

Abstract
The failure of therapeutic agents to cross the blood-brain barrier (BBB) has been a major impediment in the treatment of neurological disorders and brain tumors. We have addressed this issue using an immunoliposome nanocomplex (designated scL) that delivers therapeutic nucleic acids across the BBB into the deep brain via transcytosis mediated by transferrin receptors. We validated brain delivery of payloads after systemic administration by monitoring uptake of fluorescently labeled payloads and by confirming up- or down-modulation of specific target gene expression in the brain, mainly in neuronal cells. As proof of concept for the therapeutic potential of our delivery system, we employed scL delivering an siRNA targeting tumor necrosis factor alpha to suppress neuroinflammation and neuronal apoptosis and to protect mice in lethal endotoxemia triggered by bacterial lipopolysaccharide. Brain delivery of therapeutic payloads via scL has major implications for the development of treatments for neurological disorders and brain tumors.
Keywords: CNS drug delivery, crossing blood-brain barrier, neuroinflammation, receptor-mediated transcytosis, siRNA delivery, targeted nanocomplex, therapeutic nucleic acids
Graphical Abstract
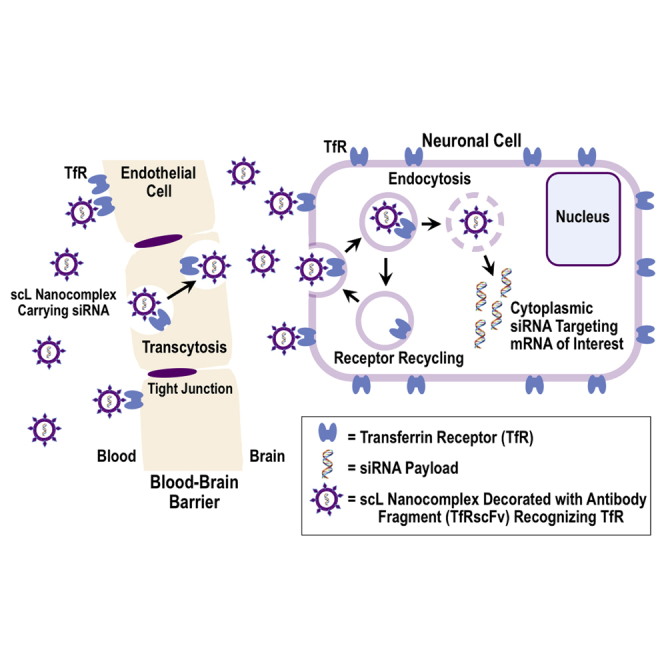
Chang and colleagues have developed an immunoliposome nanocomplex that, after its systemic administration, actively delivers nucleic acid therapeutics across the BBB into the deep brain via transcytosis mediated by transferrin receptors. The therapeutic potential for neurological disorders and neuroinflammation was demonstrated in mice with lethal endotoxemia triggered by bacterial lipopolysaccharide.
Introduction
The blood-brain barrier (BBB), comprised of brain capillary endothelial cells forming extremely tight junctions, is a highly selective diffusion barrier that protects brain parenchyma from blood-borne pathogens, toxins, and other potentially harmful substances in the blood.1 The entry of many therapeutic molecules from blood to brain is also significantly blocked by the BBB.1, 2 Despite major advances in the understanding of the molecular pathways involved in neurological disorders, the use of therapeutic agents with the potential to act on these pathways has often been significantly limited by their inabilities to traverse the BBB.3 As a result, many current drugs only produce minimal impact and marginal benefit in neurodegenerative processes. Similarly, the BBB contributes to primary and metastatic brain tumors being particularly refractory to chemo- and bio-therapeutics.
