

Abstract
Background
The blood-brain barrier (BBB) has been hypothesized to play a role in migraine since the late 1970s. Despite this, limited investigation of the BBB in migraine has been conducted. We used the inflammatory soup rat model of trigeminal allodynia, which closely mimics chronic migraine, to determine the impact of repeated dural inflammatory stimulation on BBB permeability.
Methods
The sodium fluorescein BBB permeability assay was used in multiple brain regions (trigeminal nucleus caudalis (TNC), periaqueductal grey, frontal cortex, sub-cortex, and cortex directly below the area of dural activation) during the episodic and chronic stages of repeated inflammatory dural stimulation. Glial activation was assessed in the TNC via GFAP and OX42 immunoreactivity. Minocycline was tested for its ability to prevent BBB disruption and trigeminal sensitivity.
Results
No astrocyte or microglial activation was found during the episodic stage, but BBB permeability and trigeminal sensitivity were increased. Astrocyte and microglial activation, BBB permeability, and trigeminal sensitivity were increased during the chronic stage. These changes were only found in the TNC. Minocycline treatment prevented BBB permeability modulation and trigeminal sensitivity during the episodic and chronic stages.
Discussion
Modulation of BBB permeability occurs centrally within the TNC following repeated dural inflammatory stimulation and may play a role in migraine.
Keywords: Headache, migraine, minocycline, trigeminal, blood-brain barrier, glia
Background
Changes in cerebral blood flow that occur during the prodrome phase of migraine prompted the hypothesis that migraine patients have a breakdown in the blood- brain barrier (BBB) (1). This hypothesis, from the 1970s, was based on the idea that if the BBB is impaired ictally or interictally in migraineurs, then the brain parenchyma would be exposed to peripheral vasoactive and neurogenic compounds or immune and inflammatory cells (2). Although the critical role of the BBB in traumatic brain injury, stroke, and neurodegeneration has become greatly appreciated in scientific literature, little evidence has been reported for its role in the development and induction of migraine pain (3,4). With a growing list of therapeutic options targeting the BBB and the development of animal models of trigeminal pain, investigation into how BBB disruption is involved in migraine stands to have great potential value to the future of migraine research and treatment (5–7).
The BBB is a specialized neurovascular unit formed by a complex association of endothelial cells, pericytes, astrocytes, neurons, an extracellular matrix, and a basal lamina, which surround blood capillaries within the brain. This neurovascular unit contains numerous transport mechanisms that modulate the passage of small molecules, ions, nutrients, cells, and other compounds into and out of the brain parenchyma (8). Astrocytic endfeet, located on the outer surface of the basal lamina, and microglia, regulate many aspects of the BBB including the rigidity of tight junctions to modulate BBB permeability (9–11). The growing evidence that glia play a significant role in migraine suggests that BBB permeability may contribute to this disorder (12,13).
Some clinical evidence suggests a potential role of the BBB in migraine. Ictal and interictal serum levels of S100B, a marker for astrocyte activation, and MMP- 9, a matrix metalloproteinase that digests endothelial cell basal lamina and causes BBB breakdown, are increased in migraine patients (14,15). Two functional MMP-2 gene polymorphisms are also associated with migraine patients with aura (16). White matter brain lesions found in MRI images of migraine patients are proposed to be focal disruptions of the BBB (17,18). Gadolinium enhancement MRI analysis has also demonstrated BBB breakdown in some, but not all, clinical studies, as described by Edvinsson and Tfelt-Hansen (2008), who argue that there is a lack of evidence to support BBB changes in migraine (19–21). A recent study also found that permeability to 11C-dihydroergotamine, a radioligand of dihydroergotamine, was unchanged in migraineurs during GTN-induced headaches, suggesting no change in permeability (22). However, peripheral nerve injury models of chronic pain do cause disruptions in the BBB and blood-spinal cord barrier, suggesting that nociceptor activation in the trigeminal system may induce BBB changes (23,24).
To explore this, we developed a rat model of trigeminal allodynia that closely mimics clinical observations in chronic migraine patients (chronic trigeminal allodynia, phonophobia, increased sensitivity to migraine triggers, mitochondrial dysfunction, and similar efficacious responses to migraine treatments) (7,25–30). With this rat model, we simulate the putative dural nociceptor activation that is thought to occur during a migraine attack by infusing an inflammatory soup (IS) outside of the BBB onto the dura through an affixed cannula three times per week for a total of 10 infusions (7,31). This repeated dural stimulation, representing the repeated episodic nature of migraine attacks, produces an acute phase of trigeminal allodynia in the rats. After receiving five or more infusions, baseline morning trigeminal sensitivity develops, representing a transition from an episodic to a chronic state of trigeminal sensitivity, which is similar to patients who transition from episodic to chronic migraine (32). After 10 infusions, they completely transition to a state of chronic trigeminal sensitivity that is maintained, outlasting the final infusion by upwards of three months; at this stage, they are considered transitioned rats. This model presents the opportunity to investigate the role of BBB permeability during both the episodic stage (following the second infusion, when dural stimulation only features an acute effect on trigeminal sensitivity, but does not induce long-lasting sensitivity) and the chronic stage (one week after the 10th infusion, when the animals have developed long-lasting trigeminal sensitivity).
The purpose of this study was to investigate whether BBB permeability changes occur in a rat model of trigeminal allodynia that closely mimics chronic migraine. We examined the trigeminal nucleus caudalis (TNC), which is a critical brainstem nucleus that is the primary input of dura-innervating afferents of the trigeminal ganglion; the periaqueductal grey (PAG), due to its role in descending pain modulation; the sub-cortex, which is a gross anatomical region of the brain containing numerous nuclei such as the thalamus and amygdala that are involved in migraine; and the frontal cortex as a control, since it is minimally responsible for trigeminal sensory information processing. We also analyzed the cortex directly below the dural stimulation site (infusion cortex). Although this cortex is not stimulated with the IS, we examined this area to identify any changes in BBB permeability induced by repeated inflammatory stimulation of the dura. We then investigated the potential for minocycline, a tetracycline-derived antibiotic with anti-inflammatory properties that prevents glial activation, to prevent BBB breakdown (33,34). Minocycline has been shown to reduce allodynia in rodent neuropathic pain models and burn injury models, and has been investigated for its potential use in migraine, providing evidence for an interesting anti-nociceptive therapeutic (35–37).
Methods
Male Sprague Dawley adult rats (250–300 g, Charles River N = 82) were individually housed in a temperature-controlled environment, under a 12-hour light/dark cycle, and allowed access to food and water ad libitum. All procedures performed on the animals were approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. ARRIVE guidelines were considered and followed throughout all experimental planning. All efforts were made to minimize animal numbers and suffering.
Implantation of the cannula
Surgical procedures were previously described (7). Briefly, after two weeks of habituation, rats were anesthetized with isoflurane (3% induction, 1.5% maintenance) mixed with compressed air. A 3 mm wide craniotomy above the superior sagittal and transverse sinus junction was performed to expose the dura, and a plastic cap with a stainless steel cannula was secured to the skull with small screws and dental cement (26 gauge, Plastics One Inc., Roanoke, VA, USA). The cannula was sealed with a replaceable custom-cut obdurator that extended just beyond the internal end of the cannula above the dura. This prevented scar tissue formation over and into the cannula. Rats recovered for one week, during which trigeminal pressure thresholds were monitored to ensure return to pre-surgery baseline. Any rats that did not return to pre-surgery sensory thresholds were not included in the study. All animals that returned to pre-surgery sensory thresholds were randomly assigned to the treatment groups.
Infusion of inflammatory soup or saline
Rats were infused onto the dura through the cannula with an inflammatory soup (IS) (1 mM histamine, serotonin, bradykinin, and 0.1 mM prostaglandin E2 in 0.9% sterile saline, pH 7.0 (Sigma Aldrich, St Louis, MO) or saline in a square Plexiglas chamber (17.5 × 18 × 12 in) that allowed for free movement during the infusion. Thus, the IS or saline was infused over the dura and outside the blood-brain barrier. Polyethylene tubing (PE50) was connected to a microinfusion pump (WPI Inc, Sarasota, FL, USA) and the cannula, allowing for a steady infusion of 25 μl of IS or saline over 5 min at a rate of 5 μl/min.
Tactile sensory testing
Rats, tested during the day, were trained and acclimated to an atraumatic plastic tube restraint (inner diameter 8 cm, length 25 cm) before and after cannula implantation, and entered uncoaxed to prevent them from walking away during testing.
