

Abstract
The occurrence of illegal medicines is a well-established global problem and concerns mostly small molecules. However, due to the advances in genomics and recombinant expression technologies there is an increased development of polypeptide therapeutics. Insulin is one of the best known polypeptide drug, and illegal versions of this medicine led to lethal incidents in the past. Therefore, it is crucial for the public health sector to develop reliable, efficient, cheap, unbiased and easily applicable active pharmaceutical ingredient (API) identification and quantification strategies for routine analysis of suspected illegal insulins. Here we demonstrate that our combined label-free full scan approach is not only able to distinguish between all those different versions of insulin and the insulins originating from different species, but also able to chromatographically separate human insulin and insulin lispro in conditions that are compatible with mass spectrometry (MS). Additionally, we were also able to selectively quantify the different insulins, including human insulin and insulin lispro according to the validation criteria, put forward by the United Nations (UN), for the analysis of seized illicit drugs. The proposed identification and quantification method is currently being used in our official medicines control laboratory to analyze insulins retrieved from the illegal market.
Keywords: Illegal insulin, LC–DAD–MS/MS, Screening method, Quantification method
1. Introduction
Insulin, primary for the treatment of type 1 diabetes and occasional for type 2 diabetes, is one of the best known biopharmaceuticals. The polypeptide, endogenously produced in the pancreas, contains two chains (alpha and beta) linked by two inter-disulphide bonds and one intra-disulphide bond on the alpha chain. Initially, diabetic patients were treated with insulin originating from the pancreas of pigs or cows. Nowadays, insulin is mainly being produced by recombinant technologies for Escherichia coli or yeasts. The polypeptide is first synthetized and processed by the cells to proinsulin prior to posttranslational cleavage, with the release of the bio-active C-peptide, to mature insulin [1]. Besides human insulin, several recombinant analogues of human insulin are now being accepted by several medicines authorities.
Recent estimation from the WHO states that 347 million people worldwide have already been diagnosed with diabetes [2]. As this number is still increasing, it can result in a deficit of insulin availabilities for some parts of the world [3]. These patients might, due to insulin shortage or due to socio-economic circumstances, be drawn to the illegal market to meet their vital needs. The spurious/falsely-labelled/falsified/counterfeit (SFFC) medicines, available on the illegal market, are not subjected to the strict quality norms formulated in pharmacopoeias and therefore could harbour some serious health risks, since massive hospitalisations and at least one mortal incident have been reported in connection with the use of counterfeit insulin [4], [5]. Therefore, it is pivotal for the public health sector to develop reliable, efficient, cheap and easily applicable active pharmaceutical ingredient (API) identification and quantification strategies for routine analysis of suspected illegal insulins.
The identification of insulin has been well documented in the literature and mainly focuses on immunological assays. Since these methods also have some inherent disadvantages [6], [7], [8], [9], [10], alternative instrumental analytical methods including ultra-high performance liquid chromatography–diode array detection (UHPLC–DAD) [11], capillary electrophoresis (CE) [12], [13], [14] and micellar electrokinetic chromatography (MEKC) have been developed [15], [16], [17]. The main drawback of these methodologies is that identification solely relies on spectral information which could result in undesired false positives. Therefore, mass spectrometry (MS)-based methods have been developed [18], [19], [20], [21], [22], [23], [24], [25], [26]. The most effective and sensitive methods developed employ an immunopurification step prior to liquid chromatography–tandem mass spectrometry (LC–MS/MS) [8], [19], [20], [24], [26]. Although this methodology seems to work well for the different insulins, the application is quite limited due to the lengthy sample preparation. Alternatively, in 2014 a very sensitive and specific LC–MS/MS method for human insulin and 5 recombinant analogues was described [25]. However, the method employed a selected reaction monitoring (SRM) mode and is consequently too restricted for counterfeit analysis. Hence modifications and API-related impurities, including proinsulin and the bio-active C-peptide will not be detected. Moreover, another recently developed sensitive methodology utilising immunoaffinity purification, followed by full scan LC–MS/MS and ion mobility (LC–IM–MS/MS) on those 6 different peptides, supplemented with bovine and porcine insulin was published [26]. The authors showed that it was possible to distinguish between all different insulins by combining IM–MS/MS. However, this method is quite expensive, not only due to the equipment necessary for correct identification, but also due to the methodology that requires the use of a labelled insulin standard even for identification. In order to simplify the identification approach, we set out to develop a combined LC-MS/MS coupled to a DAD (LC–DAD–MS/MS) methodology to screen for human insulin, 6 recombinant insulin analogues, and porcine and bovine insulin.
2. Materials and methods
2.1. Standards and reagents
The reference standard of the human C-peptide (Batch 123M4758V, purity ≥95%) was purchased from Sigma–Aldrich (St. Louis, USA). Recombinant 6×histidine tagged proinsulin (batch MJL0714091, purity ≥95%) was bought from R&D systems (Minneapolis, USA). Detemir (batch 4), bovine insulin (batch 3), porcine insulin (batch 3), insulin aspart (batch 2), insulin lispro (batch 1), and insulin glargine (batch 1) were European pharmacopoeia reference standards. Insulin detemir (batch DR78773) and insulin degludec (batch DP52825) were kindly provided by Novo Nordisk® (Copenhagen, Denmark) and insulin glulisine (batch HOl 000-WS-05) was kindly provided by Sanofi (Paris, France). Commercial available insulins (Humuline® NPH (lot: C290162), Humuline® regular (lot: C324652), Humalog® (lot: C368234), Lantus® (lot: 4F031B), Apidra® (lot: 4F181A) and NovoRapid® (lot: DS6N161)) were purchased from a legal pharmacy.
Acetonitrile was ultra-liquid chromatography (ULC)-MS grade and purchased from Biosolve (Valkenswaard, the Netherlands). Water was obtained using a milliQ-Gradient A10 system (Millipore, Billerica, MA, USA). Sodium chloride (batch K45393104, purity≥99.5%), glycerol (batch S23772), sodium dihydrogen phosphate (batch K93151845), di-sodium hydrogen phosphate (batch A0252879), zinc acetate (batch A0180402) and analytical grade formic acid were bought from Merck (Darmstadt, Germany). Phenol (batch A0258794) was purchased from Acros organics (Thermo Fisher Scientific, Geel, Belgium).
