

ABSTRACT
The alarmone ppGpp is a critical regulator of virulence gene expression in Francisella tularensis. In this intracellular pathogen, ppGpp is thought to work in concert with the putative DNA-binding protein PigR and the SspA protein family members MglA and SspA to control a common set of genes. MglA and SspA form a complex that interacts with RNA polymerase (RNAP), and PigR functions by interacting with the RNAP-associated MglA-SspA complex. Prior work suggested that ppGpp indirectly exerts its regulatory effects in F. tularensis by promoting the accumulation of polyphosphate in the cell, which in turn was required for formation of the MglA-SspA complex. Here we show that in Escherichia coli, neither polyphosphate nor ppGpp is required for formation of the MglA-SspA complex but that ppGpp promotes the interaction between PigR and the MglA-SspA complex. Moreover, we show that polyphosphate kinase, the enzyme responsible for the synthesis of polyphosphate, antagonizes virulence gene expression in F. tularensis, a finding that is inconsistent with the notion that polyphosphate accumulation promotes virulence gene expression in this organism. Our findings identify polyphosphate kinase as a novel negative regulator of virulence gene expression in F. tularensis and support a model in which ppGpp exerts its positive regulatory effects by promoting the interaction between PigR and the MglA-SspA complex.
IMPORTANCE In Francisella tularensis, MglA and SspA form a complex that associates with RNA polymerase to positively control the expression of key virulence genes. The MglA-SspA complex works together with the putative DNA-binding protein PigR and the alarmone ppGpp. PigR functions by interacting directly with the MglA-SspA complex, but how ppGpp exerts its effects was unclear. Prior work indicated that ppGpp acts by promoting the accumulation of polyphosphate, which is required for MglA and SspA to interact. Here we show that formation of the MglA-SspA complex does not require polyphosphate. Furthermore, we find that polyphosphate antagonizes the expression of virulence genes in F. tularensis. Thus, ppGpp does not promote virulence gene expression in this organism through an effect on polyphosphate.
KEYWORDS: MglA, SspA, gene regulation, ppGpp
INTRODUCTION
Francisella tularensis is a Gram-negative, intracellular pathogen and the causative agent of tularemia, a potentially fatal disease. F. tularensis mostly infects rodents and other small mammals but can also infect humans. Humans can become infected via multiple routes, including through an arthropod vector and through ingestion, but the most severe form of the disease occurs following inhalation of aerosolized bacteria (1). F. tularensis is a highly infectious pathogen, with the most virulent strains having an infectious dose of as few as 10 bacteria (2). Due to its highly infectious nature, its ability to cause severe disease, and its ability to be easily aerosolized, F. tularensis had been developed by several countries as a bioweapon, leading the CDC to list F. tularensis as a category A select agent (3).
The ability of F. tularensis to replicate within macrophages is key to its ability to cause disease. Prominent among those factors required for intramacrophage growth and virulence are the components of a type VI secretion system that are encoded on the so-called Francisella pathogenicity island (FPI) (4–8). All of the virulence genes on the FPI, as well as many other genes in F. tularensis, are positively regulated by three key regulators called MglA, SspA, and PigR (also known as FevR) (9–13). MglA and SspA are members of the stringent starvation protein A (SspA) family of proteins and form a heteromeric complex that binds to RNA polymerase (RNAP) (11, 14, 15). Interaction between the MglA-SspA complex and RNAP is thought to be essential for these proteins to exert their regulatory effects (11). Furthermore, interaction between PigR and the RNAP-associated MglA-SspA complex is necessary in order for PigR, MglA, and SspA to function as positive regulators (16). Although PigR, MglA, and SspA have been shown to occupy the promoters of both regulated and nonregulated genes, a small 7-bp sequence element is both necessary and sufficient for conferring control by PigR (and thus presumably by MglA and SspA as well) (17). This sequence, which is referred to as the PigR response element, is found ∼6 bp upstream of the −35 element of target promoters and could function as a binding site for PigR (17).
The small molecules guanosine pentaphosphate (pppGpp) and guanosine tetraphosphate (ppGpp) (referred to collectively here as [p]ppGpp), also appear to be critical for virulence gene expression in F. tularensis (13). (p)ppGpp, which is referred to as an alarmone, is produced in response to a variety of stress signals (18). In Escherichia coli, (p)ppGpp is produced by the proteins RelA and SpoT (reviewed in reference 18). RelA is a monofunctional enzyme that synthesizes (p)ppGpp in response to amino acid starvation and thus mediates the so-called stringent response, a process in which the increase in (p)ppGpp leads to inhibition of rRNA transcription, increased expression of amino acid biosynthesis genes, and a reduction in protein synthesis (19–21). SpoT is a bifunctional enzyme, capable of both degrading and synthesizing (p)ppGpp, which responds to conditions of carbon, phosphate, and fatty acid limitation (20). The effects of ppGpp on transcription in E. coli are potentiated by the small RNAP-associated protein DksA (22, 23). Recently, ppGpp has been shown to bind to two distinct sites on E. coli RNAP, referred to as sites 1 and 2 (24–26). At high concentrations of ppGpp, binding to site 2, which is at the RNAP-DksA interface, is thought to account for most of the effects of ppGpp on transcription initiation (26). (p)ppGpp exerts regulatory effects on hundreds of genes in E. coli (27, 28) and has also been shown to regulate virulence gene expression in several pathogens, including Legionella pneumophila (29), Salmonella spp. (30, 31), Vibrio cholerae (32), Mycobacterium tuberculosis (33), Pseudomonas aeruginosa (34), and enterohemorrhagic E. coli (35) (reviewed in reference 36).
In addition to its effects on transcription, (p)ppGpp is also known to directly bind to target enzymes to influence their activity, allowing (p)ppGpp to directly control many cellular processes (reviewed in reference 37). In E. coli it is well established that (p)ppGpp inhibits the activity of polyphosphate phosphatase (PPX), the enzyme responsible for breaking down polyphosphate (reviewed in reference 38), and that relA spoT mutant cells that can no longer synthesize (p)ppGpp do not contain detectable amounts of polyphosphate (Fig. 1) (39, 40). Polyphosphate is a chain of tens to hundreds of inorganic phosphate molecules that is synthesized in response to stress conditions, including starvation (38), by the enzyme polyphosphate kinase (PPK) (40). Polyphosphate has been shown to be important for the production of many virulence factors, including those involved in motility and biofilm formation in P. aeruginosa (41–43). Moreover, ppk, the gene encoding PPK, has also been found to be important for the virulence of several pathogens, including Mycobacterium tuberculosis (44, 45), Salmonella spp. (46), Helicobacter pylori (47), Vibrio cholerae (48), Shigella flexneri (46), and F. tularensis (49). Thus, at least in principle, the effects of (p)ppGpp on the virulence of a particular organism could be accounted for solely by its effects on the abundance of polyphosphate in the cell.
