

Abstract
Transforming growth factor β (TGF-β)/Smad3 signaling plays a role in tissue fibrosis. We report here that Erbb4-IR is a novel long non-coding RNA (lncRNA) responsible for TGF-β/Smad3-mediated renal fibrosis and is a specific therapeutic target for chronic kidney disease. Erbb4-IR was induced by TGF-β1 via a Smad3-dependent mechanism and was highly upregulated in the fibrotic kidney of mouse unilateral ureteral obstructive nephropathy (UUO). Silencing Erbb4-IR blocked TGF-β1-induced collagen I and alpha-smooth muscle actin (α-SMA) expressions in vitro and effectively attenuated renal fibrosis in the UUO kidney by blocking TGF-β/Smad3 signaling. Mechanistic studies revealed that Smad7, a downstream negative regulator of TGF-β/Smad signaling, is a target gene of Erbb4-IR because a binding site of Erbb4-IR was found on the 3′ UTR of Smad7 gene. Mutation of this binding site prevented the suppressive effect of Erbb4-IR on the Smad7 reporter activity; in contrast, overexpression of Erbb4-IR largely inhibited Smad7 but increased collagen I and α-SMA transcriptions. Thus, kidney-specific silencing of Erbb4-IR upregulated renal Smad7 and thus blocked TGF-β/Smad3-mediated renal fibrosis in vivo and in vitro. In conclusion, the present study identified that Erbb4-IR is a novel lncRNA responsible for TGF-β/Smad3-mediated renal fibrosis by downregulating Smad7. Targeting Erbb4-IR may represent a precise therapeutic strategy for progressive renal fibrosis.
Keywords: Erbb4-IR, TGF-β/Smad3, Smad7, fibrosis
Feng et al. characterize a novel Smad3-dependent long non-coding RNA Erbb4-IR and further demonstrate its role in promoting renal fibrosis in a mouse unilateral ureteral obstruction model. Erbb4-IR binds and represses the promoter of Smad7, thereby enhancing TGF-β1/Smad3-mediated fibrogenesis. Erbb4-IR may represent a precision therapeutic target for chronic kidney disease.
Introduction
Fibrosis is a hallmark and common pathway leading to end-stage organ diseases, including chronic kidney disease (CKD). Transforming growth factor β1 (TGF-β1), a well-studied pro-fibrogenic cytokine, exerts its fibrogenic effect on the progression of CKD by activating the downstream Smad signaling pathway in both experimental animal models and human kidney diseases.1, 2, 3 It is now clear that TGF-β1 activates Smad3 to mediate fibrosis, whereas overexpression of Smad7 prevents renal fibrosis in vitro and in vivo.2, 3 Although Smad3 is a key transcription factor in response to many fibrogenic mediators, a lesson learned from gene knockout mice suggests that targeting Smad3 may cause autoimmune disease by impairing immunity.4 Thus, alternative approaches to specifically inhibit TGF-β1-mediated tissue fibrosis should be achieved by targeting the downstream effector genes of Smad3 that are responsible for fibrosis.
Emerging evidence shows that non-coding RNAs (ncRNAs) play an important role in the development of kidney diseases, including diabetic nephropathy.5, 6, 7 The identification of Smad3-related microRNAs (miRNAs) in nephropathy is supposed to be a specific therapy for treating renal fibrosis in chronic kidney disease, including diabetic nephropathy.8, 9, 10, 11, 12, 13, 14 However, off-target effect, avoidance from internal nucleases, and toxicity for miRNA therapy hinder their clinical application. This is because the role of miRNA is nonspecific and acts as a cofactor instead of downstream effector in the signaling pathways involved.15 Since the discovery of Xist, a long non-coding RNA (lncRNA) required for mammalian X chromosome inactivation,16 many lncRNAs have been reported to play regulatory roles in the pathophysiological progress of tumors, autoimmune diseases, and cardiovascular diseases.17, 18, 19, 20, 21 In kidney diseases, some lncRNAs have also been reported.21 For example, PVT1 is the first identified lncRNA-associated kidney disease, which is associated with end-stage renal disease in type 1 and type 2 diabetes.22 However, the underlying mechanisms of lncRNAs in the pathogenesis of kidney disease remain largely unclear. Thus, identification and characterization of kidney-disease-associated lncRNAs may represent a promising research strategy for resolving renal disorder and may lead to the development of precision therapies for kidney diseases.
By using RNA sequencing (RNA-seq), we identified a number of Smad3-dependent lncRNAs that participated in the renal fibrogenesis and inflammation in mouse models of ureteral obstructive nephropathy (UUO) and anti-glomerular basement membrane glomerulonephritis (anti-GBM GN).23 Compared to the wild-type mice, 151 lncRNAs are Smad3 dependent.23 Interestingly, a Smad3-dependent lncRNA, Arid2-IR, has been reported to specifically regulate renal inflammation, but not fibrosis.24 In this study, we characterized and investigated another novel lncRNA, np_5318, which is largely upregulated in the UUO kidney and is identified as a direct Smad3 target gene.23 Here, we characterized that lncRNA np_5318 is a 2,310-nt noncoding RNA located within the intron region between the first and second exons of Erbb4 on chromosome 1 of the mouse genome; thus, we named this novel lncRNA as Erbb4-IR. In the present study, we also investigated the functional role and mechanisms of Erbb4-IR in the pathogenesis of renal fibrosis. The therapeutic potential of Erbb4-IR-targeted therapy on renal fibrosis was also explored.
Results
Erbb4-IR Is a Novel lncRNA Specific for Renal Fibrosis
By using RNA-seq, we previously found that a potential lncRNA transcript (np_5318) was significantly upregulated in the UUO-injured kidney.23 In this study, we further characterized lncRNA np_5318 by obtaining its full-length sequence via rapid amplification of cDNA ends (RACE). As shown in Figure S1, np_5318 is 2,310 nt in length and is located within the intron region between the first and second exons of Erbb4 on chromosome 1 of a mouse (Chr1: 69026879–69029188) (Figures 1A and 1B). Therefore, it is designated as Erbb4-IR. The sequence of Erbb4-IR is partially conserved among species compared by the ECR browser (Figure S2).25 By using the coding potential calculator (CPC) and coding potential assessment tool (CPAT) analysis,26, 27 scores of −1.23878 and 0.046 were obtained, respectively. Thus, Erbb4-IR was confirmed and classified as a noncoding RNA. Interestingly, we found that Erbb4-IR was progressively increased in the kidney after UUO-induced injury (Figures 1C and 1D), but not in both ischemic and cisplatin-induced acute kidney injury (Figure S3), suggesting that Erbb4-IR may function to induce renal fibrosis. By in situ hybridization, Erbb4-IR was specifically expressed by tubular epithelial cells (TECs) and elongated fibroblast-like cells with both cytoplasmic and nuclear patterns within areas of severe tubulointerstitial fibrosis (Figure 1C). In addition, upregulation of Erbb4-IR was also found in some glomerular parietal epithelial cells and podocytes as well as vascular cells (Figure 1C).