Various strategies have been employed to address this lack of access to the brain by therapeutics, including direct intraventricular injection,4 intrathecal or intranasal administration to avoid the BBB,5, 6 or invasive strategies involving mechanical or chemical disruptions of the BBB.7, 8, 9, 10 However, disruption of BBB to allow entry of therapeutic agents would concurrently compromise the normal protective role of the BBB with potential for brain toxicity or secondary infections.11, 12, 13 Moreover, many of these approaches are not amenable to the multiple doses of medicines to treat chronic disorders that require long-term interventions.2
Therapies based on small interfering RNA (siRNA) possess notable potential advantages over other therapeutic approaches. However, the realization of the promise of siRNA therapeutics requires that the siRNA be protected from degradation/clearance and delivered into the cytoplasm of relevant target cells to silence their targeted mRNAs. It is crucial to develop a non-invasive, effective, and clinically feasible means to transport therapeutic agents including siRNAs across the BBB. One approach has been the design of a delivery system that mimics neurotropic viruses by using short synthetic peptide derived from rabies virus glycoprotein conjugated to cell-penetrating peptides.14, 15 This strategy successfully demonstrated the gene silencing by an siRNA in animals with traumatic brain injury having a partially disrupted BBB.15 Another strategy takes the advantage of endogenous transport machinery to gain access to the brain. Due to the low permeability of the BBB, the brain parenchyma depends on different molecular transport systems to maintain homeostasis.16 Non-invasive strategies can exploit these intrinsic mechanisms to transport macromolecules into the brain using a natural process known as a receptor-mediated transcytosis (RMT). Examples of receptors engaging in RMT across the BBB include those for transferrin (Tf), insulin, and insulin-like growth factors.3, 4, 17, 18, 19 The Tf/Tf receptor (TfR) system, which naturally mediates cellular uptake of iron carried by Tf, has been focused upon as a pathway capable of carrying macromolecules into the brain.20, 21 However, despite the perceived potential of anti-TfR antibody-therapeutic conjugates and promising data in animals, no BBB-traversing agent using this approach has been approved as a drug for human use.16, 22
Employing a single-chain fragment from the variable region of an anti-human TfR monoclonal antibody (TfRscFv), we have developed a BBB-crossing liposome (designated scL). Compared to the full-length TfR monoclonal antibody, TfRscFv has advantages including the superior stability as well as a smaller size amenable to less cumbersome manufacturing than an intact IgG.16 In order to secure high concentrations of drug reaching the brain, scL is made of cationic lipids forming a small unilamellar liposome capable of efficient encapsulation of various payloads. The scL nanocomplex is approximately 100 nm in diameter, including its targeting moiety and payload. We have confirmed the scL can be used for delivery of various nucleic acids (e.g., oligonucleotide [ODN], siRNA,23 plasmid DNA,24 or cDNA) into the brain. We have examined the delivery of payload and efficacy of target gene modulation in the brain after the systemic (intravenous) administration of scL-based nanocomplex. Here, we demonstrate not only delivery of the nucleic acid-based payloads to the brain but also document their ability to modulate neuronal gene expression and apoptosis and to protect mice from otherwise fatal neuroinflammation triggered by bacterial lipopolysaccharide (LPS).
Results
In Vivo Brain Targeting via Systemically Delivered scL-Based Nanocomplex
To demonstrate the improved delivery of payload into the brain, we compared the transport of fluorescently labeled ODNs in vivo by intravenously injecting either 6-carboxyfluorescein phosphoramidite (6FAM)-conjugated ODN encapsulated in scL nanocomplex with a targeting moiety (scL-6FAM-ODN), nanocomplex without a targeting moiety (Lip-6FAM-ODN), or free 6FAM-ODN without encapsulation in nanocomplex (all at 100 μg of 6FAM-ODN/mouse). Twenty-four hours after a single bolus injection intravenously, the fluorescent signal was assessed in brains using the Maestro in vivo imaging system. With scL-6FAM-ODN treatment, an intense fluorescent signal was evident throughout the brain, especially in the cerebellum, cerebral cortex, and hippocampus regions (Figure 1). Accumulation of 6FAM-ODN is maximized by encapsulation in scL, with the fluorescent signal being significantly stronger with scL-6FAM-ODN than with either non-targeting Lip-6FAM-ODN nanocomplex or free 6FAM-ODN (Figures 1A and 1B).
Figure 1.

Systemic Administration of scL Nanocomplex Results in Localization of Payloads in Mouse Brain
(A and B) Mice were intravenously injected with controls or scL carrying fluorescence-labeled oligonucleotide (6FAM-ODN, green). (A) Maestro imaging of brain at 24 hr post-injection. The fluorescence image is overlaid on the bright-field image in mid-sagittal view. Representative images from untreated (UT) mice are shown along with those from mice injected with free 6FAM-ODN, untargeted Lip-6FAM-ODN, and scL-6FAM-ODN targeting the TfR. (B) The fluorescence signal in mouse brains was quantified. Data are represented as mean ± SD (n = 2 to 7 mice per group). *p = 0.004. (C and D) Time course study of the systemic delivery of scL carrying fluorescent-labeled oligonucleotide (Cy5-ODN, red) to mouse brain. (C) Maestro imaging of serial coronal sections of the brain at the indicated time after intravenous injection of scL-Cy5-ODN are shown along with brains from UT mice. (D) Quantitation of Cy5 fluorescence in (C) was plotted as a function of time post-administration of the scL-Cy5-ODN.
To understand the distribution of ODNs delivered by the scL nanocomplex, we evaluated the coronal sections of brain at a various time after the systemic administration. In this experiment, BALB/c mice received a single tail-vein injection of scL nanocomplex carrying cyanine 5 (Cy5)-labeled ODN (at 25 μg of Cy5-ODN/mouse) and fluorescent signal examined in the coronal sections of the brain every 6 hr following the injection. A strong fluorescent signal was seen in several areas of mouse brain (e.g., hippocampus, cortex, and cerebellum) (Figure 1C). As early as 6 hr post-injection, a significant presence of Cy5 fluorescence was observed with the signal peaking at 12 hr and decreasing but still detectable by 48 hr post-injection (Figures 1C and 1D). Taken together, these results demonstrate that the scL-based nanocomplex has the ability to deliver payloads across the BBB after systemic administration in mice.