FIG 1.
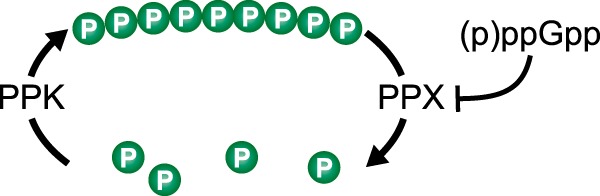
Control of polyphosphate abundance in E. coli. Polyphosphate is synthesized by polyphosphate kinase (PPK) and broken down by polyphosphate phosphatase (PPX). (p)ppGpp influences the abundance of polyphosphate by inhibiting PPX activity.
In F. tularensis, (p)ppGpp controls the expression of many of the same genes that are controlled by PigR, MglA, and SspA (13). Studies in F. tularensis suggest that (p)ppGpp promotes the interaction between PigR and the RNAP-associated MglA-SspA complex (13), and recent work has revealed that ppGpp can bind directly to the MglA-SspA complex to mediate its effects (15). However, other recent findings suggested that (p)ppGpp might exert its effects on virulence gene expression indirectly by promoting the accumulation of polyphosphate, which in turn was required for MglA and SspA to form a complex (50). Here we present evidence that polyphosphate is not required in order for MglA and SspA to interact. Moreover, we show that polyphosphate antagonizes the expression of genes that are positively regulated by (p)ppGpp, PigR, MglA, and SspA. Our findings have important implications for how (p)ppGpp exerts its regulatory effects in F. tularensis.
RESULTS
(p)ppGpp promotes the interaction between PigR and the MglA-SspA complex in E. coli.
Prior work suggested that in F. tularensis (p)ppGpp exerts its effects on virulence gene expression by promoting the accumulation of polyphosphate, which in turn, is required for MglA and SspA to form a complex (50). To test whether or not the interaction between MglA and SspA depends (directly or indirectly) on the ability of cells to synthesize (p)ppGpp, we took advantage of a two-hybrid assay we had used previously to detect the interaction between MglA and SspA in E. coli (11, 16, 51). In this assay, MglA from F. tularensis is fused to a monomeric DNA-binding protein called Zif and SspA from F. tularensis is fused to the ω subunit of E. coli RNAP (Fig. 2A). The MglA-Zif and SspA-ω fusion proteins are then synthesized in an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible fashion in cells of an E. coli reporter strain in which a Zif binding site is positioned upstream of a test promoter that drives expression of lacZ. Interaction between the RNAP-bound SspA-ω fusion protein and the DNA-bound MglA-Zif fusion protein activates transcription from the test promoter, resulting in an increase in lacZ expression (11, 16). To determine whether (p)ppGpp modulates the interaction between MglA and SspA, we first constructed a relA spoT mutant derivative of our E. coli reporter strain that can no longer synthesize (p)ppGpp (indicated as ppGpp0) (39). The results depicted in Fig. 2B show that the MglA-Zif fusion protein interacted with the RNAP-tethered SspA-ω fusion protein in both wild-type (WT) and ppGpp0 cells of the reporter strain. Note that although the absolute amount of reporter gene expression was slightly higher in WT cells than in ppGpp0 cells, the magnitude of MglA-Zif dependent activation from the test promoter was identical (∼18-fold) in both WT and ppGpp0 cells of the reporter strain (Fig. 2B). These findings indicate that in E. coli, formation of the MglA-SspA complex does not require ppGpp.
FIG 2.

(p)ppGpp does not influence formation of the MglA-SspA complex but does promote the interaction between PigR and the MglA-SspA complex in E. coli. (A) Schematic representation of E. coli two-hybrid assay used in panel B. Interaction between the MglA-Zif and SspA-ω fusion proteins stimulates transcription from the test promoter driving expression of lacZ. (B) Bacterial two-hybrid assay of the ability of MglA and SspA to interact with one another in cells of the reporter strain that synthesizes ppGpp (indicated WT) and in cells of the reporter strain that cannot synthesize ppGpp (indicated ppGpp0). (C) Schematic representation of E. coli bridge-hybrid assay used in D. Interaction between the PigR-Zif fusion protein and the complex formed between MglA and the SspA-ω fusion protein stimulates transcription from the test promoter that drives expression of lacZ. (D) Bacterial bridge-hybrid assay of the ability of PigR to interact with the MglA-SspA complex in cells of the reporter strain that synthesizes ppGpp (indicated as WT) and in cells of the reporter strain that cannot synthesize ppGpp (indicated as ppGpp0). Assays in panels B and D were performed with cells of the E. coli reporter strain KDZif1ΔZ (indicated as WT) or cells of the E. coli reporter strain ARZif1ΔAZT (indicated as ppGpp0). Cells containing compatible plasmids directing the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible synthesis of the specified proteins were grown in the presence of IPTG at the indicated concentration and then assayed for β-galactosidase activity.
Because in F. tularensis (p)ppGpp promotes the interaction between PigR and the RNAP-associated MglA-SspA complex (13), we sought to determine whether we could recapitulate the effect of (p)ppGpp on this interaction in E. coli. To do this, we used a modified version of our two-hybrid assay that permits the detection of interactions among three proteins (13, 16). In the version of this bridge-hybrid assay used here, PigR from F. tularensis is fused to Zif and SspA from F. tularensis is fused to the ω subunit of E. coli RNAP (Fig. 2C). MglA from F. tularensis is then synthesized, together with the SspA-ω and PigR-Zif fusion proteins, in cells of the same E. coli reporter strain employed in our two-hybrid assay. In these cells, the complex formed between MglA and the SspA-ω fusion protein becomes tethered to RNAP through the ω moiety of the SspA-ω fusion. Interaction between the RNAP-bound MglA-SspA complex and the DNA-bound PigR-Zif fusion protein then activates transcription from the test promoter in the E. coli reporter strain (13, 16). In WT cells of the reporter strain, the PigR-Zif fusion protein interacted with the RNAP-tethered complex formed between MglA and the SspA-ω fusion protein to activate transcription from the test promoter (Fig. 2D). In the ppGpp0 cells of the reporter strain, PigR did not detectably interact with the MglA-SspA complex (Fig. 2D). Taken together, our findings indicate that in E. coli, just as in F. tularensis, (p)ppGpp promotes the interaction between PigR and the MglA-SspA complex but is not required for MglA and SspA to interact with one another.
Polyphosphate is not required for formation of the MglA-SspA complex or the PigR-MglA-SspA complex in E. coli.