Figure 1.

Characterization of Erbb4-IR in Renal Fibrosis
(A) A full-length of Erbb4-IR (2,310 bp) amplified by PCR is detected by electrophoresis in 1% agarose gel. Marker: 1 kb DNA ladder. (B) Gene location of Erbb4-IR at the intron region between exon 1 and exon 2 in chromosome 1 of the mouse genome. (C) In situ hybridization shows that Erbb4-IR is weakly expressed by TECs in the normal kidney but is dramatically upregulated in the UUO kidney on day 7 (×400). Erbb4-IR is strongly expressed by interstitial fibroblasts, injured TECs, mainly in the nucleus. In addition, upregulation of Erbb4-IR is also found in some glomerular parietal epithelial cells, podocytes, vascular cells, and interstitial fibroblasts in the areas of severe renal fibrosis. (D) Real-time PCR detects the upregulation of Erbb4-IR in the kidney during progression of UUO. Each bar represents the mean ± SEM for groups of 6 mice. **p < 0.01 and ***p < 0.001 compared with sham-operated mice; ##p < 0.01 compared with UUO kidney at 1 day after operation. g, glomerulus; scale bars, 50 μm.
Erbb4-IR Is Induced by TGF-β1 Specifically via a Smad3-, but not a Smad2-, Dependent Mechanism
Because Erbb4-IR was markedly upregulated in TECs of the UUO kidney according to the result of in situ hybridization (Figure 1C), the specificity of Erbb4-IR in tubular epithelial cells under a fibrotic condition was further examined in cultured mouse tubular epithelial cells (mTECs). The expression of Erbb4-IR was upregulated by TGF-β1 stimulation in a time- and dose-dependent manner, being significant at 1 hr (Figures 2A and 2B), in which TGF-β1-induced Smad3 activation was evident at 15 min (Figure S4). Real-time PCR revealed that upregulation of Erbb4-IR was associated with induction of collagen I and alpha-smooth muscle actin (α-SMA) transcriptions in the TGF-β1-treated mTECs (Figures 2C and 2D). Interestingly, the addition of pro-inflammatory cytokine interleukin-1β (IL-1β) did not alter Erbb4-IR expression in mTECs (Figure 2E). These results prompted a pro-fibrogenic function of Erbb4-IR during renal fibrosis in response to TGF-β1, rather than a downstream mediator related to renal inflammation.
Figure 2.

Erbb4-IR Expression Is Positively Regulated by TGF-β1, but Not by IL-1β, and Is Upregulated prior to Upregulation of Collagen I and α-SMA in mTECs In Vitro
(A and B) Real-time PCR shows that TGF-β1 (2 ng/mL) induces expression of Erbb4-IR in mTECs in a (A) time- and (B) dose-dependent manner, being significant at 1, 12, and 24 hr after TGF-β1 stimulation. (C and D) Real-time PCR detects that TGF-β1 (2 ng/mL) induces (C) collagen I and (D) α-SMA mRNA expression in a time-dependent manner, being significant at 3 hr onward. (E) Erbb4-IR expression in response to IL-1β (10 ng/mL). Note that the addition of IL-1β does not alter Erbb4-IR in mTECs. Each bar represents the mean ± SEM for at least 3 independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with time 0 or time-matched control.
We next examined the mechanism, whereby TGF-β1 induces Erbb4-IR expression. Because Smad3 is capable of binding to the upstream sequence of Erbb4-IR (np_5318), as indicated by our previous study,23 here we investigated whether TGF-β1-induced Erbb4-IR is Smad3-dependently expressed by using the Smad3 inhibitor SIS3. As shown in Figures 3A and 3B, the addition of SIS3 was able to block TGF-β1-induced Smad3 phosphorylation and therefore dramatically inhibited the expression of Erbb4-IR. TGF-β1-induced Erbb4-IR expression via a Smad3-dependent mechanism was further confirmed in mouse embryonic fibroblasts (MEFs) lacking Smad3. We showed that deletion of Smad3 abolished Erbb4-IR induction in response to TGF-β1 (Figure 3C). In contrast, deletion of Smad2 from MEFs enhanced TGF-β1-induced Erbb4-IR expression (Figure 3D). Thus, TGF-β1 induces Erbb4-IR in a Smad3-dependent but Smad2-independent manner.
Figure 3.

Erbb4-IR Expression Is Positively Regulated by TGF-β1 via Smad3-, but Not Smad2-, Dependent Mechanism In Vitro
(A) Western blot analysis shows that the addition of a Smad3 inhibitor (SIS3) inhibits TGF-β1 (2 ng/mL) induced p-Smad3 in mTECs at 1 hr. (B) Real-time PCR shows that the addition of SIS3 inhibits TGF-β1 (2 ng/mL) induced expression of Erbb4-IR in mTECs at 1 hr. (C and D) Real-time PCR results show that the addition of TGF-β1 (2 ng/mL) induces Erbb4-IR expression in both of the Smad3 (C) and Smad2 (D) wild-type (WT) MEFs (upper panels), which is blunted in Smad3 (C) but enhanced in Smad2 (D) knockout (KO) MEFs, as shown in each lower panel. Each bar represents the mean ± SEM for at least 3 independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with either DMSO control or time-matched control; #p < 0.05 and ##p < 0.01 compared with TGF-β1+DMSO control.
Erbb4-IR Is a Downstream Effector of TGF-β1/Smad3-Mediated Renal Fibrosis In Vitro
Because the induction of Erbb4-IR was associated with the transcription of collagen I and α-SMA via a Smad3-dependent mechanism in TGF-β1-treated mTECs (Figures 2 and 3), we further tested our hypothesis that Erbb4-IR is a downstream effector of TGF-β1/Smad3 to induce renal fibrosis. This was tested in mTECs by knocking down Erbb4-IR. As shown in Figure 4, real-time PCR and western blot analysis revealed that small interfering RNA (siRNA) mediated knockdown of Erbb4-IR was able to reduce TGF-β1-induced collagen I and α-SMA expressions in mTECs. Our results suggest a pathogenic role of the TGF-β1/Smad3/Erbb4-IR axis in renal fibrogenesis. Interestingly, treatment with TGF-β1 or silencing of Erbb4-IR did not alter the expression of the host gene Erbb4 in vitro (Figure S5), indicating that Erbb4-IR is a novel lncRNA independent to its host gene Erbb4.
