging experiments in Figure 1B–F were conducted on 3 female and 2 male mice. For 2 of these females, we verified at the conclusion of the imaging experiment that Ins2, Arx and Ucn3 were reduced (Figure S1A) in line with the rest of the observations we reported. We have therefore observed no indication that sex was associated with the effects of artemether, but did not formally test this. Animals were maintained in group-housing on a 12-h light/12-h dark cycle with free access to water and standard rodent chow. All animal procedures were approved by the UC Davis Institutional Animal Care and Use Committees and performed in compliance with the Animal Welfare Act and the ILAR Guide to the Care and Use of Laboratory Animals.
Primary cell cultures
Primary islets were cultured in RPMI (5.5 mM glucose, 10% FBS, pen/strep) under 5% CO2 at 37° C in 10 cm petri dishes (i.e. not tissue culture treated). Islets for microscopy experiments were cultured overnight on uncoated #1.5 glass-bottom 35 mM culture dishes (MatTek Corporation, Ashland, MA).
METHOD DETAILS
Islet isolation
Islets were isolated by injecting collagenaseP (0.8 mg/mL in HBSS; Roche Diagnostics) (Invitrogen) via the common bile duct while the ampulla of Vater was clamped. The entire pancreas was collected following the injection of 2 mL collagenase solution and, after addition of an additional 2 mL of collagenase solution, was incubated at 37°C for 13 min. Pancreata were dissociated by gentle manual shaking followed by three washes with cold HBSS containing 5% NCS. The digested suspension was passed through a nylon mesh (pore size 425 μm; Small Parts Inc.), and islets were isolated by density gradient centrifugation on a Histopaque gradient (1.077 g/mL density; Sigma) for 20 min at 1400 × g without brake. Islets were collected from the interface, washed once with cold HBSS containing 5% NCS, and hand-picked several times under a dissecting microscope prior to culture in RPMI (5.5 mM glucose, 10% FBS, pen/strep).
Quantitative PCR
Islets were treated as indicated and then collected in Trizol reagent. RNA was isolated according to standard protocol and converted into cDNA using the High Capacity cDNA Archive Kit (Thermo Fisher Scientific, Waltham, MA) per the manufacturer’s instructions. Primers for quantitative PCR are listed in Table S1.
Western blot
Islets were treated for 72 hr with 10 μM artemether, 750 nM thapsigargin, or DMSO (control). Thirty μl of sample treatment buffer (50 mM Tris (pH 7.5), 100 mM dithiothreitol, 2% (weight/volume) sodium dodecyl sulfate, 0.1% (weight/volume) bromophenol blue, and 10% (weight/volume) glycerol) was added to 50 islets. Antibody 9661 (Cell Signaling Technologies; diluted 1:6000) was used to detect cleaved caspase 3 and was visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA). Total protein in each lane was checked with Ponceau S solution.
3D Imaging of intact islets
To determine if alpha to beta cell transdifferentiation occurred during artemether treatment, we cultured Ins1-H2b-mCherry x Gcg-Cre x Rosa26-stop-YFP triple transgenic islets as described above on uncoated #1.5 glass-bottom 35 mm culture dishes (MatTek Corporation, Ashland, MA) in RPMI (10% FBS, 5.5 mM glucose, pen/strep) and followed them over a period of 72 hr in the presence or absence of 10 μM artemether. The live islets were allowed to attach onto the glass bottom overnight and imaged in x, y, and z on a Nikon A1R+ confocal microscope using a 40x objective the next day before treatment (time = 0) and consecutively every 24 hr for 3 days after the addition of artemether. Media was also refreshed at 24 hr intervals with artemether or DMSO for control islets. For the 48 hr washout experiment, the islets were treated similarly, washed out 3x with fresh RPMI at the end of the 24 or 72 hr artemether treatments, and cultured for another 48 hr in RPMI before imaging. Z stacks of 50 micron thick were captured for each islet in resonant scanning mode. Artemether-induced beta cell death ex vivo was determined by the addition of 500 nM of the nuclear dead cell marker Sytox Blue (Thermo Fisher Scientific, Waltham, MA) 30 minutes prior to imaging. STZ-induced beta cell death was documented after 6 hr of stimulation with 5 mM Streptozotocin (EMD Millipore, Billerica, MA).