Our findings with our E. coli two-hybrid and bridge-hybrid assays do not support a model in which (p)ppGpp modulates the interaction between MglA and SspA indirectly through an effect on polyphosphate accumulation, as had previously been suggested (50). In order to explicitly test whether polyphosphate influences the ability of MglA and SspA to interact with one another, we made a version of our E. coli two-hybrid reporter strain that could no longer synthesize polyphosphate (50). This strain harbors a deletion of ppk and thus lacks PPK, the enzyme responsible for the synthesis of polyphosphate in E. coli (reviewed in reference 38). Using a version of our two-hybrid system in which SspA is fused to Zif and MglA is fused to ω (Fig. 3A), we found that MglA and SspA interact with one another equally well in WT cells and in Δppk mutant cells (Fig. 3B). These findings indicate that polyphosphate is not required in order for MglA and SspA to interact with one another in E. coli.
FIG 3.

Polyphosphate does not influence formation of the MglA-SspA complex or the PigR-MglA-SspA complex in E. coli. (A) Schematic representation of E. coli two-hybrid assay used in panel B. Interaction between the SspA-Zif and MglA-ω fusion proteins stimulates transcription from the test promoter that drives expression of lacZ. (B) Bacterial two-hybrid assay of the ability of MglA and SspA to interact with one another in cells of the reporter strain that can synthesize polyphosphate (indicated as WT) and in cells of the reporter strain that cannot synthesize polyphosphate (indicated as Δppk). (C) Schematic representation of bacterial bridge-hybrid assay used in panel D. Interaction between the PigR-Zif fusion protein and the complex formed between SspA and the MglA-ω fusion protein stimulates transcription from the test promoter that drives expression of lacZ. (D) Bacterial bridge-hybrid assay of the ability of PigR to interact with the MglA-SspA complex in cells of the reporter strain that can synthesize polyphosphate (indicated as WT) and in cells of the reporter strain that cannot synthesize polyphosphate (indicated as Δppk). Assays in panels B and D were performed with cells of the E. coli reporter strain KDZif1ΔZ (indicated as WT) or cells of the E. coli reporter strain ARZif1ΔKZ (indicated as Δppk). Cells containing compatible plasmids directing the IPTG-inducible synthesis of the specified proteins were grown in the presence of IPTG at the indicated concentration and then assayed for β-galactosidase activity.
We next asked whether polyphosphate influences the ability of PigR to interact with the MglA-SspA complex in E. coli. To do this we employed a version of our bridge-hybrid assay in which PigR is fused to Zif, MglA is fused to ω, and SspA is provided in its native form (Fig. 3C). The results shown in Fig. 3D reveal that PigR interacts with the MglA-SspA complex equally well in WT cells and in Δppk mutant cells. Altogether, our findings indicate that polyphosphate is not required for formation of the MglA-SspA complex, nor is it required for PigR to interact with the MglA-SspA complex in cells of E. coli.
Polyphosphate kinase functions to represses virulence gene expression in F. tularensis.
If formation of the MglA-SspA complex does not require polyphosphate and if polyphosphate does not promote the interaction between PigR and the MglA-SspA complex, then polyphosphate would not be expected to be required for the expression of genes that are positively regulated by (p)ppGpp/PigR/MglA/SspA in F. tularensis. To test this prediction, we constructed a mutant of the live vaccine strain of F. tularensis (LVS) in which the ppk gene (FTL_0554) (52) was deleted (LVS Δppk). We then measured the expression of the MglA-regulated iglA virulence gene in LVS WT cells, in LVS ΔpigR mutant cells (used as a negative control), and in LVS Δppk mutant cells that contained the empty vector pF2. We also measured the expression of the iglA gene in LVS Δppk mutant cells that contained the plasmid pF2-PPK-V, encoding PPK with a C-terminal vesicular stomatitis virus glycoprotein (VSV-G) epitope tag. The results depicted in Fig. 4A show that, consistent with previous findings, the abundance of the iglA transcript was ∼20-fold lower in cells of the LVS ΔpigR mutant strain than in WT cells. Surprisingly, the abundance of the iglA transcript was ∼5-fold higher in cells of the LVS Δppk mutant strain than in WT cells. Furthermore, ectopic expression of ppk in cells of the LVS Δppk mutant strain resulted in an ∼10-fold decrease in expression of the iglA gene (Fig. 4A). Consistent with these findings, Western blotting revealed that the abundance of the product of the (p)ppGpp/PigR/MglA/SspA-regulated iglC virulence gene (which is in the same operon as iglA) was higher in cells of the LVS Δppk mutant strain than in WT cells and that the effect of the Δppk mutation on IglC abundance could be complemented by providing ppk in trans (Fig. 4B). Thus, consistent with our prediction, these findings indicate that polyphosphate kinase is not required for the expression of genes that are positively regulated by (p)ppGpp/PigR/MglA/SspA in F. tularensis. Furthermore, they suggest that under the conditions of our experiments, polyphosphate antagonizes virulence gene expression in F. tularensis.
FIG 4.
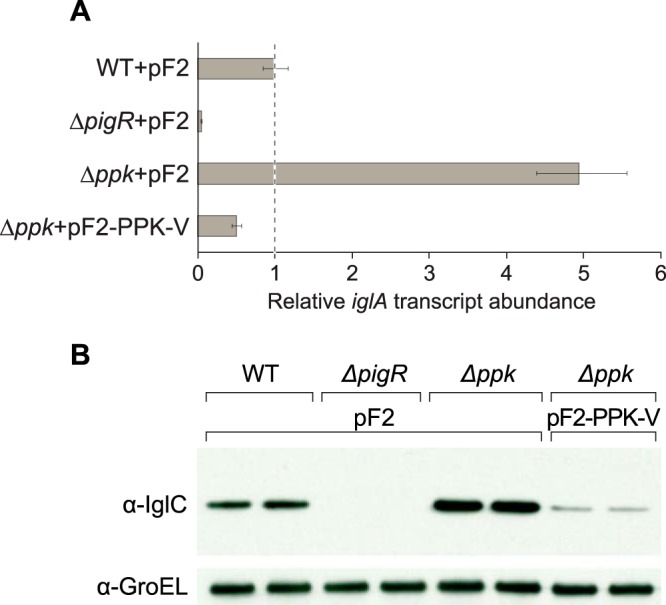
Polyphosphate kinase antagonizes virulence gene expression in F. tularensis. (A) Polyphosphate kinase functions to repress expression of the FPI iglA gene. The relative abundance of the iglA transcript was determined in cells of the indicated strains containing the specified plasmids by quantitative reverse transcriptase PCR (qRT-PCR). pF2 is an empty vector control, whereas pF2-PPK-V directs the synthesis of an epitope-tagged version of PPK. The figure shows data from a representative experiment with biological duplicates. Transcripts were normalized to tul4, and error bars represent ±1 standard deviation from the value (calculated using the mean threshold cycle). (B) Effect of polyphosphate kinase on the abundance of IglC. The abundance of IglC in the indicated strains containing the specified plasmids was determined by Western blotting with an antibody against IglC. (Note that the gene encoding IglC is in the same operon as iglA.) An antibody against GroEL was used as a loading control. Duplicate biological samples were tested for each strain and a representative data set is shown. Plasmids are as in panel A.