Figure 4.

Knockdown of Erbb4-IR Suppresses TGF-β1-Induced Collagen I and α-SMA in mTECs In Vitro
(A) Real-time PCR analysis shows that siRNA-mediated knockdown of Erbb4-IR significantly inhibits Erbb4-IR expression in mTECs with or without 1 hr TGF-β1 (2 ng/mL) stimulation compared with NCs. (B and C) Knockdown of Erbb4-IR significantly suppresses the mRNA expression levels of collagen I (B) and α-SMA (C) in TGF-β1-stimulated mTECs at 6 hr, as shown by real-time PCR. (D) Western blot analysis demonstrates that knockdown of Erbb4-IR significantly reduces protein expressions of collagen I and α-SMA protein levels in the TGF-β1 (2 ng/mL) treated mTECs at 24 hr when compared with the NCs. Each bar represents the mean ± SEM for at least 3 independent experiments.*p < 0.05, **p < 0.01, and ***p < 0.001 compared with control cells without TGF-β1 treatment. #p < 0.05, ##p < 0.01, and ###p < 0.001 compared with control cells (NCs) treated with TGF-β1.
Silencing of Erbb4-IR Effectively Attenuates Progressive Renal Fibrosis in a Mouse Model of UUO
To confirm the pathogenic role and therapeutic target of Erbb4-IR in renal fibrosis, plasmids containing small hairpin RNA (shRNA) targeting Erbb4-IR (Erbb4-IR shRNA) were specifically transfected into the injured kidney immediately after induction of UUO by using an ultrasound-microbubble delivery system, as previously described.8, 9, 10, 11, 12, 13, 14, 24 In situ hybridization and real-time PCR detected that Erbb4-IR expression was dramatically increased in the UUO kidney on day 7, which was significantly suppressed by treatment with Erbb4-IR shRNA (Figures 5A and 5C). Results of Masson’s trichrome staining showed that treatment with Erbb4-IR shRNA significantly reduced the severity of tubulointerstitial fibrosis as compared to control UUO kidneys treated without or with empty vector (EV) (Figures 5B and 5D). Examinations by immunohistochemistry, real-time PCR, and western blot analysis also revealed that treatment with Erbb4-IR shRNA significantly suppressed UUO-induced renal fibrosis, including collagen I and α-SMA mRNA expression and extracellular matrix accumulation (Figures 6 and 7). Our findings demonstrated the pathogenic role of Erbb4-IR in renal fibrosis and the potential of Erbb4-IR as a therapeutic target in renal fibrosis.
Figure 5.

Kidney-Specific Ultrasound-Microbubble-Mediated Erbb4-IR shRNA Transfer Effectively Reduces Erbb4-IR and Renal Fibrosis in UUO-Injured Mice In Vivo
(A) In situ hybridization shows that ultrasound-microbubble-mediated Erbb4-IR shRNA transfer effectively inhibits expression of Erbb4-IR on day 7 UUO-injured kidney. (B) Masson’s trichrome staining shows that kidney-specific knockdown of Erbb4-IR prevents fibrosis of the UUO-injured kidney on day 7. (C) Real-time PCR shows that Erbb4-IR is markedly induced in the UUO kidney on day 7, which is significantly reduced by ultrasound-microbubble-mediated Erbb4-IR shRNA transfer. (D) Quantification of Masson’s trichrome-positive area of collagen-like matrix deposition at the day 7 kidneys. Renal fibrosis is largely ameliorated by knockdown of Erbb4-IR. Each bar represents the mean ± SEM for groups of 6 mice. *p < 0.05 and ***p < 0.001 compared with sham-operated mice. #p < 0.05 and ###p < 0.001 compared with EV-treated UUO kidney. g, glomerulus; scale bar, 50 μm.
Figure 6.

Kidney-Specific Knockdown of Erbb4-IR Inhibits Collagen I Expression in the UUO Kidney at Day 7
(A) Immunohistochemistry. (B) Western blot analysis. Each lane represents one animal. (C) Real-time PCR analysis. Results show that kidney-specific knockdown of Erbb4-IR suppresses collagen I mRNA and protein expression in the UUO kidney. Each bar represents the mean ± SEM for groups of 6 mice. ***p < 0.001 compared with sham-operated mice. ##p < 0.01 and ###p < 0.001 compared with the EV-treated UUO kidneys. Scale bars, 50 μm.
Figure 7.

Kidney-Specific Knockdown of Erbb4-IR Inhibits α-SMA Expression in the UUO Kidney at Day 7
(A) Immunohistochemistry. (B) Western blot analysis. Each lane represents one animal. (C) Real-time PCR analysis. Results show that kidney-specific knockdown of Erbb4-IR suppresses α-SMA mRNA and protein expression in the UUO kidney. Each bar represents the mean ± SEM for groups of 6 mice. ***p < 0.001 compared with sham-operated mice. #p < 0.05 and ###p < 0.001 compared with the EV-treated UUO kidneys. Scale bars, 50 μm.
Inhibition of Smad7 Gene Transcription Is a Mechanism by Which Erbb4-IR Promotes TGF-β1/Smad3-Mediated Renal Fibrosis In Vitro and In Vivo
We next examined the potential mechanism by which Erbb4-IR mediates renal fibrosis. Interestingly, the addition of TGF-β1 activated Smad3 signaling, including phosphorylation of TGF-β receptor I (TβRI) and Smad3, whereas silencing of Erbb4-IR specifically blocked TGF-β1-induced Smad3 signaling when compared to the nonsense-treated control (NC) (Figures 8A–8C). Similarly, knockdown of Erbb4-IR from the UUO kidney also significantly suppressed phosphorylation of TβRI and Smad3 (Figures 9A–9C). These observations implied a potential regulatory role of Erbb4-IR in the activation of TGF-β/Smad3 signaling at the transcriptional level. Because Erbb4-IR was found to be highly expressed in nucleus rather than cytoplasm in TGF-β1-stimulated mTEC (Figure S6), its transcriptional regulatory role was further emphasized by a predicted Erbb4-IR binding site on the 3′ UTR of the Smad7 genomic sequence suggested by Freiburg RNA Tools (Figure S7).28 We then tested a hypothesis that Erbb4-IR may positively regulate TGF-β/Smad3 signaling by targeting Smad7, an inhibitor of TGF-β/Smad signaling. As shown in Figures 8A, 8D, and 8E, silencing of Erbb4-IR significantly increased Smad7 mRNA and protein expression in mTECs in response to TGF-β1, demonstrating that Erbb4-IR may target Smad7 to promote activation of TGF-β/Smad3 signaling (Figure 8A). This was further confirmed by dual-luciferase report assay, in which Erbb4-IR overexpression significantly inhibited the transcriptional activity of Smad7 (Figure 9A). In contrast, this inhibitory effect was prevented when the Erbb4-IR binding site was mutated (Figure 9A), revealing the binding specificity of Erbb4-IR on the 3′ UTR of Smad7 gene. To further demonstrate the negative regulatory role of Erbb4-IR in Smad7 expression, we performed a gain-of-function study by overexpressing Erbb4-IR in mTECs. Results shown in Figures 9B–9E revealed that overexpression of Erbb4-IR substantially suppressed Smad7 transcription, which was associated with a marked increase in collagen I and α-SMA mRNA expression, confirming a pathogenic role of Erbb4-IR in renal fibrosis by targeting Smad7.