Glucose uptake
To measure glucose uptake, we incubated freshly isolated intact islets from Ins1-H2b-mCherry reporter mice overnight on uncoated #1.5 glass-bottom 35 mm culture dishes (MatTek Corporation, Ashland, MA) in RPMI (10% FBS, 5.5 mM glucose, pen/strep). The next day, Z-stacks of islets were continuously acquired as the non-hydrolysable glucose analog 6-NBDG (6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-Deoxyglucose; Thermo Fisher Scientific, Waltham, MA) was added at a final concentration of 0.3 mM, using a Nikon A1R+ confocal microscope in resonant scanning mode. The relative rate of glucose uptake was determined by drawing ROIs of individual Ins1-H2b-mCherry+ beta cells that either had or had not taken up glucose. In parallel, islets were cultured for 72 more hr in media with or without 10 μM artemether. Media and artemether were refreshed every 24 hr. Islets were allowed to attach to the glass-bottom culture dishes for the last 24 hr before applying 6-BNDG and imaging as above.
Insulin secretion
Static insulin secretion experiments were carried out on 10 wild type mouse islets per well in Krebs Ringer Buffer (KRB). Mouse islets were isolated the day prior to the secretion assay, cultured overnight in RPMI (5.5 mM glucose, 10% FBS, pen/strep). Human islets were cultured overnight in CMRL (Thermo Fisher Scientific, Waltham, MA) after receipt prior to the experiment. Islets were transferred to KRB containing 5.5 mM glucose an hr before the start of the assay. Islets were picked to a 24-wells plate for final secretion in 10% of the final assay volume. The other 90% of volume contained concentrated treatment compounds in KRB (glucose, exendin-4, KCl) as indicated. From the same batch of islets, two pools were cultured in the presence or absence of 10 μM artemether for 72 hr, with the media refreshed every 24 hr. They were then subjected to the same secretion assay as above and in the continued presence of artemether in the case of artemether-treated islets. Insulin was measured by radioimmunoassay (EMD Millipore).
Calcium responses in intact islets
We used islets from a triple transgenic offspring of a cross between Ins1-H2b-mCherry, Rosa26-stop-GCaMP6 and Ucn3-Cre to label the mature beta cell lineage (van der Meulen et al., 2017). Live islets were cultured overnight after the islet prep, placed on 35 mm dishes with glass bottom (#1.5; MatTek Corporation), allowed to attach overnight and imaged in x, y, z and t on a Nikon A1R+ confocal microscope using a 40x objective with a long working distance. Similar to the glucose uptake experiments, two additional sets of islets were cultured in the presence or absence of 10 μM artemether for 72 hr prior to measurement of calcium activity, with islets transferred to fresh media and artemether every 24 hr. Treatments were continuously perfused over the islets using a Masterflex peristaltic pump at 2.5 mL per minute. Each protocol concluded with a 30 mM potassium chloride pulse to demonstrate viability and responsiveness of the islets throughout the treatment. Individual islets in individual z-planes were defined as regions of interest (ROI) and the green fluorescence intensity within the ROIs was plotted over time as a measure of calcium activity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Data were analyzed by ANOVA followed by Holm-Sidak’s multiple comparisons test or by t-test and are represented as mean ± SEM, with n defined in the corresponding figure legend. Differences were considered significant when p<0.05. Statistics were computed using Prism (GraphPad Software, La Jolla, CA).
Counting algorithm
We developed a Matlab algorithm to count the beta cells in Ins1-H2B-mCherry x Gcg-Cre x Rosa26-stop-eYFP islets. This takes the NIS Elements file of the islet imaged in 3D as its input and uses a combination of automated thresholding and size and shape exclusions to distinguish the red nuclei from the background. Recorded measurements are written to a csv file containing all measurements along with the sample information. We observed a close correlation between manual and automated counts of beta cells (<5% difference). YFP+ alpha cells and YFP+/mCherry co-positive transdifferentiated cells were counted manually.