Polyphosphate does not appear to exert its regulatory effects through substrate competition.
RelA in F. tularensis is unusual in that it can only synthesize guanosine pentaphosphate (pppGpp) from ATP and GTP, unlike its counterpart in E. coli that can synthesize both pppGpp and ppGpp from ATP and GTP or ATP and GDP, respectively (53). Furthermore, F. tularensis is distinct from E. coli in that it does not appear to encode pppGpp phosphohydrolase, the enzyme principally involved in converting pppGpp to ppGpp in E. coli (54). However, PPX from E. coli has been shown to be capable of converting pppGpp to ppGpp (54). We therefore reasoned that in F. tularensis, PPX might be the enzyme chiefly responsible for converting any pppGpp that is made to ppGpp. If ppGpp were the only species that could influence gene expression in F. tularensis (see for example 55), then polyphosphate might effectively repress the expression of ppGpp/PigR/MglA/SspA-regulated genes by competing with pppGpp for the available PPX in the cell, thus reducing the intracellular concentration of active ppGpp (Fig. 5A). According to this model, in cells of the LVS Δppk mutant strain, ppGpp would be more abundant than in WT cells because there would be no polyphosphate available to compete with pppGpp for the available PPX. To test this possibility, we insertionally inactivated ppx in cells of our LVS Δppk mutant strain. The results depicted in Fig. 5B indicate that the abundances of the iglA transcript were similar in cells of the LVS Δppk and LVS Δppk ppx mutant strains, being ∼11-fold and ∼12-fold higher, respectively, than that found in WT cells. (Note that we typically find that the effect of the Δppk mutation on iglA expression is 2- to 4-fold greater in cells that do not contain plasmids than in cells that do [cf. Fig. 4B and 5B].) These findings suggest that polyphosphate does not antagonize virulence gene expression in F. tularensis through an effect on PPX substrate competition.
FIG 5.
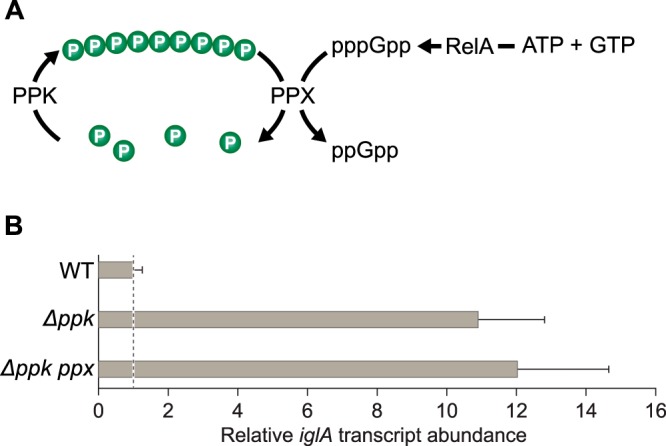
Polyphosphate does not exert its negative regulatory effects through substrate competition. (A) Substrate competition model to account for the negative regulatory effects of polyphosphate. (B) Inactivation of ppx in LVS Δppk mutant cells has no effect on the expression of the FPI iglA gene. The relative abundance of the iglA transcript was determined in cells of the indicated strains by qRT-PCR. The figure shows data from a representative experiment with biological duplicates. Transcripts were normalized to tul4, and error bars represent ±1 standard deviation from the value (calculated using the mean threshold cycle).
Polyphosphate may exert its regulatory effects through the Lon protease.
It is well established that in E. coli the activity of the Lon protease is dependent upon polyphosphate (56). To test whether polyphosphate might repress virulence gene expression in F. tularensis by influencing the activity of the Lon protease, we created a mutant of LVS in which the lon gene was deleted (LVS Δlon) and asked whether the effects of a lon deletion phenocopied those of a ppk deletion. The results depicted in Fig. 6A show that the abundance of the iglA transcript was ∼20-fold higher in LVS Δppk mutant cells and ∼11-fold higher in LVS Δlon mutant cells, compared to that in WT LVS cells. We also found that the abundance of the (p)ppGpp/PigR/MglA/SspA-regulated FTL_1219 transcript was ∼10-fold higher in LVS Δppk mutant cells and ∼7-fold higher in LVS Δlon mutant cells, compared to that in WT LVS cells (Fig. 6A). In support of the idea that polyphosphate exerts a portion of its regulatory effects through Lon, the abundance of the iglA and FTL_1219 transcripts was similar in LVS Δppk mutant cells and in cells of an LVS Δppk Δlon double mutant (Fig. 6A). Consistent with the results of our analyses of the iglA transcript, Western blotting revealed that the product of the iglC gene (IglC) was higher in LVS Δppk mutant cells, in LVS Δlon mutant cells, and in cells of an LVS Δppk Δlon double mutant, than in WT cells (Fig. 6B). Furthermore, IglC was slightly less abundant in LVS Δlon mutant cells than in LVS Δppk mutant cells, or in cells of the LVS Δppk Δlon double mutant (Fig. 6B). These findings suggest that, like polyphosphate, the Lon protease can antagonize the expression of (p)ppGpp/PigR/MglA/SspA-regulated genes (although its effect appears somewhat reduced compared to that of polyphosphate). These findings support the possibility that in F. tularensis, polyphosphate exerts its negative effects on the expression of (p)ppGpp/PigR/MglA/SspA-regulated genes at least in part by stimulating the activity of the Lon protease.
FIG 6.

Both polyphosphate kinase and the Lon protease antagonize virulence gene expression in F. tularansis. (A) Effect of polyphosphate kinase and the Lon protease on expression of the iglA and FTL_1219 genes. The relative abundance of the iglA and FTL_1219 transcripts was determined in cells of the indicated strains by qRT-PCR. The figure shows data from a representative experiment with biological duplicates. Transcripts were normalized to tul4, and error bars represent ±1 standard deviation from the value (calculated using the mean threshold cycle) (B) Effect of polyphosphate kinase and the Lon protease on IglC abundance. The abundance of IglC in cells of the indicated strains was determined by Western blotting with an antibody against IglC. An antibody against GroEL was used as a loading control. Duplicate biological samples were tested for each strain, and a representative data set is shown.
DISCUSSION
The alarmone (p)ppGpp is a critical regulator of virulence gene expression in F. tularensis and is required for the expression of those genes that are positively regulated by PigR, MglA, and SspA (13). A prior study suggested that (p)ppGpp might exert its regulatory effects in F. tularensis indirectly by promoting the accumulation of polyphosphate in the cell, which in turn was required for formation of the MglA-SspA complex (50). We find that neither (p)ppGpp nor polyphosphate is required for formation of the MglA-SspA complex in E. coli. Furthermore, we find that (p)ppGpp promotes the interaction between PigR and the MglA-SspA complex in E. coli. Finally, we have obtained evidence that in F. tularensis, polyphosphate antagonizes the expression of genes that are positively regulated by (p)ppGpp, PigR, MglA, and SspA. Our findings thus establish that (p)ppGpp does not exert its positive regulatory effects in F. tularensis by promoting the accumulation of polyphosphate.