Periorbital pressure thresholds were determined by applying von Frey monofilaments (Stoelting Co, Wood Dale, IL, USA) to both the left and right sides of the face over the medial portion of the eye. These von Frey hairs are calibrated nylon monofilaments that generate a reproducible buckling stress. Each monofilament is identified by manufacturer-assigned force values (10, 8, 6, 4, 2, 1.4, 1, 0.6, 0.4, 0.07 grams). The higher the force value on the monofilament, the stiffer and more difficult it is to bend. For each time point, the left and right threshold data were recorded separately. The von Frey stimuli were presented in sequential descending order to determine the threshold of response. After a positive response, a weaker stimulus was presented. Several behaviors presented by the rat were considered a positive response. The rat would vigorously stroke its face with the ipsilateral forepaw, quickly recoil its head away from the stimulus, or vocalize. The threshold is defined as a positive response to two of three trials of the von Frey monofilament. Results are presented as the threshold in grams ±SEM. Rats that did not respond to the 10 g stimulus were assigned 10 g as their threshold for analysis.
Blood brain barrier permeability
Sodium fluorescein (Na-fluorescein) administration is an established method to measure BBB permeability (38). Na-fluorescein (376 g/mol) was chosen for this study because it is one-third the size of albumin binding compounds such as Evans Blue (961 g/mol) that has been previously used to measure BBB permeability. Therefore, the use of Na-fluorescein allows for visualization of smaller, more focal BBB disruptions. Na-fluorescein (15% in saline, Sigma) was injected intraperitoneally (2 ml) one hour before sacrifice and perfusion. Rats were transcardially perfused with heparinized phosphate-buffered saline, pH 7.4. Brain tissue and cardiac blood were collected, stored at −20°C, and assayed for fluorescence within one week of collection. Na-fluorescein uptake into the brain was measured using a modification of a previous method (39). Brains were regionally dissected into frontal cortex, sub-cortex, infusion cortex (cortex below the area of the dura that is being stimulated by the inflammatory soup), periaqueductal grey, and trigeminal nucleus caudalis (Figure 1). Since differences in fluorescence between groups were found only in the trigeminal nucleus caudalis (TNC), we quantified that region for drug studies. To control for the effects of surgery, saline infused rats were used for quantification of the cortex directly below the infusion site.
Figure 1.
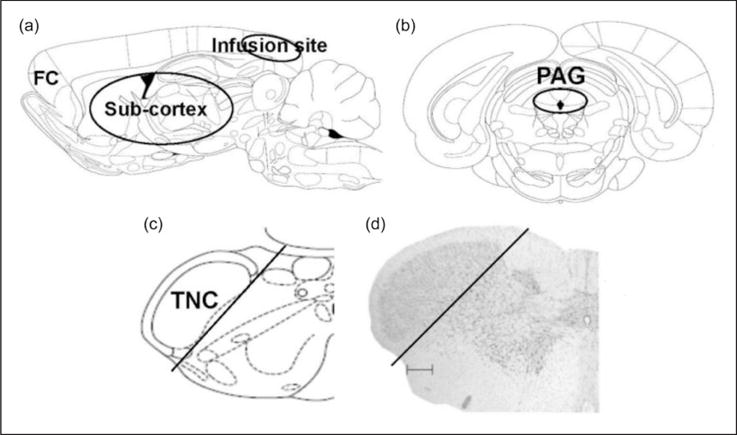
Schematic representation of dissected brain regions. (a) Sagittal view of rat brain dissected regions of frontal cortex (FC), sub-cortex and the cortex below the area of the dura that is being stimulated by the inflammatory soup (infusion site). (b) and (c) Coronal sections showing dissected regions for PAG (black circle) and TNC (black line). (d) Nissl stain of coronal section of brainstem showing TNC dissected region (black line, scale bar 300 μm). ((a), (b), and (c) are adapted from (56)).
Tissue was homogenized in 200 μl of cold 1:1 solution (7.5% trichloroacetic acid: 5N NaOH) using a dounce tissue grinder. Homogenates were plated in duplicate on a 96 well plate. Serum and sample levels of Na-fluorescein were assessed with a Cytofluor II fluorometer plate reader (excitation −485 nm, emission −530 nm; Perseptive Biosystems, Farmingham, MA). Standards (125 to 4000 mg/ml) were used to calculate the Na-fluorescein content of the samples in mg. Na-fluorescein uptake into tissue is expressed as the ratio [μg fluorescence per tissue/μg tissue]/[μg fluorescence sera/μl blood] to normalize values for blood levels of the dye at the time of tissue collection. These measurements were performed with the researcher blinded to the treatment group.
Immunohistochemistry
Rats underwent cardiac perfusion with heparinized saline followed by 4% paraformaldehyde. The brain and spinal cord were removed and fixed in paraformaldehyde. After cryoprotection treatment, all rat brains were rapidly frozen in −70°C isopentane precooled with dry ice. Serial cryostat sections (30 μm) were cut coronally through the brain stem containing the trigeminal nucleus caudalis (TNC), approximately from bregma −15.00 mm to bregma −17.50 mm by FD NeuroTechnologies (FD NeuroTechnologies Consulting & Services Inc, Ellicott City, MD) with avidin-biotin/immunoperoxidase using monoclonal anti-rat OX42 IgG (1:20,000; Accurate, Westbury, NY) and anti-rat GFAP (1:20,000; Accurate, Westbury, NY). After inactivating the endogenous peroxidase activity with hydrogen peroxidase, sections were incubated separately with avidin and biotin solutions (Vector Lab, Burlingame, CA) for blocking nonspecific binding of endogenous biotin, biotin-binding protein and lectins. Sections were then incubated free-floating for three days at 4°C in 0.01 M PBS containing 1% normal blocking serum and 0.3% Triton X-1OO (Sigma, St Louis, MO) and the specific antibody. The immunoreaction product was visualized according to the avidin-biotin complex method with the Vectastin elite ABC Peroxidase kit (Vector Lab, Burlingame, CA). After thorough rinses in distilled water, all sections were mounted on slides, dehydrated in ethanol, cleared in xylene, and coverslipped in Permount (Fisher Scientific, Fair Lawn, NJ). To check the specificity of the primary antibody, controls were made in separate sections as follows: l) replacement of the specific antibody with the isotype control IgG or serum; 2) omission of the secondary antibody; 3) Preabsorption of the specific antibody with the antigen.
Density of staining from the immunohistological images was quantified using a custom routine written in the R statistical programming language. Images were broken into three channels: Red, green, and blue. The green channel best represented the density of staining, so this color channel was used for quantification. A threshold value from 0 to 255 was selected visually to differentiate staining from unstained portions of the section. Each pixel with a color density value above this threshold was considered stained, and any pixel with a value below this was considered not stained. The relative staining values used to represent the density of staining across the whole image were calculated by adding up all of these stained pixels and dividing them by the number of unstained pixels. This analysis was performed with the researcher blinded to the treatment group. Each section and area analyzed was then normalized to saline controls as a relative level for analysis.
Drug treatment
Minocycline (Sigma, St Louis, MO) was dissolved in 5% sucrose in water and administered via oral gavage one hour before each infusion. A previous dose response using 50 and 150 mg/kg demonstrated that 150 mg/kg significantly reduced permeability. In episodic studies, minocycline (150 mg/kg) was administered one hour prior to the first and second IS infusions. Therefore, the episodic drug treatment groups were rats that received two saline infusions, two IS infusions, or two IS infusions with minocycline pretreatment. We referred to this group as “episodic” because permeability was measured one hour after the second infusion, during a time when dural stimulation results in only acute changes in trigeminal sensitivity and not long-lasting changes. In the chronic studies, minocycline (150 mg/kg) was administered one hour prior to all 10 IS infusions by a different researcher than the one who would perform the sensory testing, to ensure blinding. The chronic drug treatment groups were 10 saline infusions, 10 IS infusions, or 10 IS infusions with minocycline pretreatment. Periorbital sensory thresholds were recorded in the morning prior to drug treatment and infusion. This is the “chronic” condition, because permeability was measured one week after the last infusion when animals have developed chronic trigeminal sensitivity and have been confirmed to have sustained trigeminal sensitivity prior to analysis.
Statistical analyses
All statistical analyses were performed using SPSS version 12 (IBM, Armonk, New York). Sufficient power was confirmed with a power analysis for these studies using PASS software (NCSS, Kaysville, UT) and SPSS to limit the number of animals needed. Naive and IS- infused rats (one week after the 10th infusion) were compared at frontal cortex, sub-cortex, periaqueductal grey, and TNC using a one-way ANOVA. Saline and IS-infused rats (one week after the 10th infusion) were compared at cortex directly below the infusion site using a one-way ANOVA. The TNC of saline, IS- infused, and minocycline treated animals one hour after the second infusion were compared using a one- way ANOVA. The TNC of saline, IS infused, and minocycline treated animals one week after the 10th infusion were compared using a one-way ANOVA. Periorbital pressure thresholds were measured prior to each infusion in all groups. A one-way ANOVA was used to compare thresholds one week following the 10th infusion in all groups and one hour following the second infusion in all groups. A one-way ANOVA was used to compare immunohistological images between groups at each section level. Bonferroni post hoc tests were done where applicable.