scL-Mediated Transgene Expression In Vivo
Using a GFP expression plasmid as a payload, we wanted to confirm the enhanced delivery of payload into the mouse brain. BALB/c mice received a single intravenous injection of either GFP plasmid DNA encapsulated in scL (scL-GFP), the complex lacking a targeting moiety (Lip-GFP), or free GFP plasmid DNA (all at 30 μg of GFP plasmid DNA/mouse). Forty-eight hours after the injection, the expression of the transgene was assessed by determining the fluorescence intensity of GFP in the brain using Maestro (Figure 2A). The green fluorescence emanating from GFP was observed throughout the brain after injection of scL-GFP. Virtually no fluorescence was detected in brains of mice that received either non-targeting Lip-GFP nanocomplex or free GFP plasmid DNA. These results are similar to those of Figure 1, where 6FAM-ODN was used as a payload. However, unlike the fluorescence from 6FAM-ODN, the signal when using the GFP gene as payload requires escaping from the endosomal compartment followed by transcription into GFP mRNA and its translation into protein.
Figure 2.

Systemic Administration of scL-Based Nanocomplex Carrying the GFP Gene Results in Transgene Expression in Mouse Brain
(A) Mice were intravenously injected once with controls or scL carrying GFP plasmid DNA (30 μg of GFP plasmid DNA/mouse). Maestro imaging of mouse brain was performed at 48 hr after the single tail-vein injection. The fluorescence image is overlaid on the bright-field image in mid-sagittal view. (B–D) Other mice were repeatedly intravenously injected with scL-GFP (at 30 μg of GFP plasmid DNA/injection/mouse), i.e., every 12 hr for a total of three injections. (B) Maestro imaging of serial coronal sections of mouse brain was performed at 24, 36, and 48 hr after the last injection. (C) GFP fluorescence quantitated and plotted as a function of time. Data are represented as mean ± SD (n = 2 mice per each time point). (D) Higher-resolution immunofluorescence microscopy confirmed GFP expression in hippocampus (CA1) and cortex of mouse brain at 48 hr after the last injection of scL-GFP. Red arrows, blood vessels; white arrows, neurons; yellow arrows, possible neurons, glia, or cross-section of vessels. Scale bar, 75 μm.
To further understand the cellular distribution of GFP reporter protein expression in mouse brain, we evaluated the coronal sections of brain at a various time after the systemic administration. In this experiment, BALB/c mice received an intravenous injection of scL-GFP nanocomplex (at 30 μg of GFP plasmid DNA/injection/mouse) every 12 hr for a total of three injections. The expression of GFP was assessed by Maestro imaging at 24, 36, and 48 hr after the last intravenous injection (Figure 2B). GFP expression was evident in the serial coronal sections of brains of scL-GFP-treated mice. A time course showed that, relative to the level in the untreated brain, the expression of GFP gradually increased out to 48 hr after the last injection (Figure 2C). Further immunofluorescence analysis of mouse brain at 48 hr after the last injection of scL-GFP showed abundant green fluorescence in brain regions of hippocampus and cortex of mice treated with scL-GFP (Figure 2D), indicating that TfR-targeted nanocomplex traversed the BBB to deliver the genetic payload that is expressed in adult neurons. In the deep brain, GFP fluorescence was observed in neuronal cells and blood vessels in the hippocampus and cortex.
Using another TfR-targeting nanocomplex, we further verified the brain targeting in rats to attest the crossing of the BBB being mediated by TfR. In this experiment, we have observed a similar finding in rat brains when rat Tf was utilized as a targeting moiety (in the TfL nanocomplex) instead of TfRscFv (in the scL nanocomplex). This change in the targeting moiety on the nanocomplex was necessitated by the fact that our scFv recognizes the mouse TfR but not the rat TfR. GFP cDNA (100 μg cDNA/injection) encapsulated in either TfL or Lip (lacking a targeting moiety) was administered as a single bolus injection via a catheterized jugular vein in anesthetized rats. After 36 hr, GFP cDNA delivered by non-targeted Lip produced no detectable GFP-positive cells in the brain (Figure 3, top panel). In contrast, delivery of GFP cDNA injected as the TfL-GFP nanocomplex yielded abundant GFP expression in the cortex (Figure 3, middle panel). GFP-positive cells exhibited a morphological phenotype of neurons. Immunohistochemistry using interneuron marker parvalbumin and Nissl staining revealed the neuronal network in the cortex. Punctate parvalbumin staining was observed around GFP-positive neurons, which are also labeled via Nissl staining (Figure 3, bottom panel). These results confirm the crossing of the BBB being mediated by TfR and the feasibility of using TfR-targeting nanocomplexes (both scL and TfL) for gene delivery to neurons in the brain. More significantly, the delivered payloads, both plasmid DNA and cDNA, were intact to be expressed via transcription and translation once inside neurons and not trapped in endosomes.
Figure 3.