Bioinformatics
Read SRA files were pulled from GEO Datasets GSE73727 and GSE84714, and converted into fastq format using the NCBI SRA toolkit. Sequence files were then aligned using STAR (Dobin et al., 2013) with default parameters to the Gencode GRCh37 24 human genome. Bigwigs were generated using STAR’s wiggle output option and UCSC’s Genome Utilities. Gene-level quantification was performed on all samples’ sorted BAM files using featureCounts (Liao et al., 2014) default parameters, counted by Gencode defined exons, and aggregated to the gene level. Differential expression analyses were performed with edgeR (Robinson et al., 2010) using the generalized linear model approach and maximum likelihood method testing. RPKM values were generated using edgeR’s rpkm function.
Supplementary Material
Movie S1. 3D reconstructions of islets from Ins1-H2b-mCherry x Gcg-Cre x Rosa26-stop-YFP triple transgenic reporter mice. All beta cells are marked by nuclear mCherry expression, the alpha cell lineage is labelled by YFP, alpha to beta transdifferentiated cells (yellow arrows) express continue to express YFP while they acquire a red nucleus. Note that the same islets are shown before and after 72 hr culture with or without 10 μM artemether, as indicated. Related to Figure 1.
Movie S2. Fluorescent glucose (6-NBDG) uptake in Ins1-H2b-mCherry islets over time. Glucose uptake is recorded in islets of the same animal recorded before and after 72 hr culture with or without artemether, as indicated. Related to Figure 3.
Movie S3: Calcium responses of beta cells within intact islets in response to successive stimulation with high glucose and 30 mM KCl, recorded from Ins1-H2b-mCherry x Ucn3-Cre x Rosa26-stop-GCaMP6s triple transgenic reporter mice. Related to Figure 4.
Highlights.
○ Artemether does not induce the transdifferentiation of α cells into β cells
○ High doses of artemether dedifferentiate islet cells without inducing death
○ Artemether not only inhibits Arx and Gcg, but inhibits Ins2 > 100-fold
○ Artemether suppresses glucose uptake and prevents insulin secretion
Acknowledgments
The research described in this paper was funded by an Individual Biomedical Research Award from the Hartwell Foundation and a Career Development Award from the Juvenile Diabetes Research Foundation (2-2013-54) to MOH. SL was supported by an NSF Bridge to Doctorate Program and the UC Davis Training Program in Molecular and Cellular Biology (funded in part by T32GM007377 from NIH-NIGMS). GMN was supported by a NIGMS-funded Pharmacology Training Program (T32GM099608). We thank Dr. Maike Sander for constructive feedback on the final draft of this manuscript, Dr. Aldrin Gomes for assistance with the caspase 3 Western Blot and Jessica Huang for assistance in the quantification of alpha cell numbers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, TvdM, SL, MOH; Methodology, SL, MOH; Software, TvdM, MWA; Formal Analysis and Data Curation, TvdM, SL, AMM, MOH; Investigation, TvdM, SL, EN, CJD, GMN, MOH; Writing, TvdM, MOH; Review and Editing, TvdM, SL, EN, CJD, GMN, AMM, MOH; Visualization, MOH; Supervision, Project Administration and Funding Acquisition, MOH.
Supplemental Information
Supplemental Experimental Procedures, 2 Figures, 1 Table, and 3 Movies can be found online.