Results
We measured BBB permeability of the trigeminal nucleus caudalis (TNC), periaqueductal grey (PAG), sub-cortex, cortex directly below the area of the dura that was being stimulated by the inflammatory soup (infusion cortex), and frontal-cortex in 82 adult male Sprague Dawley rats while monitoring the development of trigeminal pain during 3–4 weeks of inflammatory dural stimulation (Figure 1). The effect of minocycline was assessed via its ability to block pain and prevent BBB disruption.
Region-specific changes in blood-brain barrier permeability occur following the transition to chronic trigeminal sensitivity
Sodium fluorescein, a small molecule that only crosses into the brain parenchyma when the BBB is disrupted, was administered systemically one week after receiving the 10th IS infusion to measure changes in BBB permeability during the chronic stage of inflammatory dural stimulation, in which the rats have transitioned to a sustained chronic state of trigeminal sensitivity. Sodium fluorescein (376 g/mol) was used as opposed to Evans blue (961 g/mol) because it has a smaller molecular weight, and thus can serve as a much more sensitive marker for changes in BBB permeability.
Fluorescence-based analysis with a plate reader revealed a significant 7.9 fold increase in BBB permeability in the TNC of rats that received 10 IS infusions (7.9±0.6) compared to naive rats (1.0±0.4) and rats that received 10 saline infusions (1.3±0.4) (p < 0.001, ANOVA) (Figure 2(a)). No significant difference in BBB permeability was seen in the sub-cortex between rats who received 10 IS infusions (2.1±0.6) and naive rats (2.4±0.8) (Figure 2(b)). No significant difference in BBB permeability was seen in the frontal-cortex between rats that received 10 IS infusions (4.3±1.7) and naive rats (5.8±3.3) (Figure 2(b)). No significant difference in BBB permeability was seen in the PAG between rats that received 10 IS infusions (5.3±0.5) and naive rats (7.2±1.3) (Figure 2(c)). Importantly, no significant difference in BBB permeability was seen in the infusion cortex (the cortex below the area of the dura that is being stimulated) between rats that received 10 IS infusions (7.0±1.7) and rats that received 10 saline infusions (8.3±3.6) (Figure 2(d)). These data show that repeated inflammatory stimulation of the dura induces chronic increases in BBB permeability only in the TNC, and not within the PAG, sub-cortex, frontal-cortex, or cortex below the area of the dura that is being stimulated by the inflammatory soup.
Figure 2.
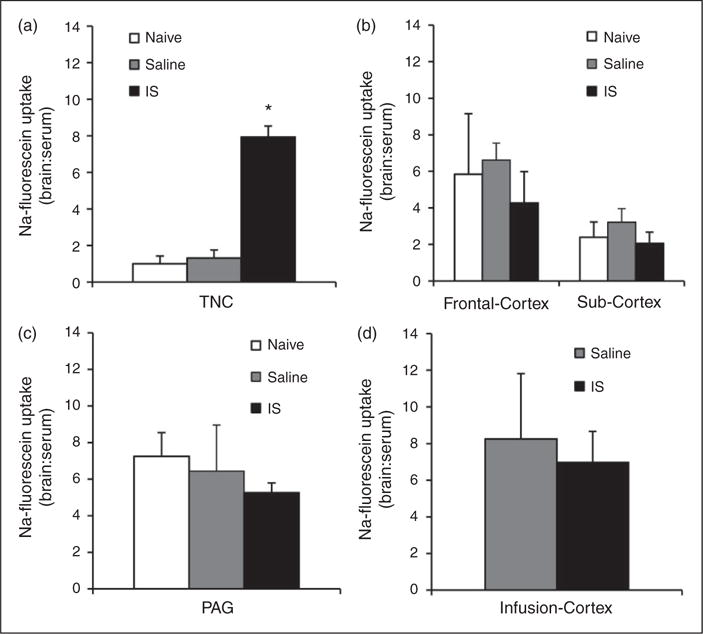
BBB permeability in multiple brain regions during the chronic stage of inflammatory dural stimulation. (a) BBB permeability (as measured by the ratio of fluorescence from sodium fluorescein uptake in the brain region in comparison to serum levels) in naive (N = 8), saline–infused (N = 7), and IS infused (N = 8) rats in the TNC. (b) BBB permeability in naive (N = 8 for each group), saline infused (N = 8 for each group), and IS infused (N = 8 for each group) rats in the frontal cortex and sub-cortex. (c) BBB permeability in naive (N = 7), saline infused (N = 7), and IS infused (N = 7) rats in the PAG. (d) BBB permeability at the site of the infusion (infusion- cortex) in saline (N = 8) and inflammatory soup infused (N = 7) rats.
TNC: trigeminal nucleus caudalis; PAG: periaqueductal grey; IS: inflammatory soup; BBB: blood-brain barrier. *p < 0.05. BBB permeability is expressed as brain:serum ratio.
Blood-brain barrier permeability is increased in the trigeminal nucleus caudalis during the episodic stage of inflammatory dural stimulation
To measure changes in BBB permeability during the episodic stage (following the second infusion, when the IS only elicits an acute period of trigeminal sensitivity that is not sustained the following day), sodium fluorescein fluorescence was measured in the TNC one hour after the second infusion. Analysis revealed a significant 4.5-fold increase in BBB permeability in the TNC one hour after receiving the second IS infusion (6.5±1.8) compared to rats that received two saline infusions (1.4±0.5) (p < 0.05, ANOVA) (Figure 3(a) – 2 IS (one hour)). Rats receiving two IS infusions also experienced increased trigeminal sensitivity one hour after the infusion, with periorbital pressure thresholds significantly falling from 9.0±0.4 g to 3.3±0.4 g (p < 0.001, ANOVA) (Figure 3(b)). To determine if these episodic changes are long lasting, like those observed when analyzing the chronic stage, sodium fluorescein fluorescence was measured in the TNC one week following the second infusion (i.e. animals only received two IS infusions). At this time point, BBB permeability was not sustained (1.4±0.2), returning to that of the saline control (Figure 3(a) – 2 IS (one week)). Periorbital thresholds also returned to baseline levels one week following the second IS infusion (9.5±0.5 g) (Figure 3(b)).
Figure 3.
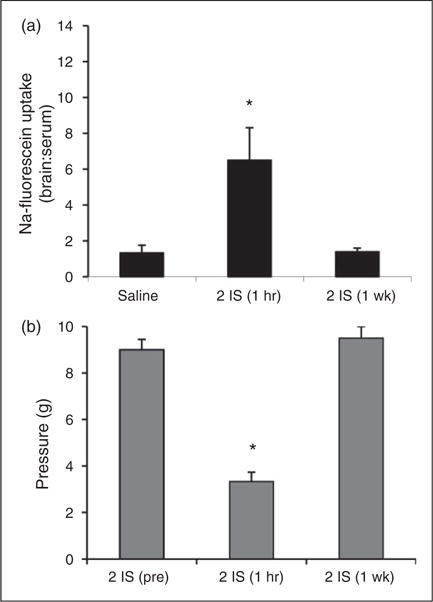
BBB permeability of the TNC and trigeminal sensitivity during the episodic stage of inflammatory dural stimulation. (a) BBB permeability (as measured by the ratio of fluorescence from sodium fluorescein uptake in the brain region in comparison to serum levels) in the TNC of saline-infused rats (saline, N = 8), one hour following the second IS infusion (2 IS (one hour), N = 8), and one week following the second IS infusion (2 IS (one week), N = 8). (b) Periorbital von Frey thresholds (g) of rats prior to their second infusion (2IS (pre)), one hour post their second infusion (2 IS (one hour)), and one week post their second infusion (2IS (one week)) (N = 8) *p < 0.05. BBB permeability is expressed as brain:serum ratio.