Figure 8.

Knockdown of Erbb4-IR Suppresses TGF-β/Smad3 Signaling by Increasing Smad7 Expression in mTECs In Vitro
(A–D) Representative western blots and quantitative analysis of western blots for Smad7 (A and D), phosphorylated TGF-β receptor I (p-TβRI) (A and B), and p-Smad3 (A and C). (E) Real-time PCR analysis for Smad7 mRNA expression. Results show that knockdown of Erbb4-IR significantly increases constitutive Smad7 mRNA at 6 hr and protein expression at 24 hr, which is associated with inhibition of TGF-β/Smad3 signaling by largely reducing p-TβRI and p-Smad3 at 24 hr after TGF-β1 (2 ng/mL) stimulation. Each bar represents the mean ± SEM for at least three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with untreated or blank control cells. #p < 0.05, ##p < 0.005, and ###p < 0.001 compared with TGF-β1-treated control (NC) or pcDNA3.1 EV-transfected cells.
Figure 9.

Overexpression of Erbb4-IR Suppresses Smad7 Promoter Activities and Transcription, Which Is Associated with Upregulation of Collagen I and α-SMA mRNA in mTECs In Vitro
(A) Dual luciferase assay shows the inhibitory effect of Erbb4-IR binding on Smad7 3′ UTR reporter activity, which is prevented by mutation of the Erbb4-IR binding site in a pGL3 mutant. (B) Transfection of Erbb4-IR full-length sequence containing pcDNA3.1 plasmid (Erbb4-IR) largely increased the Erbb4-IR expression in mTECs compared with the pcDNA3.1 EV by real-time PCR. (C–E) Real-time PCR analysis shows that overexpression of Erbb4-IR significantly decreases the mRNA expression level of (C) Smad7 but increases fibrotic markers (D) collagen I and (E) α-SMA in mTEC at 24 hr. Each bar represents the mean ± SEM for at least three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control EV. ##p < 0.01 compared with pcDNA3.1 EV-transfected cells.
The regulatory effect of Erbb4-IR on TGF-β/Smad signaling was also observed in the UUO kidney. Western blot and real-time PCR detected that knockdown of renal Erbb4-IR with shRNA largely increased renal Smad7 mRNA and protein expressions (Figures 10A, 10D, and 10E), which was associated with inactivation of TGF-β/Smad3 signaling, including a marked inhibition of renal TGF-β1 expression, phosphorylation of TβRI and Smad3, and phosphorylated Smad3 nuclear translocation (Figures 10A–10C and 11). Thus, Erbb4-IR may inhibit the transcription of Smad7 to promote TGF-β/Smad3-mediated renal fibrosis in vitro and in vivo.
Figure 10.

Kidney-Specific Knockdown of Erbb4-IR Increases Smad7 but Inhibits TGF-β/Smad3 Signaling in the UUO Kidney on Day 7
(A–D) Representative western blots and quantitative analysis of western blots for Smad7 (A and D), p-TβRI (A and B), and p-Smad3 (A and C). Each lane represents one animal. (E) Real-time PCR analysis for Smad7 mRNA expression. Results show that kidney-specific knockdown of Erbb4-IR significantly increases renal Smad7 mRNA and protein expression, which is associated with a marked inhibition of TGF-β/Smad3 signaling by largely reducing p-TβRI and p-Smad3. Each bar represents the mean ± SEM for groups of 6 mice. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to sham-operated mice. #p < 0.05 compared to EV-treated UUO kidney.
Figure 11.

Kidney-Specific Knockdown of Erbb4-IR Suppresses TGF-β1 Expression and p-Smad3 Nuclear Translocation in the UUO Kidney on Day 7
(A) Representative immunohistochemical staining of TGF-β1 in the kidney. (B) Representative immunohistochemical staining of p-Smad3 nuclear translocation in the kidney. (C) Quantitative analysis of TGF-β1 expression detected by immunohistochemistry. (D) Real-time PCR analysis of TGF-β1 mRNA expression. (E) Quantitative analysis of nucleated p-Smad3-positive cells. Each bar represents the mean ± SEM for groups of 6 mice. ***p < 0.001 compared to sham-operated mice. ##p < 0.05 and ###p < 0.001 compared to EV-treated UUO kidney. Scale bars, 50 μm.
Discussion
By using RNA-sequencing analysis, we previously detected that a Smad3-dependent lncRNA transcript np_5318 is upregulated in the UUO-injured kidney, which is inhibited in mice lacking Smad3 or overexpressing Smad7.23 In this study, we further characterized the lncRNA np_5318 by obtaining its full-length sequence via RACE. We revealed that lncRNA_5318 is 2,310 nt in length and is located within the intron region between the first and second exons of Erbb4 on chromosome 1 of a mouse (Chr1: 69026879–69029188). We therefore named lncRNA np_5318 as Erbb4-IR. Interestingly, no Smad3-binding site is found on the Erbb4 promoter nor untranslated regions by using the ECR browser (data not shown).25 This may explain that addition of TGF-β1 or knockdown of Erbb4-IR did not alter the expression of Erbb4 and suggest that Erbb4-IR is a novel gene that exerts its profibrotic function independently of its host gene Erbb4.
In this study, we revealed that Erbb4-IR is a novel lncRNA related to renal fibrosis but not inflammation. This was supported by the findings that Erbb4-IR was induced by a pro-fibrotic growth factor TGF-β1 but not by a pro-inflammatory cytokine IL-1β. Furthermore, expression of Erbb4-IR was markedly upregulated in the UUO kidney associated with progressive fibrosis but not in mouse models of acute kidney injury. Thus, Erbb4-IR may function specifically for chronic kidney disease associated with fibrosis. The fibrogenic role for Erbb4-IR in chronic kidney disease was further confirmed by the ability of silencing Erbb4-IR to inhibit renal fibrosis in the UUO kidney and mTECs in response to TGF-β1.