2.2. Sample set of suspected illegal and legal insulins
The sample set of suspected illegal insulins consisted of 20 samples which were seized by inspectors from the Belgium Federal Agency for Medicinal and Health Products (FAMHP). The confiscation of the samples took place between 2010 and 2015. Due to the confidential nature of the inspection, it is not possible to give actual information about each sample individually. Syringes noted to exclusively contain insulin and one unlabelled vial, shown previously, by classical peptide mapping, to be positive for porcine insulin, were subjected to analysis.
Aliquots of legal pharmaceutical preparations were encoded by a technician who did not participate in that particular part of the data analysis. These blind samples were used to challenge our identification methods and quantification methods.
2.3. Sample preparation
2.3.1. Screening method
Standard stock solutions (0.3 mg/mL) were made in water containing 1% formic acid. For the determination of the retention time and the determination of diagnostic ions, working solutions of 0.05 mg/mL were made in acidified water. For validation of the screening method, peptides were diluted into the chosen matrices (see Section 2.6).
2.3.2. Sample set
The legal pharmaceutical insulin solutions and the suspected illegal syringe solutions were acidified with 1% formic acid prior to the centrifugation step preceding the LC–MS/MS analysis. The unlabelled vial, containing lyophilised powder, was reconstituted to a final volume of 500 μL of water supplemented with 1% formic acid. Again the solution was centrifuged previous to mass spectroscopic analysis.
2.3.3. Quantification method
For the generation of the calibration curves for quantitative analysis by MS, standard stock solutions were diluted into 6 different concentrations in 1% formic acid in water. The selected concentration interval of the 10 times diluted samples (50–200 µg/mL initial insulin concentration for bovine insulin, porcine insulin, insulin glargine, insulin glulisine, insulin aspart and 100–200 µg/mL for insulin detemir and insulin degludec) for the validation of our MS based quantification methodology corresponds to less than 6 international unit (IU) of insulin/mL. This is at least 16×less than the most common concentration of insulin concentration available in the market (100 IU/mL). Additionally, since UV in less sensitive than MS, we selected the concentration interval of 100–200 µg/mL.
2.3.4. Quantification of the sample set
The reconstituted powder and the blind encoded aliquots of the legal insulin solutions (see Section 2.3.2) were diluted 10 times prior to LC–MS/MS analysis. The samples were subsequently further diluted with water and acidified in 1% formic acid until a concentration within the interval of the calibration line was obtained.
2.4. Instrumental conditions
2.4.1. Screening method
The acidified polypeptide solutions were subjected to analysis on a Dionex UltiMate 3000 Rapid Separation LC (RSLC) system (Thermo Scientific, Sunnyvale, CA, USA) coupled to an amaZon™ speed ETD mass spectrometer (Bruker Daltonics, Bremen, Germany). The instrument system was calibrated using the manufacturer's calibration mixture, and the mass accuracy was determined to be <0.1 Da during the period of analysis. A sample volume of 1 μL was injected onto the system. The chromatographic separation was performed at 45 °C on an Acquity UPLC CSH C18 Column (150 mm×2.1 mm, 1.7 µm) (Waters, Milford, MA, USA) with a mobile phase consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The optimized elution method, with a constant flow rate of 0.35 mL/min, employed an isocratic run at 2% B for 1.8 min, followed by a linear gradient to 24% B at 2 min, an increase to 27% B at 11.5 min, an additional increase to 80% B at 13.0 min prior to a 2 min isocratic elution at 15 min. The column was subsequently rinsed for 2 min at 99% B and a 3 min equilibration with 2% B. The mass spectrometer settings were similar to what has been described in [27]. Briefly, the mass spectrometer was operated in positive electrospray ionization (ESI+) mode, with a spray voltage of 4.5 kV and end plate voltage of 500 V. The nebulizer was set to 2 bar and desolvation gas temperature was 250 °C at a flow rate of 10 L/min. MS spectra were obtained within a mass range of 500–1500 m/z and the smart parameter setting (SPS) was set to 1050 m/z. For MS/MS precursor selection, the most intense ion was isolated above the absolute intensity of 2.5% and 5% relative intensity threshold. The ion charge control (ICC) was set to 190 000 with a maximum accumulation time of 50 ms. Collision Induced Dissociation (CID) was performed using helium as collision gas. The fragmentation amplitude was set to 100% using SmartFrag™ Enhanced for amplitude ramping (80%–120%). Fragmentation time was set to 32 ms. The MS and MS/MS queries were performed using Compass® Data Analysis 4.2 and BioTools® 3.2 (Bruker Daltonics, Bremen, Germany) software.
2.4.2. Quantification method
For quantification by MS of all insulins, except human insulin and insulin lipro, extracted ion chromatograms were analyzed and processed using Compass® Data Analysis 4.2. The same software and Hystar® 3.2 (Bruker Daltonics, Bremen, Germany) were used for quantification of human insulin and insulin by the output of the DAD since no baseline separation could be observed in the extracted ion chromatogram. The same elution conditions were used as in Section 2.4.1. However, since a DAD detector is less sensitive than an MS detector, 5 μL was injected. The insulins were detected at 220 nm.
2.5. Creation of an insulin LC–MS/MS database
The insulin database was created with pure standard solutions. Stock solutions (0.3 mg/mL) were made in pure water and working solutions (0.05 mg/mL) were made in 1% formic acid in water for each substance. An in-house database, comprising the 9 different insulins (Table 1), was built by analyzing standard solutions in triplicate. The database contained theoretical monoisotopic exact masses, retention time, MS spectrum, MS/MS spectrum and diagnostic ions for each insulin. Theoretical monoisotopic exact masses based on their molecular formula were taken from the literature [25], [26], [28].
Table 1.
Characteristics of the different polypeptides.