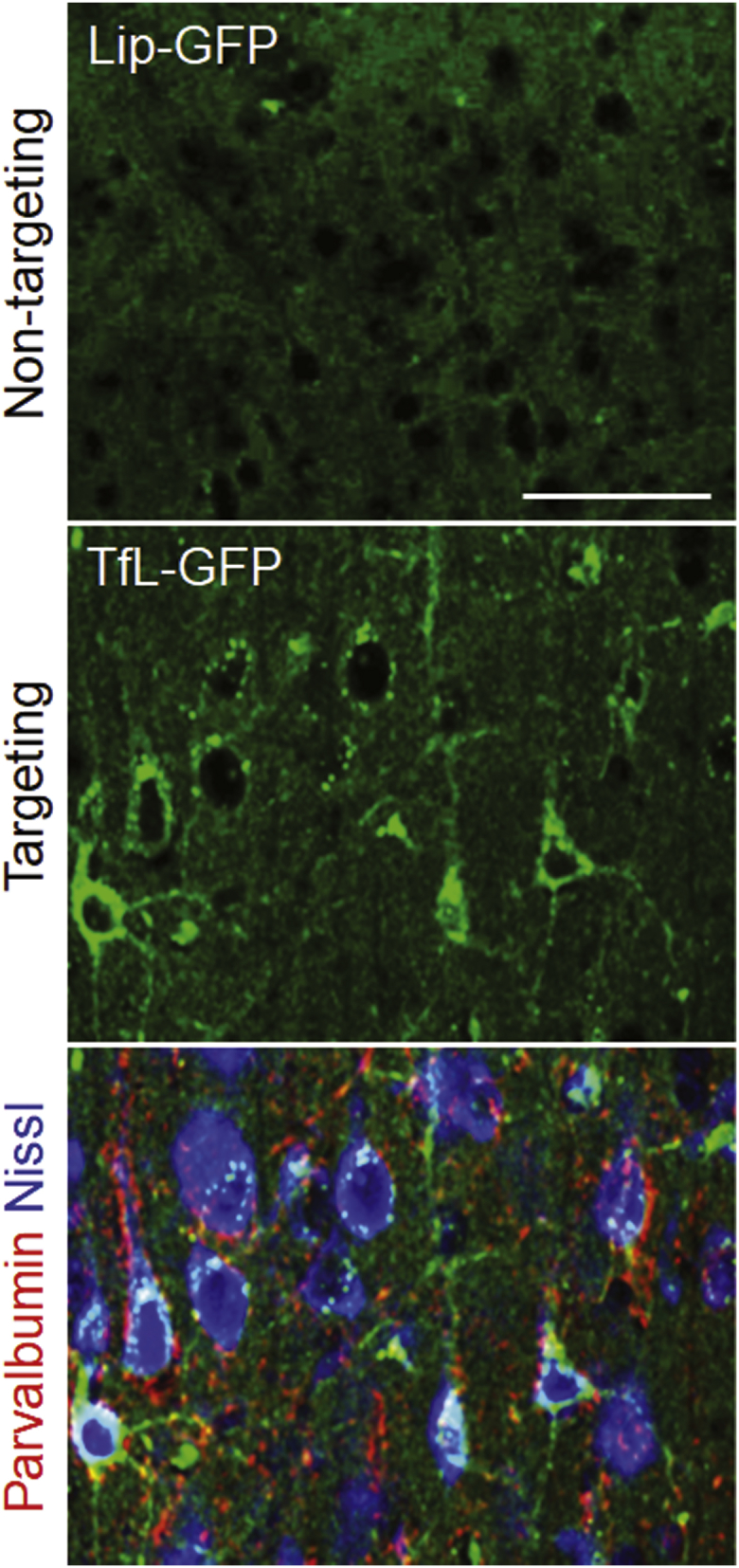
TfL-Mediated Systemic Delivery of GFP cDNA to Rat Brain Resulted in Expression of Exogenous GFP in Neuronal Cells in the Cortex
Rats were intravenously injected with non-targeted Lip-GFP or targeted TfL-GFP carrying GFP cDNA (100 μg of GFP cDNA/rat). Immunofluorescence imaging of GFP expression at 36 hr post-injection. Parvalbumin (red); Nissl staining (blue). Scale bar, 50 μm.
Silencing of SOD1 in Mouse Brain by scL-Based Nanocomplex
To investigate the therapeutic applicability of scL-based nanocomplexes for neurological disorders, we examined the silencing of a disease-related gene by scL-delivered siRNA in mouse brain. An siRNA targeting mouse copper/zinc superoxide dismutase 1 (SOD1) was used for these in vivo studies. The mutation of SOD1 is a hallmark of amyotrophic lateral sclerosis (ALS) and silencing of mutant SOD1 holds potential in the treatment of ALS. BALB/c mice were intravenously injected with scL-encapsulated SOD1 siRNA (scL-siSOD), unencapsulated siSOD, or control siRNA encapsulated in scL at 180 μg of siRNA/injection, and brains examined for SOD1 mRNA and SOD1 enzyme activity. Twenty-four hours after a single injection, about 15% reduction of SOD1 mRNA was observed in brains of mice treated with scL-siSOD compared to untreated controls (Figure 4A). However, no significant change was observed in brains of mice treated with control siRNA encapsulated in scL. Similarly, SOD1 enzyme activity in the brain was decreased in mice treated with scL-siSOD by >30% compared to the untreated controls, while no significant change was detected in brains of mice given control siRNA encapsulated in scL (Figure 4B). We also monitored for changes in the SOD1 protein level by western blot analysis (Figure 4C) and confirmed a significant silencing of SOD1 protein levels (∼54.7% reduction) 24 hr after treatment with scL-siSOD nanocomplex. No significant reduction of SOD1 protein levels was observed with control control siRNA encapsulated in scL treatment. These results demonstrate that scL enables the systemic delivery of a therapeutic siRNA to the brain and the down-modulation of a target gene in the brain. This ability to modulate gene expression using an siRNA was not dependent upon any disruption of the BBB. Rather, the delivery system utilizes the brain’s own TfR-mediated RMT process and thus holds promise for the use of siRNA-based therapeutics to treat chronic neuronal disorders like ALS.