We also identified that Erbb4-IR was a downstream effector of TGF-β/Smad3 signaling. This was supported by the findings that treatment with a Smad3-specific inhibitor (SIS3) was able to prevent TGF-β1-induced Erbb4-IR expression in mTECs and deletion of Smad3 abolished upregulation of Erbb4-IR in TGF-β1-treated MEF cells. Unexpectedly, deletion of Smad2 enhanced Erbb4-IR expression in MEF cells in response to TGF-β1, revealing a TGF-β/Smad3-dependent regulatory mechanism in Erbb4-IR induction. These findings are coincident with our previous findings that Smad2 and Smad3 play reciprocal roles in regulating TGF-β1 target genes, such as miR-192, miR-21, and miR-29 during renal fibrosis.8, 9, 10 The differential regulatory roles of Smad2 and Smad3 on TGF-β1-induced Erbb4-IR transcription may account for our previous findings that Smad3 is pathologic, whereas Smad2 is protective in renal fibrosis.29, 30, 31
Interestingly, the present study found that Erbb4-IR was not only induced by TGF-β/Smad3, but also functioned to promote TGF-β/Smad3-mediated renal fibrosis by targeting Smad7. Thus, Erbb4-IR may function as an integrated effector molecule in the positive feedback loop of TGF-β/Smad signaling to augment the fibrosis response. In general, TGF-β1 mediates tissue fibrosis by activating Smad3, which is inhibited by its inhibitor Smad7.2, 3 Many studies show that deletion of Smad3 protects against renal fibrosis,32, 33, 34 whereas deletion of Smad7 promotes, but overexpression of Smad7 inhibits renal fibrosis in a number of kidney disease models.35, 36, 37, 38, 39, 40 In the present study, we found that Erbb4-IR may execute its suppressive effects on Smad7 via directly binding on the 3′ UTR of the Smad7 genomic sequence. This was confirmed by mutation of the Erbb4-IR binding site to prevent the inhibitory effect of Erbb4-IR on the Smad7 reporter activity. The functional importance for Erbb4-IR in promoting TGF-β/Smad3-mediated renal fibrosis by targeting Smad7 is also supported by gain-of-function and loss-of-function assays, in which overexpression of Erbb4-IR inhibited but knockdown of Erbb4-IR significantly increased Smad7 transcription. Therefore, TGF-β1/Smad3 signaling may induce Erbb4-IR to further amplify its fibrotic response by inhibiting the Smad7-dependent negative feedback loop of TGF-β/Smad signaling.
Results from this study also demonstrated that targeting Erbb4-IR may be a specific and effective therapy for chronic kidney disease associated with progressive renal fibrosis. Indeed, Erbb4-IR is a fibrotic lncRNA specifically upregulated in the fibrotic kidney but absent in kidney disease with acute renal inflammation. This finding suggests a therapeutic potential of anti-Erbb4-IR specifically for renal fibrosis. However, it should be noted that lncRNAs are usually expressed at a lower level and often in a tissue-specific manner, which may hinder their general application clinically. In addition, most of lncRNAs between the mouse and human are not conserved, which also hampers translation of the finding of lncRNAs from the mouse to the human. Nevertheless, evidence for the importance of lncRNAs is continuously growing and identification of mouse orthologs of human lncRNAs may help in the elucidation of their biological role clinically.
In conclusion, as summarized in Figure 12, Erbb4-IR is a novel lncRNA specifically upregulated in the kidney with progressive fibrosis. Erbb4-IR is induced by TGF-β1 via a Smad3-dependent manner. Erbb4-IR also acts as a positive regulator to further promote TGF-β1/Smad3-mediated renal fibrosis by targeting Smad7. Thus, inhibition of Erbb4-IR may represent a novel and precise therapeutic strategy for renal fibrosis.
Figure 12.
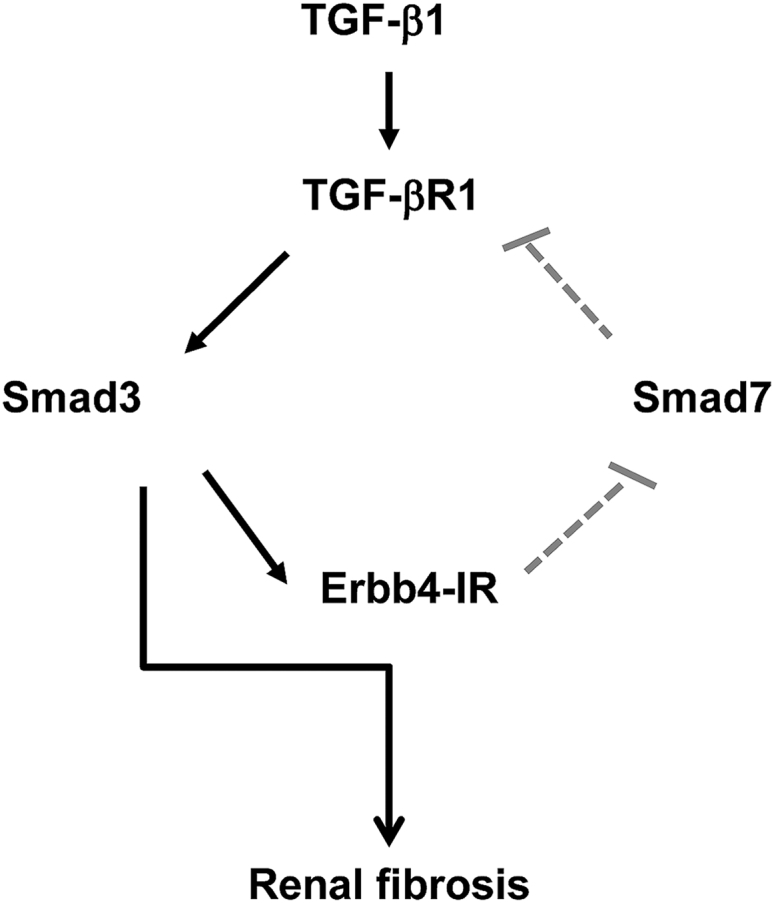
Potential Mechanisms of Erbb4-IR Promote TGF-β/Smad3-Mediated Renal Fibrosis
Activation of TGF-β/Smad3 induces Erbb4-IR to inhibit Smad7 transcription, which results in a loss of the negative feedback on TGF-β/Smad3 signaling and thus promotes renal fibrosis.