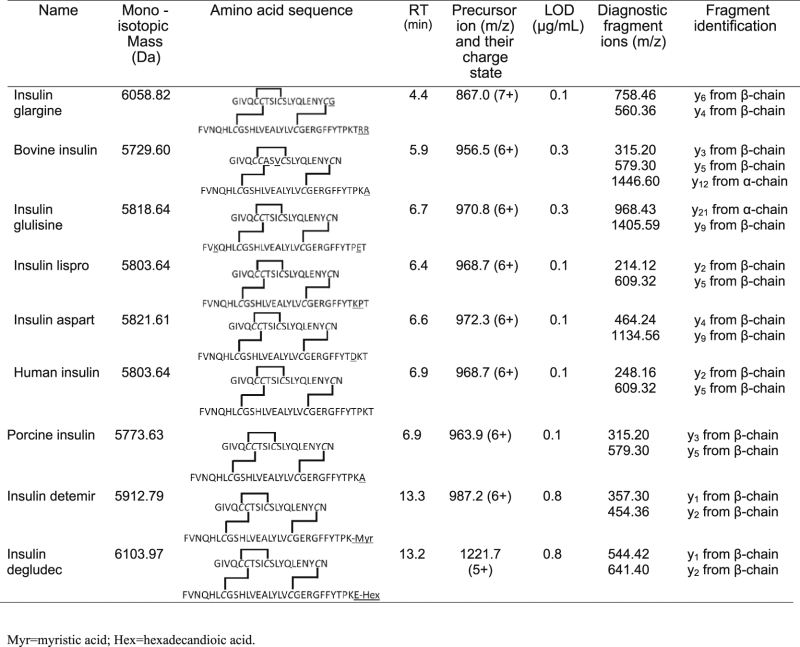 |
2.6. Validation of the screening method
A screening method should be able to identify and distinguish the different insulins from each other and from the matrix ingredients, like buffers, salts, and phenol. Furthermore, the methods used for these types of analysis should also generate no possible false positives or false negatives.
To ensure the selectivity of the screening method, the validation strategy followed was similar to the one previously described [27]. Briefly, we determined the screening detection limit (SDL), which corresponds to the lowest concentration for which it has been demonstrated that a given analyte, present in real life samples, can be detected in at least 95% of the samples. As required, the validation involved all insulins in three typical buffer matrices. Matrix 1 contained 0.9% (m/v) NaCl; matrix 2 consisted of 20 mM sodiumphosphate buffer with pH 7.2 and 5% glycerol. Matrix 3 was composed of 20 mM sodiumphosphate buffer with pH 7.2, 10 mM sodium chloride, 16 mM phenol, 1.7% glycerol and 0.6 mM zinc acetate.
In compliance with the validation procedure, the method was validated using the 9 different insulins which were analyzed before and after being spiked in the typical matrices of biopharmaceuticals.
The concentration used for validation, concurrent with an SDL of 10 µg/mL, was at least 100 times lower than the concentration of insulins present in legal commercial available products. Each insulin polypeptide was detected with its relative retention time, exact monoisotopic mass, SDL, the m/z values of the most intense precursor ion and their diagnostic fragment ions already utilised in the past [24], [25], [26] (Table 1).
2.7. Validation of the quantification method
The present method was validated by applying accuracy profiles, which are based upon the “total error” approach. Briefly stated, this approach estimates the highest error of an analytical method [29], [30], [31].
Three different solutions of each dilution were daily made and analyzed for five consecutive days. The corresponding concentrations were back-calculated using the calibration lines generated in Section 2.3.3. These calculated concentrations were then used to determine the linearity of the results, trueness, precision (repeatability and intermediated precision) and accuracy by means of an excel sheet [32] that has been successfully used by our research group [27], [32], [33], [34], [35], [36].
2.8. Forced degradation study
Standard stock solutions (0.2 mg/mL) were made in water and acidified with 0.1% formic acid and incubated at 37 °C for 7 days [17]. The possible precipitation was removed by centrifugation for 15 min at 20,238 g in a microcentrifuge, prior to injection.
3. Results and discussion
3.1. Development and optimization of the screening method
The development of the UHPLC method was based on the methods recently published for this type of polypeptide [25], [26]. However, those LC methods could not distinguish between human insulin and insulin lispro. These insulins do not differ in mass and can therefore only be distinguished from each other by the presence of their specific MS2 diagnostic ions (y2 fragment ions) [25], [26], [28]. Indeed, most of the identification methods used today are solely based on these specific fragment ions. Therefore, we set out to improve the LC-separation of these insulins in MS compatible conditions to generate an additional discrimination criterion. After using several different columns and UHPLC gradients, the best separation was obtained when using the Acquity UPLC CSH C18 Column (150 mm×2.1 mm, 1.7 µm) (Waters, Milford, MA, USA) and by expanding and modifying the gradient used by Chambers et al. [25], resulting in a total run time of 20 min. An example of the different chromatograms obtained with the optimized experimental conditions for the different insulins, proinsulin and the bio-active C-peptide is shown in Fig. 1. Based on the retention time, three different clusters can be identified, i.e., one with insulin glargine, a second cluster with bovine insulin, insulin glulisine, insulin lispro, insulin aspart, human insulin and porcine insulin and a third cluster containing insulin detemir and insulin degludec. These later can be easily separated from each other since they differ significantly in mass. To distinguish between cluster 2, containing six insulins, we combined the difference retention time, the mass of the precursor ion and at least 2 diagnostic fragment ions. Furthermore, we also observed a significant retention time difference between human insulin and insulin lispro even when we injected a mixture of these compounds. Fig. 2 illustrates the separation of human insulin and insulin lispro detected by MS (A) and by absorption at 220 nm (B).
Fig. 1.

Total ion chromatograms of a full scan mass spectrum of the 9 different insulins, proinsulin and the bio-active C-peptide. Each m/z value corresponds to the measured precursor ion with the maximum intensity.
Fig. 2.

Total ion chromatogram of a full scan mass spectrum of a 50:50 mixture of 100 µg/mL of (A) human insulin and insulin lispro and (B) the DAD chromatogram of the mixture at 220 nm.
The initial MS/MS method, described by Vanhee et al. [27], was not optimal for the fragmentation of these high molecular weight polypeptides; hence we altered the smart parameter setting (SPS) to 1050 m/z.