Figure 4.

Systemic Administration of scL-siSOD Downmodulates SOD1 in Mouse Brain
Mice were intravenously injected with scL-siSOD nanocomplexes (180 μg of siRNA/mouse). Negative control siRNA (siCTRL) was also complexed with scL (scL-siCTRL) for injection. Twenty-four hours after siRNA administration, brains were examined for SOD1 mRNA (A) and SOD1 enzyme activity (B) and compared to brains from UT mice. Data are represented as mean ± SD (n = 7 to 10 mice per group). *p < 0.05. (C) Western blot analysis assessing changes in SOD1 protein levels in mouse brain 24 hr after siRNA administration. **p < 0.001.
Suppressing Neuroinflammation by scL-Delivered Anti-TNF-α siRNA
We have further investigated the therapeutic applicability of scL to a disease model using LPS-triggered neuroinflammation in mice. In this model, tumor necrosis factor alpha (TNF-α) plays a critical role in mediating inflammation and neuronal apoptosis, and so the silencing of the TNF-α could be particularly beneficial in reducing the neuroinflammation that is triggered by LPS.
Initially, we tested whether TNF-α secretion could be suppressed using scL nanocomplex delivering anti-TNF-α siRNA (scL-siTNF). Raw 264.7 murine macrophage cells were transfected with scL-siTNF and stimulated with LPS 24 hr later to examine the effects of the transfected siRNA on the LPS response. Comparisons were made for TNF-α mRNA levels in the cell pellet as measured by RT-PCR (Figure 5A) and for secreted TNF-α level in the culture supernatant as quantitated by ELISA (Figure 5B). Both TNF-α mRNA and secreted TNF-α of cells pretreated with control siRNA encapsulated in scL increased 47-fold and 205-fold, respectively, after LPS treatment. When cells were pretreated with scL-siTNF, TNF-α mRNA and secreted protein after LPS treatment increased 24-fold and 96-fold, respectively. These values represent ∼48% and ∼53% reduction in TNF-α mRNA and TNF-α protein, respectively, in cells treated with the anti-TNF-α siRNA as a nanocomplex compared to an analogous nanocomplex carrying the control siRNA. These results support the feasibility of using scL-siTNF to suppress the production of the inflammatory cytokine.
Figure 5.

scL Suppresses TNF-α Production in Cell Culture and Delivers siRNA into Mouse Brain Cells In Vivo
(A and B) Raw 264.7 cells were transfected with siTNF or control siRNA complexed with scL followed by LPS treatment 24 hr after transfection. TNF-α mRNA levels (A) in cells and secreted TNF-α protein (B) in culture medium were assessed in triplicate 4 hr after LPS treatment. Data are represented as mean ± SD. *p < 0.001, **p < 0.05. (C) Quantitative analysis of the siTNF uptake in mouse brain cells using RT-PCR. Mice were intravenously injected with scL-siTNF nanocomplexes (180 μg of siRNA/mouse). At 24 hr after a single injection, the physical presence of siTNF was determined. Data are represented as mean ± SD (n = 2 to 3 mice per group). *p < 0.001.
Based on the promising results in cell culture, we proceeded to test whether scL could deliver siTNF to mouse brain cells in vivo and to assess the therapeutic benefit of this agent. BALB/c mice received a single intravenous injection with free siTNF or siTNF complexed with either scL or Lip (lacking a targeting moiety). Twenty-four hours after the siRNA injections, brain cells were isolated and magnetically fractionated into astrocyte, microglia, and neuron using microbeads. The physical presence of siRNA in each brain cell fraction was assessed by TaqMan small RNA assays (Figure 5C) with U6 small nuclear RNA (snRNA) used as an endogenous control. The highest level of TNF-α siRNA that accumulated in brain cells was in mice treated with the siRNA delivered via scL-siTNF. Among the major brain cell types, neurons showed the highest uptake of siTNF. A much lower accumulation of TNF-α siRNA was detected in the brain of Lip-siTNF-treated mice. These results confirm the delivery of the payload to the brain cells by the TfR-targeting scL nanocomplex.