Materials and Methods
RACE and Full-Length cDNA Generation
SMARTer RACE 5′/3′ Kit (Clontech) was used for a rapid amplification of 5′ and 3′ ends of Erbb4-IR. cDNA reverse-transcription from the kidney tissue of the UUO kidney was used as the template for RACE PCR according to the user’s manual. The gene specific primer (GSP) and nested gene specific primer (NGSP) for 5′ and 3′ RACE were as follows: GSP1-5′ race 5′-GATTACGCCAAGCTTTTCACACAACCCCAAACAAGCTGTCGGA-3′, NGSP1-5′ race 5′-GATTACGCCAAGCTTACACAACCCCAAACAAGCTGTCGGA-3′, GSP2-3′ race 5′-GATTACGCCAAGCTTTGCTGAGAAAGTTATCCTGCTCCTGG-3′, and NGSP2-3′ race 5′-GATTACGCCAAGCTTTGTTGCTGCCCCACTCTCTCCTCT-3′. The full length of Erbb4-IR cDNA was PCR synthesized with the forward primer 5′-ATGACAAAATGGAAAATTTACTCTCTGCTGC-3′ and reverse primer 5′-TTTTTTTCTTATTCACTTTACAACCAACTCAC-3′.
Bioinformatics Analysis of Erbb4-IR Sequence
The location of Erbb4-IR in the mouse genome was searched through https://blast.ncbi.nlm.nih.gov/Blast.cgi and http://www.genome.ucsc.edu/. The alignment of Erbb4-IR among multiple vertebrate genomes was blasted through the ECR browser (https://ecrbrowser.dcode.org/).25 The protein-coding potential of the Erbb4-IR sequence was evaluated by two widely used computational programs: CPC (http://cpc.cbi.pku.edu.cn/programs/run_cpc.jsp) and CPAT (http://lilab.research.bcm.edu/cpat/index.php).26, 27 For evaluation in CPC, transcripts with scores higher than 1 are predicted to be “coding”, lower than −1 are “non-coding”, and between −1 and 1 are classified as “weak non-coding” ([−1, 0]) or “weak coding” ([0, 1]). Although for CPAT the cutoff value of mouse coding probability is 0.44, transcripts with scores higher than 0.44 are classified as “coding”, whereas those lower than 0.44 are “non-coding”.
Cell Culture
The mTEC (a gift from Dr. Jeffrey B. Kopp, NIH) and MEF cells were cultured in DMEM/F12 medium (Gibco, CA), supplemented with 5% fetal bovine serum (FBS) (Gibco, CA).9, 10, 24 Cells were stimulated with or without TGF-β1 (2 ng/mL, R&D Systems, MN) for different time points. To inhibit Smad3 activity, cells were pre-treated with the Smad3 inhibitor SIS3 (Sigma-Aldrich) at dosages of 1 or 2 μM for 1 hr prior to 2 ng/mL of TGF-β1 stimulation.
Transfection of siRNA Targeting Erbb4-IR In Vitro
To examine the role of Erbb4-IR in renal fibrosis, mTECs were transfected with 100 nM Erbb4-IR siRNA (sense 5′-GCCUACAGUUUAUCCACAAdTdT-3′, anti-sense 3′-dTdTCGGAUGUCAAAUAGGUGUU-5′) or NC siRNA (sense 5′-AUGAACGUGAAUUGCUCAAUUU-3′, anti-sense 3′-dTdTUACUUGCACUUAACGAGUUAAA-5′) using Lipofectamin RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. The cells were then stimulated with TGF-β1 (2 ng/mL) for 1, 6, and 24 hr. All cells were fasted with 0.5% FBS medium for 24 hr before stimulation and maintained in medium with 0.5% FBS until the end of stimulation.
Construction of Erbb4-IR shRNA-pSuper.Puro Vector
Erbb4-IR shRNA sequences (sense 5′-AGCTTGCCTACAGTTTATCCACAAttCAAGAGATTGTGGATAAACTGTAGGCTTTTTTGAATTCC-3′, anti-sense 5′-TCGAGGAATTCAAAAAAGCCTACAGTTTATCCACAATCTCTTGAATTGTGGATAAACTGTAGGCA-3′) were annealed and cloned into pSuper.puro vector (Oligoengine, WA) at HindIII and XhoI sites.
Mouse Kidney Injury Model of UUO and Ultrasound-Mediated Gene Transfer of Erbb4-IR shRNA Plasmids
A mouse model of UUO was induced in male C57BL/6J mice at 8 weeks of age (20–22 g body weight) and Erbb4-IR shRNA expressing plasmids were transfected into the left kidney as described earlier.9, 10, 11, 12, 13 In brief, before the left ureter was ligated, groups of 6–8 mice received the mixed solution (200 μL/mouse) containing either the Erbb4-IR shRNA-pSuper.puro vector or empty pSuper.puro vector (200 μg/mouse) and lipid microbubbles (Sonovue, Bracco, Milan, Italy) at a ratio of 1:1 (v/v) via the tail vein injection, as described earlier.9, 10, 11, 12, 13, 14 Immediately after injection, an ultrasound transducer (Therasonic, Electro Medical Supplies, Wantage, UK) was directly placed on the skin of the back against the left kidney with a pulse-wave output of 1 MHz at 2 W/cm2 for a total of 5 min. Kidney tissues were harvested at day 7 after the ultrasound treatment. In addition, groups of 6–8 sham-operated and UUO mice without ultrasound treatment were used as controls. The experimental procedures were performed following the approved protocol by the Animal Experimentation Ethics Committee at the Chinese University of Hong Kong.
Real-Time PCR Analysis
Total RNA was isolated from the cultured cells and kidney tissues using Trizol (Invitrogen, CA) according to the manufacturer’s instructions. Real-time PCR was performed by SYBR Green Supermix using the CFX96 PCR System (Bio-Rad, CA), as described earlier.9, 10, 11, 12, 13, 14 The primers used in this study, including mouse collagen I, α-SMA, Smad7, TGF-β1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), are described previously.9, 10, 11, 12, 13, 14 Other primers include the following: mouse Erbb4-IR forward 5′-AACTCGCCACAGAAATCCAC-3′ and reverse 5′-ACAACCCCAAACAAGCTGTC-3′; mouse Erbb4 forward 5′-GGACGGGCCATTCCACTTTA-3′ and reverse 5′-TGGGGCCAGTTGTTTCATTG-3′. The relative level of the detected gene was normalized with the internal control GAPDH by the delta-delta Ct method and expressed as mean ± SEM.