Astrocyte and microglial activation in the trigeminal nucleus caudalis only occurs following the transition to chronic trigeminal sensitivity
To further characterize changes that may contribute to BBB modulation during the episodic and chronic stages, we investigated activation of astrocytes and microglia, two glial cell types that play a critical role in BBB maintenance, throughout the TNC every 300 μm between −13.5 to 16.2 mm posterior to bregma for at least 10 sections from each brain. Using antibody markers suggestive of astrocyte (GFAP) and microglia (OX42) activation, we investigated three areas of each section of the TNC (dorsal, medial, and ventral) along this rostral-caudal axis to determine if region-specific changes could be observed. The dorsal and ventral areas included analysis of lamina 1 and 2, while the medial area included analysis of lamina 3 and 4. Regions of these areas in the TNC are innervated by the mandibular (V3), maxillary (V2), and ophthalmic (V1) divisions of the trigeminal nerve (Figure 4) (40,41). Transitioned rats experience mechanical hypersensitivity in the periorbital region of the face, which transmits signals to the TNC via both the V1 and V2 branches of the trigeminal nerve. This region also receives input from the dura at the site of the IS infusion (7,31).
Figure 4.
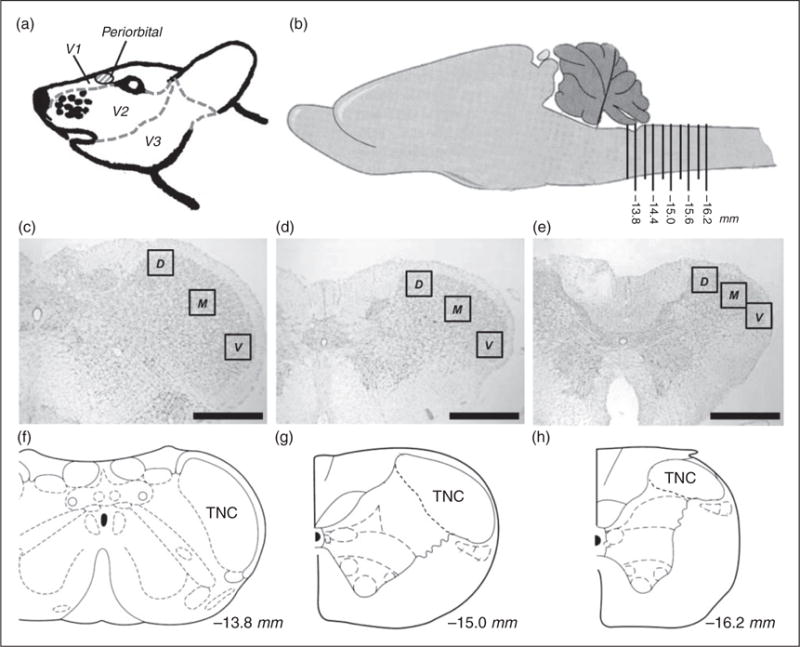
Schematic representation of regions analyzed for astrocyte and microglial activation. (a) Side-view of rat trigeminal region that is innervated by the V1, V2, and V3 branches of the trigeminal ganglion. The periorbital region where VFH testing is performed is seen in red. (b) Side-view of the rat brain featuring the region of the brainstem analyzed. Ten caudal regions, 300 μm apart each, were analyzed along the brainstem representing −13.5 to −16.2 mm posterior to bregma. (c) Nissl stain of coronal representing the second region analyzed section −13.8 mm posterior to bregma. (d) Nissl stain of coronal section representing the sixth region analyzed −15.0 mm posterior to bregma. (e) Nissl stain of coronal section representing the 10th region analyzed −16.2 mm posterior to bregma. (f) Coronal view of −13.8 mm posterior to bregma showing the TNC. (g) Coronal view of −15.0 mm posterior to bregma showing the TNC. (h) Coronal view of −16.2 mm posterior to bregma showing the TNC. Black boxes in (c), (d), and (e) represent the regions of analysis for dorsal (D), medial (M), and ventral (V) of the TNC. (Black line, scale bar 500 μm) *p < 0.05. (b), (f), (g), and (h) are adapted from (56).
Although BBB and trigeminal sensitivity changes were seen in the episodic stage, no areas of the TNC had significantly higher levels of either GFAP or OX42 staining in comparison to saline controls during this stage (Figure 5). Since there were greater changes in BBB permeability and trigeminal sensitivity during the chronic stage, we measured GFAP and OX42 staining throughout the TNC one week following the last infusion. Significant increases in GFAP staining were seen in ventral, medial, and dorsal regions throughout the rostral end of the TNC (Figure 6(a), (c), (e)). GFAP staining was significantly higher in the ventral region of the TNC from −13.5 to −15.0 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6(a)). GFAP staining was significantly higher in the medial region of the TNC from −13.5 to −15.6 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6(c)). GFAP staining was significantly higher in the dorsal region of the TNC from −13.5 to −15.0 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6(e)). OX42 staining was significantly higher in the ventral, medial, and dorsal regions throughout the caudal end of the TNC (p < 0.05, ANOVA) (Figure 6(b), (d), (f)). OX42 staining was significantly higher in the ventral region of the TNC from −14.1 to −16.2 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6(b)). OX42 staining was significantly higher in the medial region of the TNC from −14.4 to −15.9 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6(d)). OX42 staining was significantly higher in the rostral region of the TNC from −15.3 to −16.2 mm posterior to bregma (p < 0.05, ANOVA) (Figure 6 (f)).
Figure 5.
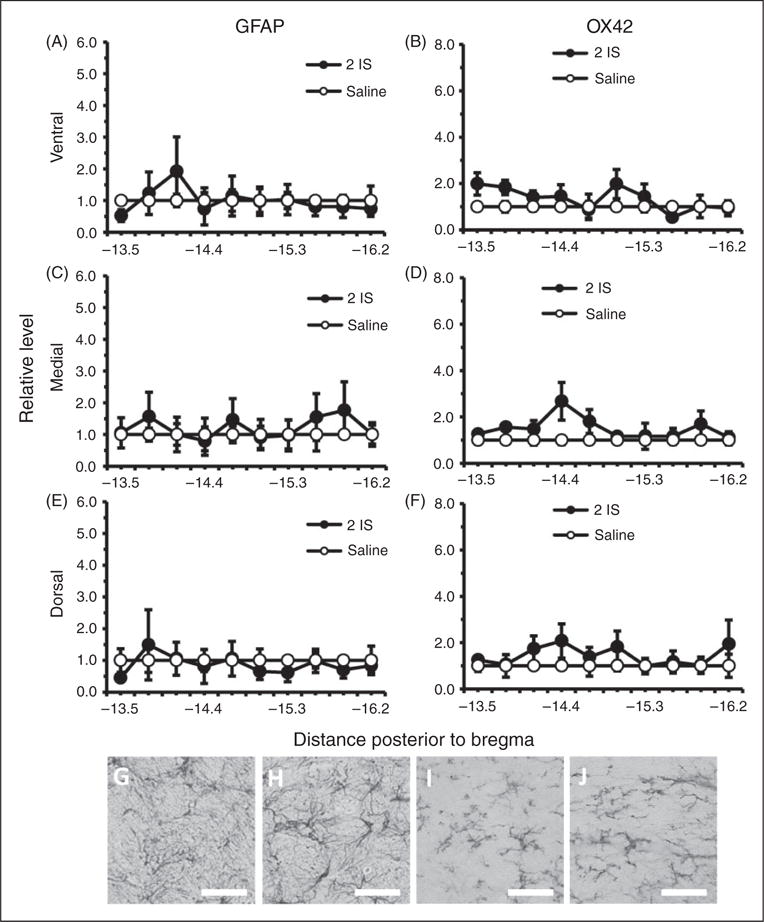
Astrocyte and microglial labeled cells in the TNC during the episodic stage of inflammatory dural stimulation. (a), (c), and (e) Glial fibrillary acid protein (GFAP) labeled astrocyte immunoreactivity relative to saline controls, within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 mm posterior to bregma) one hour following the second IS infusion (2IS) or the second saline infusion. (b), (d), and (f) Microglial activation as measured by OX42 immunoreactivity relative to saline controls within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 mm posterior to bregma) one hour following the second IS infusion (2IS) or the second saline infusion. (g) 40× representative GFAP immunoreactivity of TNC medial region −13.8 mm posterior to bregma one hour following second IS infusion. (h) 40× representative GFAP immunoreactivity of TNC medial region −13.8 mm posterior to bregma one hour following second saline infusion. (i) 40× representative OX42 immunoreactivity of TNC medial region −15.9 mm posterior to bregma one hour following second IS infusion. (j) 40× representative OX42 immunoreactivity of TNC medial region −15.9 mm posterior to bregma one hour following second saline infusion.
(Black line, scale bar 75 μm) N = 4 for all groups. GFAP: astrocyte activation marker; OX42: microglia activation marker; *p < 0.05.
Figure 6.