We had hypothesized that scL-delivered siTNF would be able to suppress TNF-α production in vivo and by doing so would impede the apoptosis and neuroinflammation triggered by LPS. To test this hypothesis, BALB/c mice were given intravenous injections of either siTNF or siCTRL encapsulated in either the TfR-targeting scL-based nanocomplex or in an analogous liposomal vector lacking a targeting moiety. After three injections at 24 hr intervals, mice were challenged with LPS 24 hr after the last injection (Figure 6A). To assess the down-modulation of the gene targeted by siTNF, the levels of TNF-α mRNA were measured in the brain (Figure 6B). One hour after LPS challenge, TNF-α mRNA level in brains of control siRNA encapsulated in scL-treated mice increased approximately 25-fold compared to those of brains from PBS-treated mice (no LPS). In contrast, with scL-siTNF treatment, the TNF-α mRNA levels in brains were reduced by nearly 57% compared to those in brains of mice treated with control siRNA encapsulated in scL. About a 30% reduction was observed in brains of mice treated with Lip-siTNF. Suppression of TNF-α production was further assessed on protein level in the brain by ELISA (Figure 6C). One hour after LPS challenge, TNF-α levels in the brain increased about 7-fold in mice given control siRNA encapsulated in scL compared to those of PBS-treated mice. Treatment with scL-siTNF suppressed TNF-α production by ∼35% in the brain compared to control siRNA encapsulated in scL treatment whereas only an ∼10% decrease of brain TNF-α was seen in the Lip-siTNF treatment group. Twenty-four hours after the LPS challenge, the survival of animals was assessed (Figure 6D). Under the current experimental conditions, 90% of the mice died from the lethal effects of LPS when the mice had been pretreated with the control siRNA in the form of control siRNA encapsulated in scL. In contrast, 90% of the animals survived when they were pretreated with scL-siTNF. Without targeted delivery, Lip-siTNF was found to be partially protective with 50% survival rate. However, this protective effect with Lip-siTNF might be due to the silencing TNF-α in peripheral macrophages rather than neuronal, considering that Lip-siTNF failed to deliver siRNA to brain cells in Figure 5C. The increased survival seen in mice pretreated with scL-siTNF clearly demonstrates the benefit of blocking TNF-α, a key mediator of inflammation, in this model of acute neuroinflammation.
Figure 6.

scL-Delivered TNF-α siRNA Protects Mice in Lethal Endotoxemia In Vivo
(A) Mice were intravenously injected with scL-siTNF nanocomplexes and challenged with LPS as indicated. PBS was injected as a control. (B and C) At 1 hr after LPS injection, the changes in the level of TNF-α mRNA (B) and TNF-α protein (C) in mouse brains were assessed by RT-PCR and ELISA. Data are represented as mean ± SD (n = 5 to 10 mice per group). *p < 0.001, **p < 0.05. (D) Survival was assessed at 24 hr after intraperitoneal injection of LPS (n = 10 mice per group). (E) Representative images of TUNEL staining of brain 24 hr after LPS injection (20× magnification). Scale bar, 200 μm.
We assumed that the survival benefit seen with scL-siTNF was due to impeding the neuronal apoptosis that accompanies LPS-triggered neuroinflammation. To examine directly the effects of TNF-α silencing on LPS-induced neuronal apoptosis, brain sections obtained from the surviving mice were assessed utilizing a TUNEL stain (Figure 6E). Brains of the mice treated with either control siRNA encapsulated in scL or Lip-siTNF showed extensive TUNEL-positive staining (brown staining of nuclei) in the hippocampus, cortex, and cerebellum areas. In contrast, brains of mice treated with scL-siTNF had very few TUNEL-positive apoptotic cells resembling brains of PBS-treated mice without LPS treatment where the brain sections appeared more blue than brown. Collectively, our results support the contention that scL nanocomplex-mediated siRNA delivery can suppress TNF-α production and reduce the lethal effects of LPS in a mouse model of neuroinflammation.
Discussion
It is clear that the human brain requires iron for numerous metabolic processes and acquires iron from the blood via TfR-mediated transcytosis of diferric Tf. Taking advantage of this Tf/TfR system, attempts to transport therapeutics across the BBB have been made (e.g., bispecific antibodies).25, 26 Although the utilization of TfR to cross the BBB has been investigated in rodents for decades, success in translating these findings to humans remains elusive.27 The issue causing some such therapeutics to fail is connected to the affinity of the antibodies being used, the pH dependence of their dissociation from the TfR, and how differences between antibodies influence the fate of both the antibody conjugate and the TfR to which it binds.27 In an in vitro model for the human BBB based on an immortalized human cerebral microvascular endothelial cell line, it has been shown that an anti-TfR antibody with appropriate affinity and pH dependence crosses this in vitro BBB model via TfR-mediated transcytosis.25 Similar considerations also come into play when using gold nanoparticles coated with the ligand Tf,28 where the density of Tf on the gold nanoparticles determines the avidity with which the particles are bound to the TfR of the BBB endothelial cells and the penetration efficiency across the BBB.