Our findings that neither (p)ppGpp nor PPK (and thus polyphosphate) are required in order for MglA and SspA to interact with one another were obtained using an E. coli-based two-hybrid assay. These findings refute the notion that (p)ppGpp exerts its regulatory effects in F. tularensis by influencing the formation of the MglA-SspA complex and run counter to those findings recently obtained using essentially the same two-hybrid system (50). Although we cannot readily explain the discrepancy between the findings reported here and those published in the competing study (50), we note that our findings in E. coli are consistent with our previous finding in F. tularensis that (p)ppGpp does not influence the amount of RNAP-associated MglA-SspA complex in the cell (13). Indeed, if (p)ppGpp were required for the accumulation of polyphosphate (as it is in E. coli) and if polyphosphate were strictly required for formation of the MglA-SspA complex, then cells that cannot synthesize (p)ppGpp would not be expected to contain any RNAP-associated MglA-SspA complex. Furthermore, our discovery that polyphosphate actually functions to repress genes that are positively regulated by (p)ppGpp shows that (p)ppGpp cannot exert its stimulatory effect on virulence gene expression in F. tularensis by promoting the accumulation of polyphosphate.
We have found that (p)ppGpp promotes the interaction between PigR and the MglA-SspA complex in both F. tularensis (13) and in E. coli (Fig. 2D). These findings are consistent with the recent discovery that ppGpp interacts directly with the MglA-SspA complex, with residues of MglA and SspA that are important for ppGpp binding being critical to their regulatory activities (15, 16). We note that if (p)ppGpp promotes the accumulation of polyphosphate in F. tularensis, as it does in E. coli (40), then (p)ppGpp would be expected to exert both positive and negative effects on virulence gene expression. Any effect of (p)ppGpp on the accumulation of polyphosphate in F. tularensis would serve to dampen the expression of virulence genes, possibly fine-tuning their expression.
Our findings indicate that PPK functions to antagonize virulence gene expression in F. tularensis, at least under the conditions of our experiments. Polyphosphate can bind directly to Lon from E. coli and stimulate its proteolytic activity (56), and we speculate that in F. tularensis polyphosphate produced by PPK exerts a portion of its negative effects on the expression of virulence genes by influencing the activity of the Lon protease. Prior work in E. coli and Pseudomonas protegens suggests that Lon degrades at least one important transcription regulator in each of these organisms (57, 58). In LVS, Lon is thought to degrade several virulence-associated factors and has been shown to be important for tolerance to certain stresses (59). Further work will be required to determine whether any of the key regulators of virulence gene expression in F. tularensis, including PigR, MglA, and SspA, serve as the substrates for Lon, or whether Lon might exert regulatory effects in F. tularensis through its ability to interact with the DNA (60, 61).
MATERIALS AND METHODS
Plasmids, strains, and growth conditions.
Francisella tularensis subsp. holarctica strain LVS and the strain LVS ΔpigR have been previously described (11, 13). All F. tularensis strains were grown with aeration at 37°C in modified Mueller-Hinton (MH) broth (Difco) supplemented with 0.1% glucose, 0.025% ferric pyrophosphate, and 2% IsoVitaleX (BD Biosciences), or on cysteine heart agar (CHA; Difco) supplemented with 1% hemoglobin solution (BD Biosciences). When appropriate, 5 μg/ml kanamycin was used for selection. The E. coli strain XL1-Blue (Stratagene) was used for plasmid construction.
Construction of LVS deletion constructs and mutant strains.
The strains LVS Δppk, LVS Δlon, and LVS Δppk Δlon contain an in-frame deletion of the ppk locus (FTL_0544), the lon locus (FTL_0894), or both the ppk and lon loci, respectively. The plasmids pEX-ppk and pEX-lon were constructed as described previously (11) and were used to generate the strains LVS Δppk and LVS Δlon using allelic exchange (62). Note that pEX-ppk and pEX-lon are derivatives of the pEX plasmid (not the pEX2 plasmid) and contain a single copy of the sacB gene (11). The strain LVS Δppk Δlon was generated from the strain LVS Δppk using allelic exchange and the plasmid pEX-lon. The strain LVS Δppk ppx was generated by insertional inactivation of the ppx locus (FTL_0612) in the strain LVS Δppk. The 5′ end of the ppx locus overlaps with the 5′ end of an istfu2 repetitive element on the opposite strand, making it difficult to obtain a deletion of ppx using allelic exchange. The plasmid pEX-ppx-frag was used to generate the strain LVS Δppk ppx. The plasmid pEX-ppx-frag is a derivative of the pEX plasmid (11) that confers resistance to kanamycin and contains a 423-bp fragment of the ppx gene. This fragment corresponds to amino acids 51 to 191 of the product of ppx and includes a stop codon after the codon for amino acid 191. The suicide plasmid pEX-ppx-frag was electroporated into cells of strain LVS Δppk. Cells were plated on CHA supplemented with hemoglobin and 5 μg/ml kanamycin to select those in which the pEX-ppx-frag plasmid had integrated into the chromosome through a single homologous recombination event. All LVS strains were confirmed by PCR and/or Southern blotting. Similar to what has been found previously with cells of a ppk mutant of F. tularensis strain SchuS4 (49), cells of our LVS Δppk mutant exhibited an ∼10-fold reduction in intracellular growth within J774 cells compared to those of WT LVS (see Fig. S1 in the supplemental material).
Plasmids for complementation analyses.
Plasmid pF2-PPK-V was used for complementation of the LVS Δppk strain, and plasmid pF2 was used as the corresponding empty vector control. pF2-PPK-V confers resistance to kanamycin and directs the synthesis of full-length LVS PPK with a vesicular stomatitis virus glycoprotein (VSV-G) epitope tag fused to its C terminus and driven from a modified groEL promoter lacking a putative upstream promoter (UP) element. The empty vector pF2 containing the modified groEL promoter and lacking a putative UP element has been previously described (11). Plasmid pF2-PPK-V was generated by cloning an EcoRI- and BamHI-digested PCR product into EcoRI-BamHI-digested plasmid pF2. The PCR product for this vector was amplified from the full-length F. tularensis ppk gene using an appropriate template, a forward primer that introduced an EcoRI cleavage site and Shine-Dalgarno sequence to the 5′ end of the LVS ppk gene, and a reverse primer which added the sequence for the VSV-G epitope tag and a BamHI site to the 3′ end of the LVS ppk gene.
Plasmids for bacterial two-hybrid and bridge-hybrid assays.
The plasmids pBR-MglA-ω, pBR-SspA-ω, pACTR-SspA-Zif, pACTR-MglA-Zif, pACTR-PigR-Zif, pACTR-AP-Zif, pCL-SspA, pCL-MglA, and pCL have been previously described (11, 13, 16).