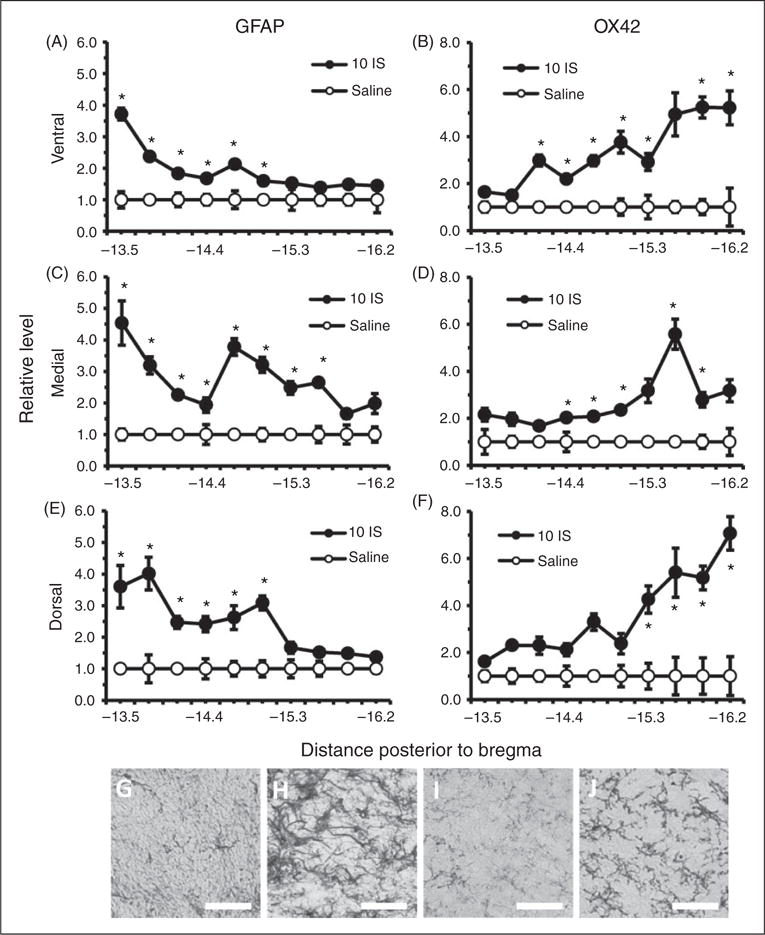
Astrocyte and microglial cells in the TNC one week following the 10th inflammatory dural stimulation. (a), (c) and (e) Glial fibrillary acid protein (GFAP) labeled astrocyte immunoreactivity relative to saline controls within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 posterior to bregma) one week following the 10th IS infusion (10IS) or the 10th saline infusion. (b), (d) and (f) Microglial staining as measured by OX42 immunoreactivity relative to saline controls within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 mm posterior to bregma) one week following the 10th IS infusion (10IS) or the 10th saline infusion. (g) 40× representative GFAP immunoreactivity of TNC dorsal region −13.5 mm posterior to bregma one week following 10th IS infusion. (h) 40× representative GFAP immunoreactivity of TNC dorsal region −13.5 mm posterior to bregma one week following 10th saline infusion. (i) 40× representative OX42 immunoreactivity of TNC ventral region −15.9 mm posterior to bregma one week following 10th IS infusion. (j) 40× representative OX42 immunoreactivity of TNC ventral region −15.9 mm posterior to bregma one week following 10th saline infusion.
(Black line, scale bar 75 μm) N = 4–5 for all groups. GFAP: astrocyte activation marker; OX42: microglia activation marker; *p < 0.05.
To attain a better understanding of the temporal changes in astrocyte and microglial activation in the TNC following inflammatory dural stimulation, we measured GFAP and OX42 staining one hour following the 10th IS infusion. This allowed us to examine the immediate effects of the inflammatory soup on astrocyte and microglial activation during the chronic stage when the trigeminal system may be sensitized to these effects. Increased GFAP and OX42 staining was seen at this time point, but it was not as widespread throughout the TNC. We found that GFAP staining was significantly increased in rostral portions of the TNC (Figure 7(a), (c), (e)). GFAP staining was increased at −13.8 to −14.1 mm posterior to bregma in the ventral and dorsal regions of the TNC, but only at −13.8 mm posterior to bregma in the medial region (p < 0.05, ANOVA) (Figure 7(a), (c), (e)). OX42 staining was increased in central portions of the TNC: −15.0 and −15.6 mm posterior to bregma in ventral, −14.7 to −15.0 mm posterior to bregma in medial, and −14.0 to −14.7 mm posterior to bregma in dorsal (p < 0.05, ANOVA) (Figure 7(b), (d), (f)).
Figure 7.
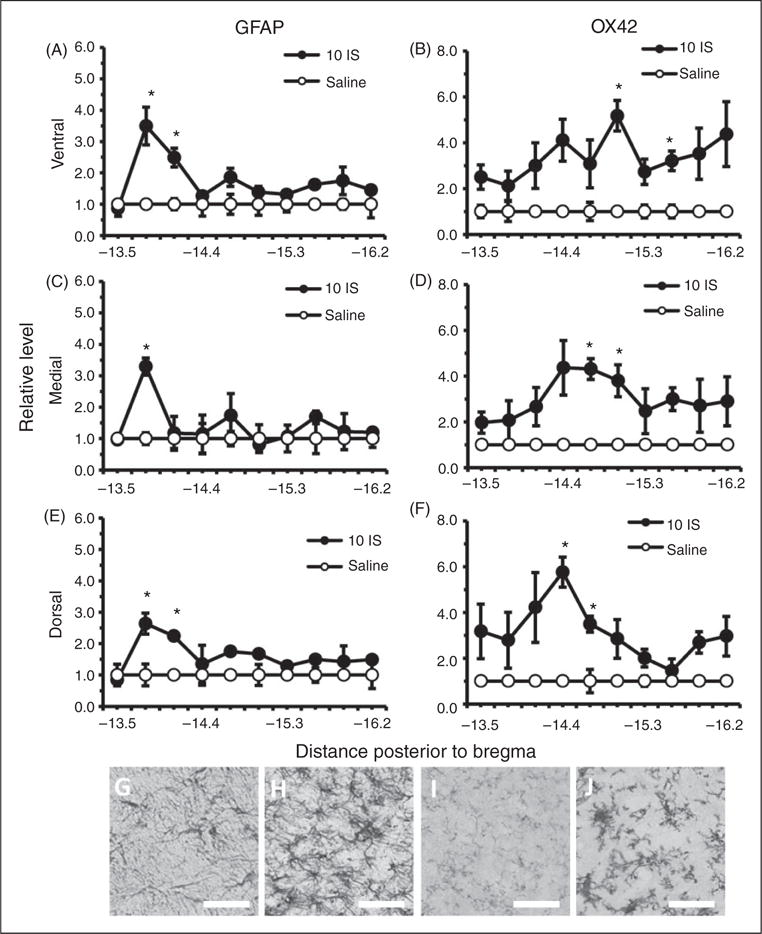
Astrocyte and microglial cells in the TNC one hour following the 10th inflammatory dural stimulation. (a), (c) and (e) Glial fibrillary acid protein (GFAP) labeled astrocyte immunoreactivity relative to saline controls within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 mm posterior to bregma) one hour following the 10th IS infusion (10IS) or the 10th saline infusion. (b), (d) and (f) Microglial staining as measured by OX42 immunoreactivity relative to saline controls within the dorsal, medial, and ventral area of the TNC at 10 regions along the rostral-caudal axis (representing −13.5 to −16.2 mm posterior to bregma) one hour following the 10th IS infusion (10IS) or the 10th saline infusion. (g) 40× representative GFAP immunoreactivity of TNC dorsal region −13.8 mm posterior to bregma one hour following 10th IS infusion. (h) 40× representative GFAP immunoreactivity of TNC dorsal region −13.8 mm posterior to bregma one hour following 10th saline infusion. (i) 40× representative OX42 immunoreactivity of TNC medial region −15.0 mm posterior to bregma one hour following 10th IS infusion. (j) 40× representative OX42 immunoreactivity of TNC medial region 15.0 mm posterior to bregma one hour following 10th saline infusion. (Black line, scale bar 75 μm) N = 4–5 for all groups.
GFAP: astrocyte activation marker; OX42: microglia activation marker; *p < 0.05.