3.2. Validation of the screening method
The parameters tested to validate the screening method included the selectivity of the method, the sensitivity of the method, and the effect of matrix interferences.
3.2.1. Selectivity of the screening method
The selectivity of the LC–MS/MS and subsequent data analysis method was confirmed by determining the retention time of each component, their corresponding MS spectrum, the presence of diagnostic ions in the MS2 spectrum originating from the precursor ions in Table 1, and at least 3 other fragment ions (cf. the identification point strategy [27]). The error tolerated on the relative retention time was ±0.2 min. Additionally, we also tolerated a 1.5 Da mass difference between the mass calculated from the multiply charged ion envelope after deconvolution and the mass calculated from the reported amino acid sequence [37]. Furthermore, a difference of 0.4 Da was tolerated for the masses of fragment ions.
3.2.2. Sensitivity of the screening method
Next, the limits of detection (LOD) were determined based on the methods described by the International Conference on Harmonization (ICH) [38], the European Pharmacopoeia [39] and the United Nations (UN) [40] (Table 1). Basically, the LOD corresponds to that amount of injected insulin that has a signal-to-noise ratio of at least 3.3 for the diagnostic ion with the lowest intensity. With the highest LOD, being 0.8 µg/mL, one can clearly state that this method is sensitive enough for the detection of those polypeptides in illegal preparations, since the concentration present in insulin syringes or pumps is generally more than 3 mg/mL. As mentioned in Section 2, our methodology incorporates a 10 times dilution for real life samples prior to MS analysis. Taking this dilution into account, our SDL corresponds to 10 µg/mL insulin.
3.2.3. Matrix effect
To evaluate the matrix effect, the 9 different peptides were prepared at a concentration of 10 µg/mL in the three different matrices, prior to a 10-fold dilution for LC–MS/MS analysis (see Section 2.6). These matrices represent the common used salts, buffers, sugars and PEG that are frequently added to the polypeptides prior to lyophilisation. These compounds had no effect on the different mass spectra and had only a limited effect on the retention time since the shift in the retention time was less than 0.2 min for the different concentrations tested. Furthermore, no wrongful identification of the different insulins occurred in those three matrices and during the validation of the quantification method.
3.3. Validation of the quantification method
First, we validated the quantification method based on the extracted ion chromatograms for the 7 different insulins. Again, we diluted the standards 10 times before the quantification, concurrent to what we would do for real-life samples. Prior to full validation, we assessed the effect of the different matrices on the area of the extracted ion chromatograms and found that matrix effect was negligible since no significant difference could be found at the highest and the lowest concentrations chosen for full validation (Supplement Table A1).
Moreover, as the different MS peaks of human insulin and insulin lispro did not drop back to the baseline and no complete separation was observed, we could not reliably quantify these polypeptides by this methodology. However, our UV data showed that between 100 and 200 µg/mL we could separate human insulin and insulin lispro to baseline. Therefore, we subsequently performed the quantification of these two polypeptides based on the UV data, using the concentration interval which showed a peak resolution higher than 1.7 and with tailing factors smaller than 2.2 for a solution containing equal amounts of both insulins. Similar to the preceding step for full MS quantification, we also assessed the effect of the different matrices on the separation and quantification of those two insulins and also here we obtained no difference (Supplementary Table A1).
3.3.1. Selectivity
Once the identity of the component is determined with LC–MS/MS, the quantification is done by integrating the area of the extracted ion chromatograms. As mentioned in Section 3.1, the proposed LC–DAD–MS/MS data was selective since no wrongful identification occurred during the validation of the screening method.
3.3.2. Linearity of the calibration line
Calibration curves were obtained as described in [27]. The results are summarized in Table 2. With all R2 values for the MS-based data above 0.967, and all p-values of the LOF (lack of fit) test higher than 0.05, we concluded that linear calibration lines were fit for purpose, within the chosen concentration ranges. The same rationale as described in [27] was used for the UV-data.
Table 2.
Overview of the calibration lines associated R2 and p-values of the lack of fit (LOF) for all insulins.
| Method | R2 | LOF |
|---|---|---|
| LC–MS/MS | ||
| Insulin aspart | 0.985511 | 0.1274 |
| Bovine insulin | 0.982149 | 0.0927 |
| Insulin degludec | 0.975789 | 0.5224 |
| Insulin detemir | 0.967552 | 0.5641 |
| Insulin glargine | 0.978510 | 0.0809 |
| Insulin glulisine | 0.972743 | 0.0564 |
| Porcine insulin | 0.975399 | 0.2840 |
| LC–DAD | ||
| Human insulin | 0.997353 | 0.4886 |
| Insulin lispro | 0.995817 | 0.7947 |
Regression lines were statistically evaluated with Excel 2010 and Statgraphics ® Centurion XIV.
3.3.3. Linearity, trueness, precision, accuracy and uncertainty assessment
The present method was validated according to ISO-17025 applying accuracy profiles which are based upon the “total error” approach. The results are given in Table 3 and Fig. 3.
Table 3.
Linearity, trueness, precision, accuracy and uncertainty of the MS-based and the UV-based (only applicable to Human insulin and insulin lispro) quantification methods.