Construction of E. coli strains for two-hybrid and bridge-hybrid assays.
The E. coli strain KDZif1ΔZ was used as the wild-type reporter strain for the two-hybrid and bridge-hybrid assays and was previously described (51). The ΔrelA spoT and Δppk derivatives of the E. coli reporter strain for the two-hybrid and bridge-hybrid assays were constructed from the strain FW102, which is the KDZif1ΔZ parent strain (51, 63). The ΔrelA spoT E. coli reporter strain contains an in-frame deletion of the E. coli relA gene, a chloramphenicol resistance cassette inserted into the spoST locus (spoS is also referred to as rpoZ and encodes the ω-subunit of RNAP), and harbors an F′ episome containing the lac promoter derivative placZif1-61, which drives expression of a linked lacZ reporter gene. This F′ episome has been previously described (51). To generate the ΔrelA spoT E. coli reporter strain, the strain relA::kan was first generated through P1-mediated transduction of the allele relA::kan from the relA mutant strain of the Keio collection (64) to the recipient strain FW102. The kanamycin resistance cassette in the relA::kan allele is flanked by FLP recognition target (FRT) sites. FLP recombinase was expressed from the plasmid pCP20 (65) in the strain FW102 relA::kan to excise the kanamycin resistance cassette and generate the ΔrelA strain, which contains an in-frame deletion of the E. coli relA gene. The λ Red recombinase system (65) was used to generate the ΔrelA rpoZ-spoT::cat strain. Specifically, PCR was used to amplify the cat gene from the pKD3 plasmid (65) using a forward primer that included the 40-bp sequence upstream of the 5′ end of rpoZ and a reverse primer that included the 3′ end of spoT and the 40 bp sequence downstream. The resulting PCR product, which contained the cat gene flanked by FRT sites and regions of homology to the 5′ end of rpoZ and the 3′ end of spoT, was electroporated into cells of the E. coli ΔrelA strain containing λ Red helper plasmids, as described in reference 65. The desired ΔrelA rpoZ-spoT::cat mutants were selected for as previously described (65). The previously described F′ episome containing the lac promoter derivative placZif1-61, which drives expression of a linked lacZ reporter gene (51), was mated into the strain ΔrelA rpoZ-spoT::cat to generate the strain ARZif1ΔAZT (referred to as the ΔrelA spoT or ppGpp0 E. coli reporter strain in the text).
The Δppk E. coli reporter strain for the two-hybrid and bridge-hybrid assays contains an in-frame deletion of the E. coli ppk locus and a chloramphenicol resistance cassette replacing the E. coli rpoZ gene (also referred to as spoS). The rpoZ::cat strain was first generated by P1-mediated transduction of the spoS3::cat allele from strain KDZif1ΔZ (51) into the recipient strain FW102 (63). P1-mediated transduction of the ppk::kan allele from the ppk mutant strain of the Keio collection (64) into the rpoZ::cat recipient strain generated the ppk::kan rpoZ::cat strain. The kanamycin resistance cassette in the ppk::kan allele is flanked by FRT sites. FLP recombinase was expressed in the ppk::kan rpoZ::cat strain to excise the kanamycin resistance cassette and generate the Δppk rpoZ::cat strain. The F′ reporter construct (51) was mated into the Δppk rpoZ::cat strain to generate the strain ARZif1ΔKZ (referred to in the text as the Δppk E. coli reporter strain). The strains ARZif1ΔKZ and ARZif1ΔAZT were confirmed by Southern blotting.
Bacterial two-hybrid and bridge-hybrid assays.
The bacterial two-hybrid and bridge-hybrid assays were performed as previously described (11, 13, 16). Cells were grown with aeration at 37°C in LB supplemented with carbenicillin (100 μg/ml), tetracycline (10 μg/ml), and IPTG at the indicated concentration for the two-hybrid assay and with carbenicillin, spectinomycin (100 μg/ml), tetracycline, and IPTG at the indicated concentration for the bridge-hybrid assay. Cells were permeabilized with CHCl3 and assayed for β-galactosidase activity as previously described (16). Assays were performed at least twice in duplicate. Duplicate measurements differed by less than 10%. Results shown are averages from a single representative experiment.
RNA isolation and qRT-PCR.
LVS cells were grown in liquid culture (50 ml) in the presence of kanamycin with aeration at 37°C until cultures reached an optical density at 600 nm (OD600) of ∼0.25 to ∼0.4. Ten milliliters of cells was harvested by centrifugation at 4,000 rpm for 20 min at 4°C. RNA was isolated using Tri-Reagent (Molecular Research Center, Inc.) as previously described (16). RNA quality was determined by gel electrophoresis. cDNA synthesis using Superscript III reverse transcriptase (Invitrogen) and quantitative reverse transcriptase PCR (qRT-PCR) were performed essentially as described previously (16, 66). The abundances of the iglA and FTL_1219 transcripts were measured relative to that of the tul4 transcript (11). qRT-PCR was performed at least twice on sets of biological triplicates. Data shown are from representative experiments.
Immunoblots.
Cell lysates were separated by SDS-PAGE on 4 to 12% Bis-Tris NuPAGE gels in MES running buffer (Life Technologies). The XCell II Blot Module (Life Technologies) was used to transfer proteins to polyvinylidene difluoride (PVDF). Membranes were probed as described previously (66), using anti-GroEL (diluted 1:30,000) or anti-IglC (diluted 1:2,000) antibodies and subsequently incubated with polyclonal goat anti-rabbit antibody conjugated to horseradish peroxidase (diluted 1:5,000; Pierce) or polyclonal goat anti-mouse antibody conjugated to horseradish peroxidase (1:5,000; Pierce), respectively. Immunoblots were visualized using chemiluminescent detection as described previously (66).
Intramacrophage growth assays.
Assays were performed with J774 murine macrophage-like cells essentially as described previously (66).
Supplementary Material
ACKNOWLEDGMENTS
We thank Tom Bernhardt for supplying P1 transducing phage, strains from the Keio collection, and plasmids for use with the λ Red recombinase system. We thank Karsten Hazlett and Dan Clemens for antibodies, Ann Hochschild for comments on the manuscript, and Renate Hellmiss for artwork.
This work was supported by National Institutes of Health grant AI081693 to S.L.D. K.M.R. was supported by NIH training grant HD055148.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00460-17.