Minocycline treatment prevents changes in blood-brain barrier permeability and periorbital sensitivity during the episodic and chronic stages of repeated inflammatory dural stimulation
Since minocycline can prevent activation of astrocytes and microglia to prevent breakdown of the BBB, rats were treated with 150 mg/kg p.o. minocycline one hour before each IS infusion (33,34). BBB permeability and trigeminal sensitivity were measured during the episodic and chronic stages of transitioning to chronic trigeminal sensitivity. Rats receiving minocycline treatment prior to each infusion (2 Mino + 2 IS (1 hr)) did not experience an increase in BBB permeability during the episodic stage, with fluorescence levels in the TNC remaining low at 1.9±0.7 (Figure 8(a)). Rats receiving this treatment also did not experience an increase in trigeminal sensitivity one hour after the second IS infusion, with periorbital thresholds (9.0±0.6 g) remaining similar to pre-infusion thresholds (9.0±0.4 g) (Figure 8(b)).
Figure 8.
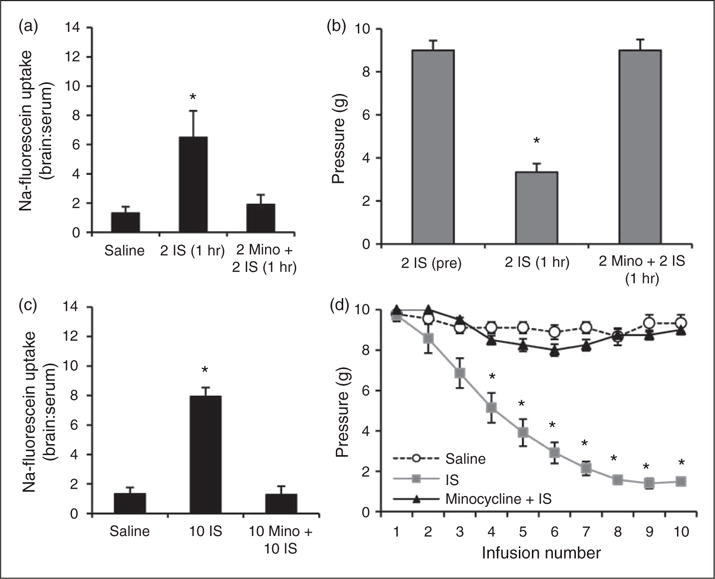
Minocycline treatment blocks changes in blood-brain barrier permeability and trigeminal sensitivity during episodic and chronic stages of inflammatory dural stimulation. (a) BBB permeability (as measured by the ratio of fluorescence from sodium fluorescein uptake in the brain region in comparison to serum levels) in the TNC one hour following the second saline infusion (N = 7), one hour following the second IS infusion (N = 7), and one hour following the second IS infusion/minocycline treatment (N = 8). (b) Periorbital von Frey Thresholds (g) for rats before their second IS infusion (N = 8), one hour after their second IS infusion (N = 8), and one hour after their second IS infusion/minocycline treatment (N = 8). (c) Changes in BBB permeability one week following the 10th saline infusion (n = 8), one week following the 10th IS infusion (10 IS, N = 8), and one week following the 10th IS infusion/minocycline treatment (10Mino + 10IS, N = 8). (d) Changes in periorbital pressure threshold across 10 infusions (three weeks) of saline, IS, or IS/minocycline treatment. Note: Normal variability with saline infusion ranges one von Frey step (i.e. 10 g to 8 g). BBB permeability is expressed as brain:serum ratio.
The 7.9-fold increase in BBB permeability in the TNC of rats following 10 IS infusions was prevented with minocycline treatment. Rats receiving minocycline treatment (10 Mino + 10 IS) had no significant increase in BBB permeability in the TNC (1.3±0.6) (Figure 8(c)). Furthermore, minocycline treatment prevented the development of chronic trigeminal pain. Rats receiving IS infusions began to develop trigeminal sensitivity after the third infusion, whereas minocycline treated rats did not (Figure 8(d)). After 10 IS infusions, rats maintained periorbital thresholds (1.6±0.2 g) that were significantly lower than those of saline-infused controls (8.8±0.3 g) (p < 0.001) (Figure 8(d)). Minocycline treatment prevented this increase in trigeminal sensitivity, with periorbital thresholds remaining similar to those of saline-infused controls (8.7±0.4 g) (Figure 8(d)). These data illustrate that minocycline treatment is effective in preventing the breakdown of the BBB in the TNC during the episodic and chronic stages of transitioning to chronic trigeminal sensitivity. In doing so, this treatment also prevents the development of periorbital sensitivity in our rat model of trigeminal allodynia.
Discussion
This study reveals a potential role of the blood-brain barrier in the development of chronic trigeminal pain following repeated inflammatory dural stimulation. Infusion of an inflammatory soup onto the dura increases BBB permeability in the TNC, and can be seen during the episodic stage (one hour after the second IS infusion) and chronic stage (one week after 10th IS infusion) of transitioning to chronic trigeminal sensitivity. Sub-cortical regions and the periaqueductal grey, which also play a role in nociception signaling and processing, do not, however, feature changes in BBB permeability. Furthermore, no BBB changes were seen in the cortex directly below the area of the dura that is being stimulated by the inflammatory soup. These data suggest that the regionally-specific increase in permeability may be due to recurrent activation of meningeal afferent fibers and not cortical activation beneath the infusion site. The ventral, medial, and dorsal areas along the entire length of the TNC had limited astrocyte and microglial activation (as measured by OX42 and GFAP staining, respectively) during the episodic stage, but widespread activation throughout the TNC during the chronic stage, presenting astrocyte and microglial activation as a potential conduit to the observed changes in BBB permeability. Microglial activation was isolated to the most caudal area of the TNC, while astrocyte activation was isolated to the more rostral portion of the TNC. Changes in BBB permeability were prevented with minocycline treatment, a tetracycline antibiotic known to block BBB breakdown via inhibition of microglial and astrocytic activation (35–37). Minocycline treatment also blocked the induction of trigeminal sensitivity during the episodic and chronic stages. These findings identify that increased BBB permeability in the TNC occurs during the onset and chronification of trigeminal pain following inflammatory dural stimulation in this rat model.
Beggs et al. (2010) also demonstrated that sciatic nerve constriction increased permeability in the BBB and blood-spinal cord barrier (23). This study found a global increase in BBB permeability one and two days after injury, with permeability returning to baseline levels after seven days. They did not, however, see initial BBB changes at six hours. These data demonstrate that peripheral nerve injury can cause transient increases in BBB permeability that are not seen initially and diminish seven days post-injury. Similarly, we found that BBB permeability returned to baseline levels seven days after the second IS infusion. Unlike Beggs and colleagues, however, we found initial (1 hr post second infusion) increases in BBB permeability. These initial changes in BBB permeability may be due to our use of sodium fluorescein (376 g/mol) instead of Evans Blue (961 g/mol), as Beggs and colleagues used. This lower molecular weight increases the sensitivity of the marker for fluctuations in permeability.
Although a similar study demonstrated that a single dural IS infusion did not modify BBB permeability at 2 or 24 hours in the TNC, the IS concentration in this study was 10–100 fold lower than that used here (42). This suggests that there may be a dose-dependent effect of the IS on BBB permeability. Furthermore, we observed chronic increases (one week post 10th infusion) in permeability. Taken together, these data suggest that prolonged changes in BBB permeability are possible, but may only occur after multiple peripheral inflammatory soup stimulations.
Our analysis of other brain regions involved in trigeminal pain processing revealed that disruption of the BBB is localized to the TNC. Subcortical regions and the PAG, which play a vital role in descending inhibition, had no changes in permeability. This is similar to the clinical findings of Schankin et al. (2016), who also did not see changes in the BBB within the brain or the raphe nucleus (22). Changes in BBB permeability may only occur in regions innervated by primary afferents, such as C-fibers, which release both substance-P and calcitonin gene-related peptide (CGRP) at the site of insult and substance-P within the dorsal horn (43,44). Release of substance-P, which induces microglial activation that leads to astrocyte activation in the TNC following stimulation of dural nociceptors, may lead to these transient changes in BBB permeability through modulation of tight junctions (11,45). Repeated dural stimulation could cause this transient mechanism to sensitize over time, leading to chronic BBB permeability changes. This sequence of activation could also sequester astrocytes from their vital role in neuronal modulation, changing neuronal excitability via microglial ATP release (45). Furthermore, mitochondrial dysfunction identified in migraineurs and this rat model could exacerbate changes in BBB permeability via the release of adenosine and reactive oxygen species (16,25,46,47).
The acute increase in BBB permeability of the TNC following repeated IS infusion also has interesting implications in that this change may transiently allow sumatriptan, a migraine therapeutic that is not permeable to the BBB in normal conditions, to gain access to central sites of action during a migraine attack (48). Although this may allow sumatriptan (295 g/mol) access, since it is smaller than sodium fluorescein (376 g/mol), it may not allow access of larger migraine therapeutics, such as CGRP antibodies or even dihydroergotamine. Although Schankin et al. (2016) found that 11C-dihydroergotamine (583.7 g/mol) did not gain access to the brain or brainstem during GTN-induced headache in migraineurs, their measurements focused on the raphe nucleus, which is more rostral than the TNC where we measured (22). Future rodent studies could examine the penetration of specific treatments to determine if larger therapies could gain central access via the TNC, while human studies could examine BBB disruption more caudally in this region of the brainstem.