In the present study, we have utilized a novel non-viral vector for systemic delivery of therapeutic nucleic acids to the brain and shown the practical utility of this approach by confirming down-modulation of targeted genes and by preventing LPS-induced neuronal apoptosis in the brains of mice. Our data strongly support the feasibility of using TfR-targeting scL-based nanocomplexes for delivery of therapeutic nucleic acid payloads across the BBB and into neurons. As demonstrated by in vivo fluorescence imaging and quantitative PCR detecting of the payloads in the brain, the scL nanocomplex, but not untargeted nanocomplex, successfully delivered payloads across the BBB. These data indicate that specific TfR binding is responsible for and critical to the observed brain-targeting capability of scL. In scL, TfRscFv facilitates the complex to undergo RMT, a pathway normally used to increase uptake of macromolecules (e.g., Tf) into the brain.20 Our TfRscFv, which is derived from a monoclonal antibody to the human TfR, has the cross-reactivity with mouse TfRs, but not with rat TfRs. We were able to test brain delivery in mice using scL nanocomplex, whereas we used rat Tf as a targeting moiety in TfL nanocomplex for brain-targeting study in rats. The fact that either anti-TfR scFv or the TfR’s ligand could serve as the targeting moiety on our nanocomplexes attests to the crossing of the BBB being mediated by TfR.
TNF-α plays a critical role in the initiation and maintenance of neuroinflammation that can lead to brain tissue destruction via apoptosis and death. Elevated levels of TNF-α are observed in a number of chronic neurological disorders, including multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, and traumatic brain injuries.22, 29, 30 In addition, the demonstrated role of the TNF-α pathway in several animal models of neuronal diseases supports an important role for TNF-α in these diseases.22 Thus, targeting TNF-α in brain offers an attractive therapeutic strategy that might slow the progression or attenuate the severity of neuronal diseases.29 Here, we have demonstrated that TNF-α siRNA complexed with scL effectively down-modulated TNF-α in vivo, blocked neuronal apoptosis in the brain, and rescued mice from the lethal effects of LPS. In this animal model, it is difficult to distinguish the effects of silencing TNF-α in the brain and peripheral organs because of the crosstalk between peripheral macrophages and microglia in the CNS. However, the presence of siTNF in the brain cells (including neurons and microglia) and the contrasts in the levels of brain apoptosis between mice treated with brain-targeting scL-siTNF nanocomplex and non-targeting Lip-siTNF nanocomplex clearly suggests the beneficial effects of the targeted delivery of siTNF to the brain. These results confirm in vivo the potential of scL-mediated systemic delivery of therapeutic nucleic acids as a novel therapeutic approach in the treatment of neurological diseases.
Whereas naked siRNA and other therapeutic nucleic acids are rapidly destroyed by serum nucleases, use of an appropriate delivery system makes possible enhanced biological stability, targeted cell uptake, and the improved pharmacokinetics. We have previously shown that the encapsulation of either antisense ODN or siRNA forming scL-based nanocomplexes greatly increased its serum stability and bioavailability in mice.31 The safety of scL-based nanocomplex has been established in the completed phase I clinical trial using scL delivering the tumor suppressor gene wtp53 (in an experimental anti-cancer agent termed SGT-53)32 and in a second phase I trial using nanocomplexes carrying the gene encoding RB94 (in a second experimental anti-cancer agent termed SGT-94).33 Thus, this method of combining liposomal encapsulation of payload with targeting the TfR may provide a safe and useful method to enhance CNS drug delivery to achieve effective gene modulation in treating acute as well as chronic neurological disorders that require longer-term interventions. In these diseases, therapies that do not jeopardize the brain by compromising the BBB would have a clear advantage.
In addition to mediating the traversing of the BBB, the TfRscFv also facilitates scL to target tumor cells via receptor-mediated endocytosis because of the elevated expression of TfR on their surface.34 In our previous studies, we have shown that human glioblastoma cells take up our nanocomplexes both in culture34 and as orthotopic xenografts in mice.35 We have demonstrated that the scL actively crosses the BBB, delivers payloads to brain tumors, and results in robust expression of payload (exogenous tumor suppressor gene wtp53) in brain tumors to chemosensitize a drug-resistant malignant glioblastoma multiforme (GBM).35, 36 This nanocomplex (SGT-53) in combination with the chemotherapeutic agent, temozolomide, is currently being tested in phase II clinical trials for a recurrent GBM (ClinicalTrials.gov Identifier NCT02340156), a disease having a grim prognosis with existing therapeutic modalities.
Because there is a differential vulnerability of neuronal subpopulations in neurodegenerative diseases,37 incorporations of additional neurotropic targeting moieties may enable the selective recognition and delivery to the susceptible subpopulation of brain cells without interfering with relatively healthy neurons. Further studies to localize the payload and to modulate gene expression in different cell types in the brain are warranted. Although further investigations are also needed to elucidate the mechanism, the payload is clearly released from the complex inside the cells in that scL-delivered plasmid DNA or siRNA are functional in vivo in gene expression or silencing, respectively. Our previous observations suggest that 6FAM-ODN moves from the endosomes to the cytoplasm and then to the nucleus with increasing times of uptake by brain tumor cells in mice after systemic administration of scL-6FAM-ODN.34, 35 The results shown here with a payload consisting of a gene encoding GFP confirm movement out of the endosomes into the nucleus (for transcription) and cytoplasmic translation of GFP mRNA.