REFERENCES
- 1.Sjostedt A. 2007. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci 1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 2.Tarnvik A, Chu MC. 2007. New approaches to diagnosis and therapy of tularemia. Ann N Y Acad Sci 1105:378–404. doi: 10.1196/annals.1409.017. [DOI] [PubMed] [Google Scholar]
- 3.Oyston PCF, Sjostedt A, Titball RW. 2004. Tularemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol 2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 4.Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KKM, Roberts MJ, Ludu JS, Letendre GW, Meierovics AI, Stephens G, Elkins KL. 2004. A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol 186:6430–6436. doi: 10.1128/JB.186.19.6430-6436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nano FE, Schmerk C. 2007. The Francisella pathogenicity island. Ann N Y Acad Sci 1105:122–137. doi: 10.1196/annals.1409.000. [DOI] [PubMed] [Google Scholar]
- 6.Barker JR, Chong A, Wehrly TD, Yu J-J, Rodriguez SA, Liu J, Celli J, Arulanandam BP, Klose KE. 2009. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74:1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, Chou S, Gonen T, Goodlett DR, Goodman AL, Mougous JD. 2014. A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16:227–236. doi: 10.1016/j.chom.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, Whitney JC, Radey MC, Peterson SB, Ruhland BR, Tran BQ, Goo YA, Goodlett DR, Dove SL, Celli J, Veesler D, Mougous JD. 2016. Secreted effectors encoded within and outside of the Francisella pathogenicity island promote intramacrophage growth. Cell Host Microbe 20:573–583. doi: 10.1016/j.chom.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lauriano CM, Barker JR, Yoon S-S, Nano FE, Arulanandam BP, Hassett DJ, Klose KE. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc Natl Acad Sci U S A 101:4246–4249. doi: 10.1073/pnas.0307690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brotcke A, Weiss DS, Kim CC, Chain P, Malfatti S, Garcia E, Monack DM. 2006. Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect Immun 74:6642–6655. doi: 10.1128/IAI.01250-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charity JC, Costante-Hamm MM, Balon EL, Boyd DH, Rubin EJ, Dove SL. 2007. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog 3:e84. doi: 10.1371/journal.ppat.0030084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brotcke A, Monack DM. 2008. Identification of fevR, a novel regulator of virulence gene expression in Francisella novicida. Infect Immun 76:3473–3480. doi: 10.1128/IAI.00430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charity JC, Blalock LT, Costante-Hamm MM, Kasper DL, Dove SL. 2009. Small molecule control of virulence gene expression in Francisella tularensis. PLoS Pathog 5:e1000641. doi: 10.1371/journal.ppat.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baron GS, Nano FE. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol Microbiol 29:247–259. doi: 10.1046/j.1365-2958.1998.00926.x. [DOI] [PubMed] [Google Scholar]
- 15.Cuthbert BJ, Ross W, Rohlfing AE, Dove SL, Gourse RL, Brennan RG, Schumacher MA. 2017. Dissection of the molecular circuitry controlling virulence in Francisella tularensis. Genes Dev 31:1549–1560. doi: 10.1101/gad.303701.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohlfing AE, Dove SL. 2014. Coordinate control of virulence gene expression in Francisella tularensis involves direct interaction between key regulators. J Bacteriol 196:3516–3526. doi: 10.1128/JB.01700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramsey KM, Osborne ML, Vvedenskaya IO, Su C, Nickels BE, Dove SL. 2015. Ubiquitous promoter-localization of essential virulence regulators in Francisella tularensis. PLoS Pathog 11:e1004793. doi: 10.1371/journal.ppat.1004793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauryliuk V, Atkinson GC, Murakami KS, Tenson T, Gerdes K. 2015. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat Rev Microbiol 13:298–309. doi: 10.1038/nrmicro3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat Rev Microbiol 6:507–519. doi: 10.1038/nrmicro1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 21.Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation, and replication by (p)ppGpp. Curr Opin Microbiol 11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Paul BJ, Berkmen MB, Gourse RL. 2005. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci U S A 102:7823–7828. doi: 10.1073/pnas.0501170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross W, Vrentas CE, Sanchez-Vazquez P, Gaal T, Gourse RL. 2013. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol Cell 50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo Y, Wang Y, Steitz TA. 2013. The structure of magic spot bound to RNA polymerase suggests how it is regulated by ppGpp. Mol Cell 50:430–436. doi: 10.1016/j.molcel.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ross W, Sanchez-Vazquez P, Chen AY, Lee J-H, Burgos HL, Gourse RL. 2016. ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol Cell 62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durfee T, Hansen A-M, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traxler MF, Summers SM, Nguyen H-T, Zacharia VM, Hightower GS, Smith JT, Conway T. 2008. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammer BK, Swanson MS. 1999. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol Microbiol 33:721–731. doi: 10.1046/j.1365-2958.1999.01519.x. [DOI] [PubMed] [Google Scholar]
- 30.Pizarro-Cerda J, Tedin K. 2004. The bacterial signal molecule, ppGpp, regulates Salmonella virulence gene expression. Mol Microbiol 52:1827–1844. doi: 10.1111/j.1365-2958.2004.04122.x. [DOI] [PubMed] [Google Scholar]
- 31.Thompson A, Rolfe MD, Lucchini S, Schwerk P, Hinton JCD, Tedin K. 2006. The bacterial signal molecule, ppGpp, mediates the environmental regulation of both the invasion and intracellular virulence gene programs of Salmonella. J Biol Chem 281:30112–30121. doi: 10.1074/jbc.M605616200. [DOI] [PubMed] [Google Scholar]
- 32.Haralalka S, Nandi S, Bhadra RK. 2003. Mutation in the relA gene of Vibrio cholerae affects in vitro and in vivo expression of virulence factors. J Bacteriol 185:4672–4682. doi: 10.1128/JB.185.16.4672-4682.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE. 2000. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol 182:4889–4898. doi: 10.1128/JB.182.17.4889-4898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erickson DL, Lines JL, Pesci EC, Venturi V, Storey DG. 2004. Pseudomonas aeruginosa relA contributes to virulence in Drosophila melanogaster. Infect Immun 72:5638–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakanishi N, Abe H, Ogura Y, Hayashi T, Tashiro K, Kuhara S, Sugimoto N, Tobe T. 2006. ppGpp with DksA controls gene expression in the locus of enterocyte effacement (LEE) pathogenicity island of enterohaemorrhagic Escherichia coli through activation of two virulence regulatory genes. Mol Microbiol 61:194–205. doi: 10.1111/j.1365-2958.2006.05217.x. [DOI] [PubMed] [Google Scholar]