Astrocyte and microglial activation was analyzed via assessment of GFAP and OX42, respectively, activation marker immunoreactivity in three areas of the TNC caudal plane (ventral, medial, and dorsal) throughout the entire rostral-caudal axis of the TNC, to better understand the potential mechanism behind BBB permeability modulation (Figures 5–7). No astrocytic or microglial activation was seen one hour after the second infusion, but BBB permeability and trigeminal sensitivity were both increased at this time point (Figure 5). Although paradoxical, this may be indicative of sub-threshold physiological changes in astrocyte and microglial activation states that are undetectable by our methodology at this early stage, but able to produce these profound changes in BBB permeability. Astrocyte and microglial activation, however, was seen throughout all regions of the TNC one week following the 10th infusion (Figure 6). Interestingly, a regionally specific characteristic of activation was found during this time point, in that astrocytic activation appeared to occur primarily in the rostral half of the TNC while microglial activation occurred in the caudal half. This spatial characteristic is reminiscent of the ellipsoidal, loculated cystic cavity that forms in models of spinal cord injury, in which the cavity is filled by activated microglia and surrounded by activated astrocytes, suggesting that similar mechanisms may be occurring from repeated and chronic activation of the trigeminal system (49). The development of this spatial characteristic may begin one hour after the last infusion during this chronic stage, radially spreading in the rostral and caudal directions and subsequently leading to astrocytic activation on the peripheral edges of this region (Figure 7). This immediate effect of activation may occur after the 10th infusion and not the second because the system could be primed and sensitized to dural stimulation following multiple IS infusions. The pattern of astrocyte and microglial activation one hour following the 10th infusion suggests that the major innervations of afferents that are sensitized to dural inflammatory stimulation may be between −14.7 and −15.0 mm posterior to bregma within the TNC. Interestingly, the diffuse nature of astrocyte and microglial activation throughout all three regions (ventral, medial, dorsal) of the TNC at each distance from bregma is reminiscent of activation patterns in the spinal cord of visceral organs, as seen with c-fos labeling following mechanical stimulation of the dura (40). Since cutaneous stimulation results in more focal c-fos labeling, our widespread astrocyte and microglial activation supports the hypothesis that this glial activation is due to dural stimulation.
Andreou et al. (2010) speculated that the application of inflammatory mediators onto the dura could locally alter BBB function, causing cortical activation at the infusion site rather than synaptic activation of second order neurons, modeling meningitis, not migraine (50). This idea stems from the cortical spreading depression (CSD) model of headache, where direct meningeal mechanical stimulation or meningeal potassium chloride administration cause neuronal activation and a spreading wave of cortical depolarization, followed by inhibition. In the CSD model, stimulating the meninges results in activation of cortex below the meninges and BBB disruptions (51). Furthermore, Zhao et al. (2016) demonstrated that PGE2 and 5-HT in the IS may permeate through the dura to gain access to the brain, suggesting that some aspects of this model are centrally- and not peripherally-mediated (52). We found, however, that there were no changes in BBB permeability in the cortex directly underneath the site of dural stimulation with the IS or in other brain regions, demonstrating that BBB permeability changes in the TNC are not centrally-mediated. Thus, this model is likely not inducing physiological changes that may be considered a model of meningitis.
Minocycline has shown promise as a neuroprotectant in clinical studies (37). With in vitro and animal studies, minocycline inhibits microglia activation, decreases iNOS activity, prevents glutamate toxicity, and prevents caspase 1 induced apoptosis (35,53–55). While these properties may all contribute to reduced BBB permeability, inhibition of microglia activation is noteworthy since we see increased microglial activation within the TNC. Supporting this, daily administration of minocycline reduced mechanical allodynia and microglia activation following spinal nerve ligation (33) and in a rodent model of sciatic nerve inflammation (36). This suggests that microglia may play a role in increasing BBB permeability following inflammatory meningeal afferent activation, since trigeminal hypersensitivity and BBB breakdown can be blocked with minocycline.
Our findings suggest that there is region-specific increases in BBB permeability isolated to the TNC in rats that have had repeated inflammatory dural stimulation. This, along with periorbital sensitivity, can be blocked with minocycline. These data complement the clinical evidence that suggests a role of the BBB in migraine, supporting the use of this model in future studies of the BBB in trigeminal pain.
Highlights.
Region-specific BBB disruption in the TNC is induced by dural stimulation.
Region-specific glial activation in the TNC is induced by dural stimulation.
Minocycline blocks the observed BBB disruption.
Minocycline blocks the trigeminal allodynia developed from this dural stimulation.
Acknowledgments
The authors would like to thank D Craig Hooper, Marzena Pedrini, Stephen D Silberstein, Jan Hoek, Erin Seifert, Davide Trotti, Lynne Kaiser, Marnie Cooper, Leyla Murphy, Imikomobong Ibia, and the Jefferson Headache Center.
Author contributions
NTF and CRM contributed equally as co-first authors by designing studies, acquiring and analyzing data, and drafting the manuscript as co-first authors. MLO designed, analyzed, supervised the study, and critically revised the manuscript. MBE made experimental, data interpretation, and editorial contributions to the manuscript. Each author gave final approval of the version to be published.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (F31-AA017852 to CRM, R01-NS061571 to MLO, NIAAA K05-AA017261 to NTF), and the Migraine Research Foundation.
Abbreviations
- BBB
Blood brain barrier
- IS
Inflammatory soup
- TNC
Trigeminal nucleus caudalis
- PAG
Periaqueductal grey
References
- 1.Harper AM, MacKenzie ET, McCulloch J, et al. Migraine and the blood-brain barrier. Lancet. 1977;1:1034–1036. doi: 10.1016/s0140-6736(77)91262-4. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Shi XQ, Echeverry S, et al. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amenta PS, Jallo JI, Tuma RF, et al. Cannabinoid receptor type-2 stimulation, blockade, and deletion alter the vascular inflammatory responses to traumatic brain injury. J Neuroinflam. 2014;11:191. doi: 10.1186/s12974-014-0191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg GA. Neurological diseases in relation to the blood–brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–1151. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Georgieva JV, Hoekstra D, Zuhorn IS. Smuggling drugs into the brain: An overview of ligands targeting transcytosis for drug delivery across the blood-brain barrier. Pharmaceutics. 2014;6:557–583. doi: 10.3390/pharmaceutics6040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oshinsky ML, Sanghvi MM, Maxwell CR, et al. Spontaneous trigeminal allodynia in rats: A model of primary headache. Headache. 2012;52:1336–1349. doi: 10.1111/j.1526-4610.2012.02247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oshinsky ML, Gomonchareonsiri S. Episodic dural stimulation in awake rats: A model for recurrent headache. Headache. 2007;47:1026–1036. doi: 10.1111/j.1526-4610.2007.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 9.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte– endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 10.Wolburg H, Neuhaus J, Kniesel U, et al. Modulation of tight junction structure in blood-brain barrier endothelial cells. Effects of tissue culture, second messengers and cocultured astrocytes. J Cell Sci. 1994;107:1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]
- 11.da Fonseca ACC, Matias D, Garcia C, et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. 2014;8:362. doi: 10.3389/fncel.2014.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricci G, Volpi L, Pasquali L, et al. Astrocyte–neuron interactions in neurological disorders. J Biol Phys. 2009;35:317–336. doi: 10.1007/s10867-009-9157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg JD, Peterlin BL, Rapoport AM. Novel approaches in migraine treatment (internet) London: Future Medicine; 2013. Microglial modulation in migraine; pp. 46–70. Available from: http://www.futuremedicine.com/doi/abs/10.2217/ebo.12.518. (accessed accessed 28 October 2015) [Google Scholar]