Western Blot Analysis
Protein from cultured cells and kidney tissues was extracted using the radio immunoprecipitation assay (RIPA) lysis buffer. Western blot analysis was performed as described earlier.9, 10, 11, 12, 13, 14 In brief, after blocking nonspecific binding with 5% BSA, membranes were then incubated overnight at 4°C with the primary antibody against collagen I (1310-01, Southern Biotech, Birmingham, AL), α-SMA (M0851, Dako, Carpentaria, CA), phosphor-TβRI (OAA100768, Avivasysbio), TβRI (sc-398, Santa Cruz Biotechnology), phosphorylated Smad3 (p-Smad3) (#8769, Cell Signaling Technology, Danvers, MA), Smad3 (511500, Invitrogen), Smad7 (sc-9183, Santa Cruz), Erbb4 (sc-283, Santa Cruz), and GAPDH (MAB374, Millipore), followed by IRDye800-conjugated secondary antibody (Rockland Immunochemicals, Gilbertsville, PA). Signals were detected using the Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE), followed by quantitative analysis using the ImageJ program. The ratio for the protein examined was normalized against GAPDH and expressed as mean ± SEM.
In Situ Hybridization
To detect the expression pattern and location of Erbb4-IR in the kidney, in situ hybridization was performed with double digoxigenin-labeled probes (Exiqon) following the established protocol, as described earlier.8, 9, 10, 11, 14, 24 The sequences of the probes are as follows: Erbb4-IR probe 5′-AATAGATATAACGACAATGTGT-3′ and scramble probe 5′-GTGTAACACGTCTATACGCCCA-3′.
Masson’s Trichrome Staining and Immunohistochemistry
To evaluate the histological damage, collagen-like matrix deposition was stained with Masson’s trichrome staining with the Trichrome stain kit (ScyTek Laboratories, West Logan, UT) according to the manufacturer’s instructions. Immunohistochemistry was performed in 4 μm paraffin-embedded tissue sections of mouse kidney tissue using a microwave-based antigen retrieval technique.41 Primary antibodies used in this study included collagen I (1310-01, Southern Biotech, Birmingham, AL), α-SMA (A2547, Sigma, St. Louis, MO), TGF-β1 (sc-146, Santa Cruz Biotechnology), and p-Smad3 (600-401-919, Rockland). The total collagen-like contents, accumulation of collagen I, α-SMA, and TGF-β1, were measured in Masson’s trichrome stained and immunostained sections by 10 random areas under high power by using a quantitative image-analysis system (Image-Pro Plus 6.5, Media Cybernetics, Silver Spring, MD) and expressed as the percent of positive area examined. The number of p-Smad3-positive cells was counted under high power fields (Χ 40) by means of a 0.0625-mm2 graticule fitted in the eyepiece of the microscope and expressed as cells per millimeters squared.
Luciferase Reporter Assay of Smad7 3′ UTR
Construction of Erbb4-IR and Smad7 3′ UTR genomic sequences overexpressing plasmids and luciferase assay were performed by Shanghai GenePharma (Shanghai, China). Full sequence of Erbb4-IR (Figure S1) was constructed onto the pcDNA3.0 vector. Smad7 3′ UTR reporter with (pGL3-Smad7) or without (pGL3 mutant) the predicted Erbb4-IR binding site (Figure S5) was constructed onto the pGL3-basic vector.
For dual luciferase reporter assay (Promega), the Smad7 3′ UTR luciferase reporter plasmid and renilla-overexpressing plasmid (as internal control) were co-transfected into 293T cells with PBS (blank control), pcDNA3.0 (empty vector), or Erbb4-IR plasmids. The luminescence in lysates of harvested cells was measured at 48 hr according to the manufacturer’s instructions. The luciferase activity (M1) and Renilla luciferase activity (M2) were measured by the GloMax-Multi Detection System (Promega). The reporter activity was represented by the ratio of M1/M2, which was the normalized luciferase activity of the experimental plasmid (pGL4-basic; Promega). Results are shown as mean ± SEM fold induction of luciferase in at least 3 independent experiments.
Overexpression of Erbb4-IR
The mTECs were transfected with 0.5 μg/mL pcDNA3.1 empty vector (EV) or plasmid containing Erbb4-IR full-length RNA sequence, as shown in Figure S1 (Erbb4-IR, constructed by PharmaGene, Shanghai, China) using lipofectamine 2000 (Invitrogen) according to the manufacturer’s manual, and the culture medium was refreshed after 4 hr. Then, the transfected cells were collected at an appropriated time point and subjected for further analysis.
Statistical Analysis
Data obtained from this study were expressed as mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by a Newman-Keuls multiple comparison test from GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
Author Contributions
M.F. and P.M.-K.T. have contributed equally to this work. H.-Y.L. and A.-P.X. designed and supervised all the experiments and contributed to manuscript preparation. M.F. and P.M.-K.T. performed experiments, analyzed data, and contributed to manuscript preparation. X.-R.H., S.-F.S., Y.-K.Y., J.X., and L.-L.L. collected animal samples and participated in animal experiments.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This study is supported by grants from the Research Grants Council of Hong Kong (GRF 14117815 and T12-402/13N) and the Science and Technology Project of Guangdong Province, China (2016A020215068).
Footnotes
Supplemental Information includes seven figures can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.09.024.
Contributor Information
An-Ping Xu, Email: anpxu@163.com.
Hui-Yao Lan, Email: hylan@cuhk.edu.hk.