- 36.Dalebroux ZD, Svesson SL, Gaynor EC, Swanson MS. 2010. ppGpp conjures bacterial virulence. Microbiol Mol Biol Rev 74:171–199. doi: 10.1128/MMBR.00046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanjee U, Ogata K, Houry WA. 2012. Direct binding targets of the stringent response alarmone (p)ppGpp. Mol Microbiol 85:1029–1043. doi: 10.1111/j.1365-2958.2012.08177.x. [DOI] [PubMed] [Google Scholar]
- 38.Rao NN, Gomez-Garcia MR, Kornberg A. 2009. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem 78:605–647. doi: 10.1146/annurev.biochem.77.083007.093039. [DOI] [PubMed] [Google Scholar]
- 39.Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M. 1991. Residual guanosine 3′, 5′-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J Biol Chem 266:5980–5990. [PubMed] [Google Scholar]
- 40.Kuroda A, Murphy H, Cashel M, Kornberg A. 1997. Guanosine tetra and pentaphosphate promote accumulation of inorganic polyphosphate in Escherichia coli. J Biol Chem 272:21240–21243. doi: 10.1074/jbc.272.34.21240. [DOI] [PubMed] [Google Scholar]
- 41.Rashid MH, Kornberg A. 2000. Inorganic polyphosphate is needed for swimming, swarming, and twitching motilities of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:4885–4890. doi: 10.1073/pnas.060030097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rashid MH, Rao NN, Kornberg A. 2000. Inorganic polyphosphate is required for motility of bacterial pathogens. J Bacteriol 182:225–227. doi: 10.1128/JB.182.1.225-227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rashid MH, Rumbaugh K, Passador L, Davies DG, Hamood AN, Iglewski BH, Kornberg A. 2000. Polyphosphate kinase is essential for biofilm development, quorum sensing, and virulence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:9636–9641. doi: 10.1073/pnas.170283397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh R, Singh M, Arora G, Kumar S, Tiwari P, Kidwai S. 2013. Polyphosphate deficiency in Mycobacterium tuberculosis is associated with enhanced drug susceptibility and impaired growth in guinea pigs. J Bacteriol 195:2839–2851. doi: 10.1128/JB.00038-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sureka K, Dey S, Datta P, Singh AK, Dasgupta A, Rodrigue S, Basu J, Kundu M. 2007. Polyphosphate kinase is involved in stress-induced mprAB-sigE-rel signalling in mycobacteria. Mol Microbiol 65:261–276. doi: 10.1111/j.1365-2958.2007.05814.x. [DOI] [PubMed] [Google Scholar]
- 46.Kim K-S, Rao NN, Fraley CD, Kornberg A. 2002. Inorganic polyphosphate is essential for long-term survival and virulence factors in Shigella and Salmonella spp. Proc Natl Acad Sci U S A 99:7675–7680. doi: 10.1073/pnas.112210499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang ZX, Zhou YN, Yang Y, Jin DJ. 2010. Polyphosphate binds to the principal sigma factor of RNA polymerase during starvation response in Helicobacter pylori. Mol Microbiol 77:618–627. doi: 10.1111/j.1365-2958.2010.07233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogawa N, Tzeng CM, Fraley CD, Kornberg A. 2000. Inorganic polyphosphate in Vibrio cholerae: genetic, biochemical, and physiologic features. J Bacteriol 182:6687–6693. doi: 10.1128/JB.182.23.6687-6693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richards MI, Michell SL, Oyston PCF. 2008. An intracellularly inducible gene involved in virulence and polyphosphate production in Francisella. J Med Microbiol 57:1183–1192. doi: 10.1099/jmm.0.2008/001826-0. [DOI] [PubMed] [Google Scholar]
- 50.Wrench AP, Gardner CL, Siegel SD, Pagliai FA, Malekiha M, Gonzalez CF, Lorca GL. 2013. MglA/SspA complex interactions are modulated by inorganic polyphosphate. PLoS One 8:e76428. doi: 10.1371/journal.pone.0076428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. 2005. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102:11082–11087. doi: 10.1073/pnas.0502663102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batten LE, Parnell AE, Wells NJ, Murch AL, Oyston PC, Roach PL. 2015. Biochemical and structural characterization of polyphosphate kinase 2 from the intracellular pathogen Francisella tularensis. Biosci Rep 36:e00294. doi: 10.1042/BSR20150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilkinson RC, Batten LE, Wells NJ, Oyston PC, Roach PL. 2015. Biochemical studies on Francisella tularensis RelA in (p)ppGpp biosynthesis. Biosci Rep 35:e00268. doi: 10.1042/BSR20150229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keasling JD, Bertsch L, Kornberg A. 1993. Guanosine pentaphosphate phosphohydrolase of Escherichia coli is a long-chain exopolyphosphatase. Proc Natl Acad Sci U S A 90:7029–7033. doi: 10.1073/pnas.90.15.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mechold U, Potrykus K, Murphy H, Murakami KS, Cashel M. 2013. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res 41:6175–6189. doi: 10.1093/nar/gkt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuroda A, Nomura K, Ohtomo R, Kato J, Ikeda T, Takiguchi N, Ohtake H, Kornberg A. 2001. Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science 293:705–708. doi: 10.1126/science.1061315. [DOI] [PubMed] [Google Scholar]
- 57.Shah IM, Wolf RE Jr. 2006. Sequence requirements for Lon-dependent degradation of the Escherichia coli transcription factor SoxS: identification of the SoxS residues critical to proteolysis and specific inhibition of in vitro degradation by a peptide comprised of the N-terminal 21 amino acid residues. J Mol Biol 357:718–731. doi: 10.1016/j.jmb.2005.12.088. [DOI] [PubMed] [Google Scholar]
- 58.Takeuchi K, Tsuchiya W, Noda N, Suzuki R, Yamazaki T, Haas D. 2014. Lon protease negatively affects GacA protein stability and expression of the Gac/Rsm signal transduction pathway in Pseudomonas protegens. Environ Microbiol 16:2538–2549. doi: 10.1111/1462-2920.12394. [DOI] [PubMed] [Google Scholar]
- 59.He L, Nair MKM, Chen Y, Liu X, Zhang M, Hazlett KRO, Deng H, Zhang J-R. 2016. The protease locus of Francisella tularensis LVS is required for stress tolerance and infection in the mammalian host. Infect Immun 84:1387–1402. doi: 10.1128/IAI.00076-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Charette MF, Henderson GW, Doane LL, Markovitz A. 1984. DNA-stimulated ATPase activity on the Lon (CapR) protein. J Bacteriol 158:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nomura K, Kato J, Takiguchi N, Ohtake H, Kuroda A. 2004. Effects of inorganic polyphosphate on the proteolytic and DNA-binding activities of Lon in Escherichia coli. J Biol Chem 279:34406–34410. doi: 10.1074/jbc.M404725200. [DOI] [PubMed] [Google Scholar]
- 62.Golovliov I, Sjöstedt A, Mokrievich A, Pavlov V. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol Lett 222:273–280. doi: 10.1016/S0378-1097(03)00313-6. [DOI] [PubMed] [Google Scholar]
- 63.Whipple FW. 1998. Genetic analysis of prokaryotic and eukaryotic DNA-binding proteins in Escherichia coli. Nucleic Acids Res 26:3700–3706. doi: 10.1093/nar/26.16.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramsey KM, Dove SL. 2016. A response regulator promotes Francisella tularensis intramacrophage growth by repressing an anti-virulence factor. Mol Microbiol 101:688–700. doi: 10.1111/mmi.13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.