References
- Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, Hadzic B, Druelle N, Napolitano T, Navarro-Sanz S, et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell. 2017;168:73–85 e11. doi: 10.1016/j.cell.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genomics. 2014;15:620. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, Downie C, Liu K, Wang J, Xing Y, et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017 doi: 10.1016/j.cmet.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014 doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, Gruss P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. doi: 10.1101/gad.269003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS genetics. 2013;9:e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res. 2007;13:7254–7263. doi: 10.1158/1078-0432.CCR-07-1598. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Huising MO, Pilbrow AP, Matsumoto M, van der Meulen T, Park H, Vaughan JM, Lee S, Vale WW. Glucocorticoids differentially regulate the expression of CRFR1 and CRFR2alpha in MIN6 insulinoma cells and rodent islets. Endocrinology. 2011;152:138–150. doi: 10.1210/en.2010-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre G, Looareesuwan S, Treeprasertsuk S, Krudsood S, Silachamroon U, Gathmann I, Mull R, Bakshi R. A clinical and pharmacokinetic trial of six doses of artemether-lumefantrine for multidrug-resistant Plasmodium falciparum malaria in Thailand. Am J Trop Med Hyg. 2001;64:247–256. doi: 10.4269/ajtmh.2001.64.247. [DOI] [PubMed] [Google Scholar]
- Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, Huber KV, Schmitner N, Kimmel RA, Romanov RA, et al. Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell. 2017;168:86–100 e115. doi: 10.1016/j.cell.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PR, Cox R, Eliasson L, Rorsman P. A K ATP channel-dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol. 2007;5:e143. doi: 10.1371/journal.pbio.0050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85:942–958. doi: 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LH, Su X. Artemisinin: discovery from the Chinese herbal garden. Cell. 2011;146:855–858. doi: 10.1016/j.cell.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- Oie HK, Gazdar AF, Minna JD, Weir GC, Baylin SB. Clonal analysis of insulin and somatostatin secretion and L-dopa decarboxylase expression by a rat islet cell tumor. Endocrinology. 1983;112:1070–1075. doi: 10.1210/endo-112-3-1070. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. beta Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017;25:727–738. doi: 10.1016/j.cmet.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol. 2013;29:81–105. doi: 10.1146/annurev-cellbio-101512-122405. [DOI] [PubMed] [Google Scholar]
- Solloway MJ, Madjidi A, Gu C, Eastham-Anderson J, Clarke HJ, Kljavin N, Zavala-Solorio J, Kates L, Friedman B, Brauer M, et al. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of alpha-Cell Mass. Cell reports. 2015;12:495–510. doi: 10.1016/j.celrep.2015.06.034. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386:399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC developmental biology. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dolleman S, Liu S, Ackermann AM, Caceres E, Hunter AE, et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metab. 2017;25:911–926.e6. doi: 10.1016/j.cmet.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Terry NA, Walp ER, Lee RA, May CL. Pancreatic alpha-cell specific deletion of mouse Arx leads to alpha-cell identity loss. PLoS One. 2013;8:e66214. doi: 10.1371/journal.pone.0066214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, McKenna LB, Bogue CW, Kaestner KH. The diabetes gene Hhex maintains delta-cell differentiation and islet function. Genes & development. 2014;28:829–834. doi: 10.1101/gad.235499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ramracheya R, Lahmann C, Tarasov A, Bengtsson M, Braha O, Braun M, Brereton M, Collins S, Galvanovskis J, et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell metabolism. 2013;18:871–882. doi: 10.1016/j.cmet.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. 3D reconstructions of islets from Ins1-H2b-mCherry x Gcg-Cre x Rosa26-stop-YFP triple transgenic reporter mice. All beta cells are marked by nuclear mCherry expression, the alpha cell lineage is labelled by YFP, alpha to beta transdifferentiated cells (yellow arrows) express continue to express YFP while they acquire a red nucleus. Note that the same islets are shown before and after 72 hr culture with or without 10 μM artemether, as indicated. Related to Figure 1.
Movie S2. Fluorescent glucose (6-NBDG) uptake in Ins1-H2b-mCherry islets over time. Glucose uptake is recorded in islets of the same animal recorded before and after 72 hr culture with or without artemether, as indicated. Related to Figure 3.
Movie S3: Calcium responses of beta cells within intact islets in response to successive stimulation with high glucose and 30 mM KCl, recorded from Ins1-H2b-mCherry x Ucn3-Cre x Rosa26-stop-GCaMP6s triple transgenic reporter mice. Related to Figure 4.