| Componds | Concentration (μg/mL) | Linearity |
Trueness (Relative bias (%)) | Precision |
Accuracy (β-Expectation tolerance limits (%)) | Uncertainty (Relative expanded uncertainty (%)) | ||
|---|---|---|---|---|---|---|---|---|
| R2 | LOF | Repeatability (RSD) | Intermediate precision (RSD) | |||||
| Insulin aspart | 50 | 0.999 | 0.320 | −10.7 | 5.73 | 5.73 | [−24.7; 3.3] | 12.07 |
| 125 | −3.27 | 2.50 | 4.47 | [−19.0; 12.5] | 10.05 | |||
| 200 | 0.65 | 2.95 | 3.30 | [−7.9; 9.2] | 7.10 | |||
| Bovine insulin | 50 | 0.999 | 0.125 | −2.40 | 8.80 | 8.80 | [−24.1; 19.3] | 18.72 |
| 125 | 2.03 | 1.56 | 3.61 | [−12.9; 17.0] | 8.21 | |||
| 200 | −4.08 | 5.46 | 6.03 | [−17.5; 9.3] | 11.51 | |||
| Insulin glargine | 50 | 0.998 | 0.451 | 1.00 | 8.43 | 10.18 | [−23.0; 25.0] | 21.56 |
| 125 | −3.29 | 5.56 | 7.12 | [−20.6; 19.9] | 15.17 | |||
| 200 | 3.52 | 2.98 | 6.12 | [−17.2; 24.2] | 14.98 | |||
| Insulin glulisine | 50 | 0.998 | 0.451 | −6.14 | 5.96 | 7.22 | [−23.2; 10.7] | 15.29 |
| 125 | −0.43 | 4.71 | 7.58 | [−20.9; 20.0] | 16.46 | |||
| 200 | −3.60 | 5.48 | 5.60 | [−16.0; 8.80] | 11.59 | |||
| Porcine insulin | 50 | 0.994 | 0.688 | −8.73 | 6.27 | 6.40 | [−24.5; 7.07] | 13.53 |
| 125 | −6.98 | 5.01 | 5.12 | [−19.6; 5.65] | 10.82 | |||
| 200 | −1.99 | 5.16 | 5.16 | [−14.6; 10.6] | 10.88 | |||
| Insulin degludec | 100 | 0.993 | 0.656 | 1.97 | 6.22 | 6.91 | [−15.9; 19.8] | 14.80 |
| 150 | −3.21 | 4.11 | 5.08 | [−17.3; 10.9] | 11.08 | |||
| 200 | 3.56 | 3.39 | 3.54 | [−5.29; 12.4] | 7.54 | |||
| Insulin detemir | 100 | 0.997 | 0.095 | −8.73 | 6.27 | 6.40 | [−24.5; 7.07] | 13.53 |
| 150 | −2.65 | 6.88 | 6.88 | [−19.5; 14.2] | 14.51 | |||
| 200 | −2.40 | 2.13 | 2.32 | [−7.62; 2.82] | 4.50 | |||
| Human insulin | 100 | 0.999 | 0.265 | −0.45 | 2.96 | 3.41 | [−9.45; 8.56] | 7.37 |
| 150 | 1.88 | 2.50 | 2.76 | [−5.21; 8.97] | 5.92 | |||
| 200 | 1.61 | 2.90 | 2.91 | [−5.53; 8.75] | 6.14 | |||
| Insulin lispro | 100 | 0.999 | 0.682 | −0.71 | 2.12 | 2.25 | [−6.36; 4.95] | 4.79 |
| 150 | −0.82 | 2.10 | 2.92 | [−9.59; 7.95] | 6.45 | |||
| 200 | −1.41 | 3.33 | 3.40 | [−9.82; 7.00] | 7.20 | |||
RSD: relative standard deviation.
Fig. 3.

Accuracy profiles of the MS-based quantification of the subset of the different insulins and the UV-based quantification of human insulin and insulin lispro with β set as 95%. Relative bias (dotted line), β-expectation tolerance limits (dashed line), acceptance limits (full line) and relative back-calculated concentrations (the following shapes:▲,◆ or ■).
3.3.3.1. Linearity
Linearity between the theoretical and measured concentration is acceptable, as R2 values were above 0.99 and the p-values of the LOF test were above 0.05 as mentioned in [27].
3.3.3.2. Trueness
The guidance put forward by the UN states that for the quantification of seized illicit drugs by MS and UV, the relative bias should not exceed 10% for the mid and highest concentration and 15% for the lowest concentration tested [40]. From Table 3 it can be concluded that the trueness for all components is acceptable since the relative bias is limited between −10.7 (lowest concentration of insulin aspart) and 3.56 for the quantification done by MS and between −0.82 and 1.88 for the quantification done by UV.
3.3.3.3. Precision
The precision of a method as defined in [40] should not exceed 15% for the highest concentrations and should not exceed 20% for the lowest concentration. Table 3 clearly demonstrates that this method results in acceptable precision for all concentrations and all components since the maximum value obtained corresponds to 10.18%.
3.3.3.4. Accuracy
Accuracy defined by the total error approach is represented by the β-expectation tolerance limits. For quantification of illegal insulins the general acceptance limits were set to [−25%; 25%]. These are the limits utilised for the quantification of 18 illegal adulterants in herbal medicines [41] and are even more stringent than what has been put forward by the UN [40]. Therefore, we reasoned that these settings are sufficient for the quantification of illicit drugs. As shown in Table 3, the β-expectation tolerance limits do not exceed the acceptance limits which means that 95% of the future measurement of unknown samples will be included within the tolerance limits. Interestingly, for human insulin and insulin lispro we achieved β-expectation tolerance values which also correspond to the criteria put forward by the European pharmacopoeia [39].
3.3.4. Limits of quantification
Based on the methods described by the International Conference on Harmonization (ICH) [38], the European Pharmacopoeia [39], and the UN [40], the LOQ was experimentally assessed by serial dilutions. The LOQ conformed to the lowest concentration of the calibration curve where the back calculated values were within ±15% of the nominal value [40], [41]. The highest LOQ for MS-based quantification of the 10 times diluted standards corresponds to 100 µg/mL (insulin detemir and insulin degludec) and the highest LOQ for UV-based quantification corresponds to 100 μg/mL (Supplementary Table A2).