- 14.Teepker M, Munk K, Mylius V, et al. Serum concentrations of s100b and NSE in migraine. Headache. 2009;49:245–252. doi: 10.1111/j.1526-4610.2008.01228.x. [DOI] [PubMed] [Google Scholar]
- 15.Gursoy-Ozdemir Y, Qiu J, Matsuoka N, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Invest. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonçalves FM, Martins-Oliveira A, Lacchini R, et al. Matrix metalloproteinase (MMP)-2 gene polymorphisms affect circulating MMP-2 levels in patients with migraine with aura. Gene. 2013;512:35–40. doi: 10.1016/j.gene.2012.09.109. [DOI] [PubMed] [Google Scholar]
- 17.Kruit MC, van Buchem MA, Launer LJ, et al. Migraine is associated with an increased risk of deep white matter lesions, subclinical posterior circulation infarcts and brain iron accumulation: The population-based MRI CAMERA study. Cephalalgia. 2010;30:129–136. doi: 10.1111/j.1468-2982.2009.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruit MC, Launer LJ, van Buchem MA, et al. MRI findings in migraine. Rev Neurol (Paris) 2005;161:661–665. doi: 10.1016/s0035-3787(05)85110-0. [DOI] [PubMed] [Google Scholar]
- 19.Cutrer FM, Sorensen AG, Weisskoff RM, et al. Perfusion-weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25–31. doi: 10.1002/ana.410430108. [DOI] [PubMed] [Google Scholar]
- 20.Arnold G, Reuter U, Kinze S, et al. Migraine with aura shows gadolinium enhancement which is reversed following prophylactic treatment. Cephalalgia. 1998;18:644–666. doi: 10.1046/j.1468-2982.1998.1809644.x. [DOI] [PubMed] [Google Scholar]
- 21.Edvinsson L, Tfelt-Hansen P. The blood-brain barrier in migraine treatment. Cephalalgia. 2008;28:1245–1258. doi: 10.1111/j.1468-2982.2008.01675.x. [DOI] [PubMed] [Google Scholar]
- 22.Schankin CJ, Maniyar FH, Seo Y, et al. Ictal lack of binding to brain parenchyma suggests integrity of the blood-brain barrier for 11C-dihydroergotamine during glyceryl trinitrate-induced migraine. Brain J Neurol. 2016;139:1994–2001. doi: 10.1093/brain/aww096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beggs S, Liu XJ, Kwan C, et al. Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol Pain. 2010;6:74. doi: 10.1186/1744-8069-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cahill LS, Laliberté CL, Liu XJ, et al. Quantifying blood- spinal cord barrier permeability after peripheral nerve injury in the living mouse. Mol Pain. 2014;10:60. doi: 10.1186/1744-8069-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fried NT, Moffat C, Seifert EL, et al. Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am J Physiol – Cell Physiol. 2014;307:C1017–C1030. doi: 10.1152/ajpcell.00332.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maxwell CR, Spangenberg RJ, Hoek JB, et al. Acetate causes alcohol hangover headache in rats. PLoS One. 2010;5:e15963. doi: 10.1371/journal.pone.0015963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshinsky ML, Murphy AL, Hekierski H, et al. Noninvasive vagus nerve stimulation as treatment for trigeminal allodynia. Pain. 2014;155:1037–1042. doi: 10.1016/j.pain.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshinsky ML, Luo J. Neurochemistry of trigeminal activation in an animal model of migraine. Headache. 2006;46:S39–44. doi: 10.1111/j.1526-4610.2006.00489.x. [DOI] [PubMed] [Google Scholar]
- 29.Vermeer LMM, Gregory E, Winter MK, et al. Exposure to bisphenol A exacerbates migraine-like behaviors in a multibehavior model of rat migraine. Toxicol Sci. 2013;137:416–427. doi: 10.1093/toxsci/kft245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wieseler J, Ellis A, Sprunger D, et al. A novel method for modeling facial allodynia associated with migraine in awake and freely moving rats. J Neurosci Methods. 2010;185:236–245. doi: 10.1016/j.jneumeth.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burstein R, Yamamura H, Malick A, et al. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–982. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 32.Manzoni GC, Camarda C, Torelli P. Chronification of migraine: What clinical strategies to combat it? Neurol Sci. 2013;34:S57–60. doi: 10.1007/s10072-013-1377-x. [DOI] [PubMed] [Google Scholar]
- 33.Guasti L, Richardson D, Jhaveri M, et al. Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol Pain. 2009;5:35. doi: 10.1186/1744-8069-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin W-J, Feng S-W, Feng Z, et al. Minocycline improves postoperative cognitive impairment in aged mice by inhibiting astrocytic activation. Neuroreport. 2014;25:1–6. doi: 10.1097/WNR.0000000000000082. [DOI] [PubMed] [Google Scholar]
- 35.Chang Y-W, Waxman SG. Minocycline attenuates mechanical allodynia and central sensitization following peripheral second-degree burn injury. J Pain. 2010;11:1146–1154. doi: 10.1016/j.jpain.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Hazey MA, Van Norman AJ, Armistead DL. Melkersson-Rosenthal Syndrome with migraine-like headaches treated with minocycline: A case report and review of the literature. W V Med J. 2008;105:15–17. [PubMed] [Google Scholar]
- 38.Hooper DC, Scott GS, Zborek A, et al. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- 39.Trout JJ, Koenig H, Goldstone AD, et al. Blood-brain barrier breakdown by cold injury. Polyamine signals mediate acute stimulation of endocytosis, vesicular transport, and microvillus formation in rat cerebral capillaries. Lab Investig J Tech Methods Pathol. 1986;55:622–631. [PubMed] [Google Scholar]
- 40.Strassman AM, Mineta Y, Vos BP. Distribution of fos-like immunoreactivity in the medullary and upper cervical dorsal horn produced by stimulation of dural blood vessels in the rat. J Neurosci. 1994;14:3725–3735. doi: 10.1523/JNEUROSCI.14-06-03725.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strassman AM, Vos BP, Mineta Y, et al. Fos-like immunoreactivity in the superficial medullary dorsal horn induced by noxious and innocuous thermal stimulation of facial skin in the rat. J Neurophysiol. 1993;70:1811–1821. doi: 10.1152/jn.1993.70.5.1811. [DOI] [PubMed] [Google Scholar]
- 42.Lundblad C, Haanes KA, Grände G, et al. Experimental inflammation following dural application of complete Freund’s adjuvant or inflammatory soup does not alter brain and trigeminal microvascular passage. J Headache Pain. 2015;16:91. doi: 10.1186/s10194-015-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Block ML, Li G, Qin L, et al. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: Substance P vs. dynorphin. FASEB. 2006;20:251–258. doi: 10.1096/fj.05-4553com. [DOI] [PubMed] [Google Scholar]
- 44.Zhu J, Qu C, Lu X, et al. Activation of microglia by histamine and substance P. Cell Physiol Biochem. 2014;34:768–780. doi: 10.1159/000363041. [DOI] [PubMed] [Google Scholar]
- 45.Pascual O, Achour SB, Rostaing P, et al. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci. 2012;109:E197–205. doi: 10.1073/pnas.1111098109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carman AJ, Mills JH, Krenz A, et al. Adenosine receptor signaling modulates permeability of the blood-brain barrier. J Neurosci. 2011;31:13272–13280. doi: 10.1523/JNEUROSCI.3337-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stuart S, Griffiths LR. A possible role for mitochondrial dysfunction in migraine. Mol Genet Genom. 2012;287:837–844. doi: 10.1007/s00438-012-0723-7. [DOI] [PubMed] [Google Scholar]
- 48.Tfelt-Hansen PC. Does sumatriptan cross the blood- brain barrier in animals and man? J Headache Pain. 2010;11:5–12. doi: 10.1007/s10194-009-0170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gwak YS, Kang J, Unabia GC, et al. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol. 2012;234:362–372. doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreou AP, Summ O, Charbit AR, et al. Animal models of headache: From bedside to bench and back to bedside. Expert Rev Neurother. 2010;10:389–411. doi: 10.1586/ern.10.16. [DOI] [PubMed] [Google Scholar]
- 51.Gupta VK. CSD, BBB and MMP-9 elevations: Animal experiments versus clinical phenomena in migraine. Expert Rev Neurother. 2009;9:1595–614. doi: 10.1586/ern.09.103. [DOI] [PubMed] [Google Scholar]
- 52.Zhao J, Bree D, Harrington MG, et al. Cranial dural permeability of inflammatory nociceptive mediators: Potential implications for animal models of migraine. Cephalalgia. doi: 10.1177/0333102416663466. Epub ahead of print 19 August 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Festoff BW, Ameenuddin S, Arnold PM, et al. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem. 2006;97:1314–1326. doi: 10.1111/j.1471-4159.2006.03799.x. [DOI] [PubMed] [Google Scholar]
- 54.Mishra MK, Basu A. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J Neurochem. 2008;105:1582–1595. doi: 10.1111/j.1471-4159.2008.05238.x. [DOI] [PubMed] [Google Scholar]
- 55.Yrjänheikki J, Keinänen R, Pellikka M, et al. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th. Cambridge, MA: Academic Press; 1998. [Google Scholar]