Supplemental Information
References
- 1.Eddy A.A. Overview of the cellular and molecular basis of kidney fibrosis. Kidney Int. Suppl. (2011) 2014;4:2–8. doi: 10.1038/kisup.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meng X.M., Tang P.M., Li J., Lan H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015;6:82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 4.Yang X., Letterio J.J., Lechleider R.J., Chen L., Hayman R., Gu H., Roberts A.B., Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatt K., Kato M., Natarajan R. Mini-review: emerging roles of microRNAs in the pathophysiology of renal diseases. Am. J. Physiol. Renal Physiol. 2016;310:F109–F118. doi: 10.1152/ajprenal.00387.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantharidis P., Wang B., Carew R.M., Lan H.Y. Diabetes complications: the microRNA perspective. Diabetes. 2011;60:1832–1837. doi: 10.2337/db11-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung A.C., Lan H.Y. MicroRNAs in renal fibrosis. Front. Physiol. 2015;6:50. doi: 10.3389/fphys.2015.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung A.C., Huang X.R., Meng X., Lan H.Y. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 2010;21:1317–1325. doi: 10.1681/ASN.2010020134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin W., Chung A.C., Huang X.R., Meng X.M., Hui D.S., Yu C.M., Sung J.J., Lan H.Y. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011;22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong X., Chung A.C., Chen H.Y., Meng X.M., Lan H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011;22:1668–1681. doi: 10.1681/ASN.2010111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong X., Chung A.C., Chen H.Y., Dong Y., Meng X.M., Li R., Yang W., Hou F.F., Lan H.Y. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56:663–674. doi: 10.1007/s00125-012-2804-x. [DOI] [PubMed] [Google Scholar]
- 12.Chung A.C., Dong Y., Yang W., Zhong X., Li R., Lan H.Y. Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Mol. Ther. 2013;21:388–398. doi: 10.1038/mt.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li R., Chung A.C., Dong Y., Yang W., Zhong X., Lan H.Y. The microRNA miR-433 promotes renal fibrosis by amplifying the TGF-β/Smad3-Azin1 pathway. Kidney Int. 2013;84:1129–1144. doi: 10.1038/ki.2013.272. [DOI] [PubMed] [Google Scholar]
- 14.Chen H.Y., Zhong X., Huang X.R., Meng X.M., You Y., Chung A.C., Lan H.Y. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 2014;22:842–853. doi: 10.1038/mt.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishida M., Selaru F.M. MiRNA-based therapeutic strategies. Curr. Anesthesiol. Rep. 2013;1:63–70. doi: 10.1007/s40139-012-0004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brockdorff N., Ashworth A., Kay G.F., McCabe V.M., Norris D.P., Cooper P.J., Swift S., Rastan S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 1992;71:515–526. doi: 10.1016/0092-8674(92)90519-i. [DOI] [PubMed] [Google Scholar]
- 17.Hajjari M., Khoshnevisan A., Shin Y.K. Molecular function and regulation of long non-coding RNAs: paradigms with potential roles in cancer. Tumour Biol. 2014;35:10645–10663. doi: 10.1007/s13277-014-2636-z. [DOI] [PubMed] [Google Scholar]
- 18.Ng S.Y., Lin L., Soh B.S., Stanton L.W. Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 2013;29:461–468. doi: 10.1016/j.tig.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 19.Wu G.C., Pan H.F., Leng R.X., Wang D.G., Li X.P., Li X.M., Ye D.Q. Emerging role of long noncoding RNAs in autoimmune diseases. Autoimmun. Rev. 2015;14:798–805. doi: 10.1016/j.autrev.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Ounzain S., Burdet F., Ibberson M., Pedrazzini T. Discovery and functional characterization of cardiovascular long noncoding RNAs. J. Mol. Cell. Cardiol. 2015;89(Pt A):17–26. doi: 10.1016/j.yjmcc.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Lorenzen J.M., Thum T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat. Rev. Nephrol. 2016;12:360–373. doi: 10.1038/nrneph.2016.51. [DOI] [PubMed] [Google Scholar]
- 22.Hanson R.L., Craig D.W., Millis M.P., Yeatts K.A., Kobes S., Pearson J.V., Lee A.M., Knowler W.C., Nelson R.G., Wolford J.K. Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes. 2007;56:975–983. doi: 10.2337/db06-1072. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q., Chung A.C., Huang X.R., Dong Y., Yu X., Lan H.Y. Identification of novel long noncoding RNAs associated with TGF-β/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 2014;184:409–417. doi: 10.1016/j.ajpath.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Q., Huang X.R., Yu J., Yu X., Lan H.Y. Long noncoding RNA Arid2-IR is a novel therapeutic target for renal inflammation. Mol. Ther. 2015;23:1034–1043. doi: 10.1038/mt.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ovcharenko I., Nobrega M.A., Loots G.G., Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41:e74. doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith C., Heyne S., Richter A.S., Will S., Backofen R. Freiburg RNA Tools: a web server integrating INTARNA, EXPARNA and LOCARNA. Nucleic Acids Res. 2010;38:W373–W377. doi: 10.1093/nar/gkq316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang F., Chung A.C., Huang X.R., Lan H.Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 30.Chung A.C., Zhang H., Kong Y.Z., Tan J.J., Huang X.R., Kopp J.B., Lan H.Y. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 2010;21:249–260. doi: 10.1681/ASN.2009010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng X.M., Huang X.R., Chung A.C., Qin W., Shao X., Igarashi P., Ju W., Bottinger E.P., Lan H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010;21:1477–1487. doi: 10.1681/ASN.2009121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato M., Muragaki Y., Saika S., Roberts A.B., Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z., Huang X.R., Lan H.Y. Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am. J. Physiol. Renal Physiol. 2012;302:F986–F997. doi: 10.1152/ajprenal.00595.2011. [DOI] [PubMed] [Google Scholar]
- 34.Zhou L., Fu P., Huang X.R., Liu F., Chung A.C., Lai K.N., Lan H.Y. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am. J. Physiol. Renal Physiol. 2010;298:F1006–F1017. doi: 10.1152/ajprenal.00675.2009. [DOI] [PubMed] [Google Scholar]
- 35.Chen H.Y., Huang X.R., Wang W., Li J.H., Heuchel R.L., Chung A.C., Lan H.Y. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60:590–601. doi: 10.2337/db10-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung A.C., Huang X.R., Zhou L., Heuchel R., Lai K.N., Lan H.Y. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transplant. 2009;24:1443–1454. doi: 10.1093/ndt/gfn699. [DOI] [PubMed] [Google Scholar]
- 37.Lan H.Y., Mu W., Tomita N., Huang X.R., Li J.H., Zhu H.J., Morishita R., Johnson R.J. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 2003;14:1535–1548. doi: 10.1097/01.asn.0000067632.04658.b8. [DOI] [PubMed] [Google Scholar]
- 38.Hou C.C., Wang W., Huang X.R., Fu P., Chen T.H., Sheikh-Hamad D., Lan H.Y. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 2005;166:761–771. doi: 10.1016/s0002-9440(10)62297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu G.X., Li Y.Q., Huang X.R., Wei L.H., Zhang Y., Feng M., Meng X.M., Chen H.Y., Shi Y.J., Lan H.Y. Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. 2014;127:195–208. doi: 10.1042/CS20130706. [DOI] [PubMed] [Google Scholar]
- 40.Dai X.Y., Zhou L., Huang X.R., Fu P., Lan H.Y. Smad7 protects against chronic aristolochic acid nephropathy in mice. Oncotarget. 2015;6:11930–11944. doi: 10.18632/oncotarget.3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lan H.Y., Mu W., Nikolic-Paterson D.J., Atkins R.C. A novel, simple, reliable, and sensitive method for multiple immunoenzyme staining: use of microwave oven heating to block antibody crossreactivity and retrieve antigens. J. Histochem. Cytochem. 1995;43:97–102. doi: 10.1177/43.1.7822770. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.