3.4. Identification of legal and suspected illegal peptide biopharmaceuticals
The described methodology was applied to the sample set (see Section 2.2). None of the 19 syringes or injectable solutions marked with “for sole use of insulin” contained the polypeptide since no peak, exceeding the background noise, was observed. This indicates that if any active pharmaceutical ingredient was present, the molecule had an m/z which was less than 500 or the insulin was present in a concentration lower than 10 µg/mL, thus well below any of the biological working concentrations. Additionally, all diluted legal polypeptide preparations, used to illustrate the applicability of the method for future illicit samples, including a Neutral Protamine Hagedorn insulin (NPH insulin) preparation, were correctly identified for the insulin moiety (Table 4). However, one positive illegal sample, purchased as CJC1295, containing porcine insulin (Fig. 4), conforms with the previous outcome of a classical peptide mapping. Interestingly, we also identified a later eluting peak with a similar m/z which could correspond to a deamidated form. Additionally, forced acid catalysed degradation, performed to induce the deamidation of A21 on porcine insulin, showed that the A21 desamido porcine insulin eluted at the same time. Furthermore, insulin aspart, bovine insulin, insulin glulisine, human insulin, insulin lispro and insulin glargine were subjected to those degradation studies and all, with the exception of insulin glargine, which does not contain an asparagine at position A21, showed the occurrence of a later eluting peak with the same m/z precursor ion (Fig. 5). These findings taken together suggest that the later eluting peak in the sample might indeed correspond to the A21 deamidated form of porcine insulin. According to the European Pharmacopoeia [39], the area of the peak due to A21 desamido porcine insulin may not be greater than 2.0% of the total area of the peaks. Furthermore, the European Pharmacopoeia also states that the sum of the areas of all the peaks, apart from those due to porcine insulin and A21 desamido porcine insulin, must not be greater than 2.0% of the total area of the peaks. Briefly stated, no desamido porcine insulin might exceed 2% of the total area of the peaks. However, the area obtained in Fig. 4 clearly demonstrates that almost 70% of the insulin present in the sample was in the deamidated form. We cannot exclude that any deamidation took place during storage; however, reference standards of porcine insulin were stored in a similar manner for even longer periods of time without the occurrence of that high amount of deamidation. This suggests that the deamidation most probably took place prior to arrival in the laboratory and might be due to incorrect preparation or handling of the insulin powder.
Table 4.
Analytical results for the identification and quantification of legal and a suspected illegal pharmaceutical preparation.
| Identified compounds | Information on secondary packaging |
Amount detected (mg/mL) | |
|---|---|---|---|
| Identity of polypeptide | Amount claimed (mg/mL) | ||
| Suspected illegal preparation | |||
| Porcine insulin | n.a. | n.a. | 0.79 |
| Illustration with legal pharmaceutical preparations | |||
| Human insulin | Humuline NPH | 3.50 | 3.32 |
| Human insulin | Humuline regular | 3.50 | 3.57 |
| Insulin lispro | Humalog | 3.50 | 3.19 |
| Insulin glargine | Lantus | 3.64 | 3.14 |
| Insulin glulisine | Apidra | 3.49 | 3.47 |
| Insulin aspart | NovoRapid | 3.50 | 3.14 |
n.a.: not analyzed.
Fig. 4.

Extracted ion chromatogram of porcine insulin at 963.9 m/z±0.5 of (A) the unlabelled vial that contains porcine insulin and (B) a reference standard that underwent forced degradation. The possible deamidated form is indicated with an *.
Fig. 5.

Extracted ion chromatograms of the different insulins and the possible A21 desamido forms (indicated with an *).
3.5. Quantification of the legal and suspected illegal insulin preparation
First, for the quantification of all non-human insulin and non-insulin lispro containing samples, we used the LC–MS/MS method. Next, the human insulin and the insulin lispro containing samples were quantified by means of the LC–DAD methodology (Table 4). All insulin quantifications were inside the limits of 25%, which is sufficient for our purposes. Moreover, the quantification of all tested human insulin and insulin lispro containing samples were in agreement with guidelines from the European Pharmacopoeia for legal insulin preparations.
However, as mentioned in the introduction, suspected illegal insulin samples could also contain degradation products, including the A21 desamido form. Although we have shown that with our methodology it is possible to separate the intact polypeptide and the suspected A21 desamido polypeptide for insulin aspart, bovine insulin, insulin glulisine, human insulin, insulin lispro, and porcine insulin, we did not observe a later eluting peak with similar m/z for insulin detemir and insulin degludec when subjected to the same treatment, suggesting that the method can not separate these insulins from their degradation products. However, several later eluting peaks appeared in the total ion chromatograms and the quantity of unmodified and/or deamidated insulin decreased compared to that prior to the treatment (Supplement Fig. 1). These findings could indicate that these insulin forms are more susceptible to acid induced degradation and additional alternative degradation reactions might have occurred. It stands to reason that alternative chromatographic methods, for instance employing mixed mode chromatography, including ion exchange chromatography coupled to MS, could be envisaged to elucidate the identity of these peaks. Clearly, it can also be stated that much more research is required to broaden our knowledge on the chemical degradation process of the available biopharmaceuticals.
4. Conclusion
A fast dilute and shoot methodology comprising LC–DAD–MS/MS was developed for the qualitative and quantitative analysis of injectable insulin preparations. Although there are already several and even more sensitive MS-based methodologies available for insulin detection, none of the LC methods reported have been able to completely separate human insulin from insulin lispro in a full scan mode. A very recent method demonstrated the separation power of MEKC–DAD, but this method is not compatible to MS and could, when only using this strategy with DAD detection, potentially result in false positives. Our method is easy and unique in that way that we were able to analyze the samples without preliminary extensive sample preparation and that we combined the selectivity of full scan MS with a powerful chromatographic separation of human insulin and insulin lispro. Furthermore, this technique also enables us to separate the intact polypeptide and the suspected A21 desamido polypeptide for insulin aspart, bovine insulin, insulin glulisine, human insulin, insulin lispro, and porcine insulin.
Moreover, we were also able to quantify the amount of unmodified insulin based on the extracted ion chromatograms. In case of human insulin and insulin lispro it was only possible to specifically quantify these compounds, after correct identification, by DAD. These methods have been validated for all insulins according to the “total error” approach. However, we cannot exclude that other deamidations of alternative modifications that co-elute and in case of quantification by MS, also resulting in the same m/z, were incorporated in the amount of unmodified insulin.
Nevertheless, this identification method, based on the retention time, the precursor ion and diagnostic ions, and the quantification method, is currently being used in our official medicines control laboratory (OMCL) to analyze insulins retrieved from the illegal market. Although not many illegal insulin-containing samples have been seized in Belgium during the last years, alerts dating from 2014 and 2015, originating from the UK, Poland, Belgium, Sweden and Argentina warn us that illegal insulin is globally available for use.
Acknowledgments
We gratefully acknowledge the Federal Agency for Medicines and Health Products (FAMHP). Furthermore we would like to thank Tim Reyns and Séverine Goscinny (WIV-ISP, Belgium) for the fruitful discussions about MS based quantification and ion mobility. We also want to acknowledge Wouter Houthoofd for the critical reading of the manuscript.
Footnotes
Peer review under responsibility of Xi׳an Jiaotong University.
Supplementary data associated with this article can be found in the online version at 10.1016/j.jpha.2016.04.006.
Appendix A. Supplementary material
Supplement Fig. 1. Total ion chromatograms of (A) insulin detemir, (B) insulin degludec. The black chromatogram corresponds to the untreated insulin and the red chromatogram corresponds to the acid and heat treated form. The TIC shown in (C) show that the later eluting peaks of detemir and degludec are not an artifact due to the treatment since the pink and red TICs show respectively the acid and heat treated human insulin and insulin degludec. The additional peaks, not present in the untreated samples, are marked with an arrow.
.
Supplementary material
.
Supplementary material
.
References
- 1.Baeshen N.A., Baeshen M.N., Sheikh A. Cell factories for insulin production. Microb. Cell Fact. 2014;13:141. doi: 10.1186/s12934-014-0141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO Fact sheet N°312 Updated, March 2016, 〈http://www.who.int/mediacentre/factsheets/fs312/en〉.
- 3.Gill G.V., Yudkin J.S., Keen H. The insulin dilemma in resource-limited countries, A way forward? Diabetologia. 2011;54:19–24. doi: 10.1007/s00125-010-1897-3. [DOI] [PubMed] [Google Scholar]
- 4.Cheng M.M. Is the drugstore safe? Counterfeit diabetes products on the shelves. J. Diabetes Sci. Technol. 2009;3:1516–1520. doi: 10.1177/193229680900300634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldschreiber P. Public health issues with counterfeit medicines. Clin. Med. 2009;9:63–64. doi: 10.7861/clinmedicine.9-1-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chambers E.E., Legido-Quigley C., Smith N. Development of a fast method for direct analysis of intact synthetic insulins in human plasma: the large peptide challenge. Bioanalysis. 2013;5:65–81. doi: 10.4155/bio.12.290. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z., Caulfield M.P., Mcphaul M.J. Quantitative insulin analysis using liquid chromatography-tandem mass spectrometry in a high-throughput clinical laboratory. Clin. Chem. 2013;59:1349–1356. doi: 10.1373/clinchem.2012.199794. [DOI] [PubMed] [Google Scholar]
- 8.Hess C., Thomas A., Thevis M. Simultaneous determination and validated quantification of human insulin and its synthetic analogues in human blood serum by immunoaffinity purification and liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2012;404:1813–1822. doi: 10.1007/s00216-012-6271-5. [DOI] [PubMed] [Google Scholar]
- 9.Thevis M., Thomas A., Schänzer W. Mass spectrometric determination of insulins and their degradation products in sports drug testing. Mass. Spectrom. Rev. 2008;27:35–50. doi: 10.1002/mas.20154. [DOI] [PubMed] [Google Scholar]
- 10.Parfitt C., Church D., Armston A. Commercial insulin immunoassays fail to detect commonly prescribed insulin analogues. Clin. Biochem. 2015;48:1354–1357. doi: 10.1016/j.clinbiochem.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Moslemi P., Najafabadi A.R., Tajerzadeh H. A rapid and sensitive method for simultaneous determination of insulin and A21-desamido insulin by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2003;33:45–51. doi: 10.1016/s0731-7085(03)00336-4. [DOI] [PubMed] [Google Scholar]
- 12.Kunkel A., Günter S., Dette C. Quantitation of insulin by capillary electrophoresis and high-performance liquid chromatography method comparison and validation. J. Chromatogr. A. 1997;781:445–455. [Google Scholar]
- 13.Ortner K., Buchberger W., Himmelsbach M. Capillary electrokinetic chromatography of insulin and related synthetic analogues. J. Chromatogr. A. 2009;1216:2953–2957. doi: 10.1016/j.chroma.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Yeh H.H., Wu H.L., Lu C.Y. Simultaneous determination of regular insulin and insulin aspart by capillary zone electrophoresis and application in drug formulations. J. Pharm. Biomed. Anal. 2010;53:145–150. doi: 10.1016/j.jpba.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 15.Haunschmidt M., Ortner K., Hainz K. Investigations on the migration behavior of insulin and related synthetic analogues in CZE, MEKC and MEEKC employing different surfactants. Electrophoresis. 2010;31:1560–1564. doi: 10.1002/elps.200900547. [DOI] [PubMed] [Google Scholar]
- 16.Deng B., Liu Z., Luo G. Rapid quantitative determination and assessment of insulin in oil formulation by micellar electrokinetic capillary chromatography. J. Pharm. Biomed. Anal. 2002;27:73–80. doi: 10.1016/s0731-7085(01)00511-8. [DOI] [PubMed] [Google Scholar]
- 17.Lamalle C., Servais A.-C., Radermecker R.P. Simultaneous determination of insulin and its analogues in pharmaceutical formulations by micellar electrokinetic chromatography. J. Pharm. Biomed. Anal. 2015;111:344–350. doi: 10.1016/j.jpba.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 18.Stöcklin R., Vu L., Vadas L. A stable isotope dilution assay for the in vivo determination of insulin levels in humans by mass spectrometry. Diabetes. 1997;46:44–50. doi: 10.2337/diab.46.1.44. [DOI] [PubMed] [Google Scholar]
- 19.Kippen A.D., Cerini F., Vadas L. Development of an isotope dilution assay for precise determination of insulin, C-peptide, and proinsulin levels in non-diabetic and type II diabetic individuals with comparison to immunoassay. J. Biol. Chem. 1997;272:12513–12522. doi: 10.1074/jbc.272.19.12513. [DOI] [PubMed] [Google Scholar]
- 20.Darby S.M., Miller M.L., Allen R.O. A mass spectrometric method for quantitation of intact insulin in blood samples. J. Anal. Toxicol. 2001;25:8–14. doi: 10.1093/jat/25.1.8. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Cabaleiro D., Van Uytfanghe K., Stove V. Pilot study for the standardization of insulin immunoassays with isotope dilution liquid chromatography/tandem mass spectrometry. Clin. Chem. 2007;53:1462–1469. doi: 10.1373/clinchem.2007.088393. [DOI] [PubMed] [Google Scholar]
- 22.Van Uytfanghe K., Rodriguez-Cabaleiro D., Stöckl D. New liquid chromatography/electrospray ionisation tandem mass spectrometry measurement procedure for quantitative analysis of human insulin in serum. Rapid Commun. Mass. Spectrom. 2007;21:819–821. doi: 10.1002/rcm.2889. [DOI] [PubMed] [Google Scholar]
- 23.Thomas A., Thevis M., Delahaut P. Mass spectrometric identification of degradation products of insulin and its long-acting analogues in human urine for doping control purposes. Anal. Chem. 2007;79:2518–2524. doi: 10.1021/ac062037t. [DOI] [PubMed] [Google Scholar]
- 24.Thomas A., Schänzer W., Delahaut P. Sensitive and fast identification of urinary human, synthetic and animal insulin by means of nano-UPLC coupled with high-resolution/high-accuracy mass spectrometry. Drug Test. Anal. 2009;1:219–227. doi: 10.1002/dta.35. [DOI] [PubMed] [Google Scholar]
- 25.Chambers E.E., Fountain K.J., Smith N. Multidimensional LC-MS/MS enables simultaneous quantification of intact human insulin and five recombinant analogs in human plasma. Anal. Chem. 2014;86:694–702. doi: 10.1021/ac403055d. [DOI] [PubMed] [Google Scholar]
- 26.Thomas A., Schänzer W., Thevis M. Determination of human insulin and its analogues in human blood using liquid chromatography coupled to ion mobility mass spectrometry (LC-IM-MS) Drug Test. Anal. 2014;6:1125–1132. doi: 10.1002/dta.1710. [DOI] [PubMed] [Google Scholar]
- 27.Vanhee C., Janvier S., Desmedt B. Analysis of illegal peptide biopharmaceuticals frequently encountered by controlling agencies. Talanta. 2015;142:1–10. doi: 10.1016/j.talanta.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 28.Jonassen I., Havelund S., Hoeg-Jensen T. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm. Res. 2012;29:2104–2114. doi: 10.1007/s11095-012-0739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez A.G., Herrador M.A. A practical guide to analytical method validation, including measurement uncertainty and accuracy profiles. Trends Anal. Chem. 2007;26:227–238. [Google Scholar]
- 30.Hubert P., Nguyen-Huu J.J., Boulanger B. Harmonization of strategies for the validation of quantitative analytical procedures: a SFSTP proposal – part III. J. Pharm. Biomed. Anal. 2007;45:82–96. doi: 10.1016/j.jpba.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 31.Feinberg M. Validation of analytical methods based on accuracy profiles. J. Chromatogr. A. 2007;1158:174–183. doi: 10.1016/j.chroma.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 32.Sacré P.Y., Deconinck E., Chiap P. Development and validation of a ultra-high-performance liquid chromatography-UV method for the detection and quantification of erectile dysfunction drugs and some of their analogues found in counterfeit medicines. J. Chromatogr. A. 2011;1218:6439–6447. doi: 10.1016/j.chroma.2011.07.029. [DOI] [PubMed] [Google Scholar]
- 33.Deconinck E., Canfyn M., Sacré P.Y. A validated GC–MS method for the determination and quantification of residual solvents in counterfeit tablets and capsules. J. Pharm. Biomed. Anal. 2012;70:64–70. doi: 10.1016/j.jpba.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Desmedt B., Rogiers V., Courselle P. Development and validation of a fast chromatographic method for screening and quantification of legal and illegal skin whitening agents. J. Pharm. Biomed. Anal. 2013;83:82–88. doi: 10.1016/j.jpba.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 35.Desmedt B., Canfyn M., Pype M. HS-GC–MS method for the analysis of fragrance allergens in complex cosmetic matrices. Talanta. 2015;131:444–451. doi: 10.1016/j.talanta.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Deconinck E., Kamugisha A., Van Campenhout P. Development of a Stationary Phase Optimised Selectivity Liquid Chromatography based screening method for adulterations of food supplements for the treatment of pain. Talanta. 2015;138:240–246. doi: 10.1016/j.talanta.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 37.B. Ma, G. Lajoie, Unit 13.10 De novo interpretation of tandem mass spectra, Curr. Protoc. Bioinforma. (2009), 〈http://dx.doi.org/10.1002/0471250953.bi1310s25〉 [DOI] [PubMed]
- 38.(CHQ2(r1), International Conference on Harmonization: Validation of Analytical Procedures: Text and Methodology (Adopted by CPMP, Issued as CPMP/ICH281/95), 1996, 〈http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf〉
- 39.European Pharmacopeia (8th ed.) Council of Europe, Strasbourg, France, 2014.
- 40.Guidance for the Validation of Analytical Methodology and Calibration of Equipment used for Testing of Illicit Drugs in Seized Materials and Biological Specimens, Laboratory and Scientific Section, United Nations Office on Drugs and Crime, Vienna, New York, 2009.
- 41.Ren Y., Wu C., Zhang J. Simultaneous screening and determination of 18 illegal adulterants in herbal medicines and health foods for male sexual potency by ultra-fast liquid chromatography-electrospray ionization tandem mass spectrometry. J. Sep. Sci. 2012;35:2847–2857. doi: 10.1002/jssc.201200280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Fig. 1. Total ion chromatograms of (A) insulin detemir, (B) insulin degludec. The black chromatogram corresponds to the untreated insulin and the red chromatogram corresponds to the acid and heat treated form. The TIC shown in (C) show that the later eluting peaks of detemir and degludec are not an artifact due to the treatment since the pink and red TICs show respectively the acid and heat treated human insulin and insulin degludec. The additional peaks, not present in the untreated samples, are marked with an arrow.
Supplementary material
Supplementary material