In summary, we have shown that scL nanocomplex provides a tool for targeted delivery of therapeutic nucleic acids to the brain in vivo. These findings indicate that therapeutics can be formulated as nanocomplexes so as to circumvent a critical barrier of CNS delivery, i.e., the BBB. The ability to actively cross the intact BBB via RMT markedly enhances the prospect of nucleic acid-based therapeutics for the treatment of neurological disorders. In principle, scL-mediated delivery can also provide a method for delivery of other therapeutic and diagnostic molecules such as chemotherapeutic drugs or imaging contrast agents across the intact BBB.
Materials and Methods
Complex Preparation
A cationic liposome comprising 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) and dioleolylphosphatidyl ethanolamine (DOPE) at a 1:1 molar ratio (Avanti Polar Lipids, Alabaster, AL), referred to as Lip in this study, was prepared using the ethanol injection method as described previously.35 In brief, both lipids in ethanol were injected quickly into 55°C water in a test tube while vortexing. The test tube was vortexed for an additional 20 min while cooling to room temperature.38 scL was prepared by mixing the Lip with a TfRscFv solution and incubated at room temperature for 10 min. Nucleic acid payloads (ODN, DNA, or siRNA) were then added to the TfRscFv-Lip solution, mixed, and incubated at room temperature for 10 min. The ratio of TfRscFv to Lip to payload can be varied, resulting in complexes with different transfection efficiencies.39 For in vitro experiments, the complex was further diluted with serum-free DMEM (Mediatech, Manassas, VA). For animal injections, dextrose (Hospira, Lake Forest, IL) was added to a final concentration of 5%. The size and zeta potential of the complex was determined by dynamic light scattering at 25°C with a Zetasizer Nano ZS System (Malvern Instruments, Malvern, UK).
ODNs and siRNAs
Scrambled ODNs (5′-CTAGCCATGCTTGTC-3′) that were labeled on the 5′ end with either 6-carboxyfluorescein phosphoramidite (6FAM-ODN) or cyanine 5 (Cy5-ODN) were synthesized and purified by reverse phase high-performance liquid chromatography (HPLC) (TriLink Biotechnologies, San Diego, CA). Pre-designed Silencer Select siRNA targeting mouse SOD1 and Silencer negative control #1 siRNA were obtained from Ambion (Austin, TX). A siRNA-targeting mouse TNF-α (5′-GACAACCAACUAGUGGUGC-3′) was synthesized at GE Dharmacon (Lafayette, CO).
In Vivo ODN Delivery and Brain Targeting
All animal experiments had been approved by the Georgetown University institutional review board. Athymic nude mice (6 weeks old, female, Harlan, Indianapolis, IN) received a single intravenous injection with 6FAM-ODN complexed with either scL (scL-6FAM-ODN), Lip (Lip-6FAM-ODN), or as free 6FAM-ODN (all at 100 μg of 6FAM-ODN/injection). Twenty-four hours after the injection, brain sagittal slices were imaged with Maestro in vivo fluorescence imaging system (CRi, Woburn, MA). The images were analyzed, and the fluorescence intensity quantified using the vendor’s software (Maestro 2.10.0). Fluorescence intensity is expressed as photons/cm2/second. In another study, BALB/c mice (6 weeks old, female, Harlan) were injected with scL-Cy5-ODN (25 μg of Cy5-ODN/injection) via the tail vein. Every 6 hr post-injection up to 48 hr, brains were harvested and cut into coronal sections ∼2 mm thick using an adult mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA). Brain sections were imaged and analyzed as described above.
In Vivo GFP Expression
BALB/c mice (6 weeks old, female) were intravenously injected with 30 μg of GFP plasmid DNA/injection encapsulated in scL (scL-GFP), Lip (Lip-GFP), or as free GFP plasmid. Forty-eight hours after a single injection, brain sagittal slices were imaged with Maestro and analyzed as described above. In another study, BALB/c mice were intravenously injected with scL-GFP (at 30 μg of GFP plasmid DNA/injection/mouse) every 12 hr for a total of three injections. Serial coronal section of brains was imaged with Maestro at 24, 36, and 48 hr after the last injection. Forty-eight hours after the last injection, brain slices were imaged by Olympus Fluoview-FV300 laser scanning confocal system (Olympus, Center Valley, PA). In another study, anesthetized Sprague-Dawley rats (5 weeks old, female, Harlan) were intravenously injected with a single bolus (1 mL) of either TfL (using Tf instead of TfRscFv as a targeting moiety) or Lip encapsulating GFP cDNA via a catheterized jugular vein (100 μg cDNA/injection). Thirty-six hours later, animals were sacrificed, and brain coronal sections were stained with mouse anti-parvalbumin antibody (Millipore, Billerica, MA) and by Nissl staining (Molecular Probes, Eugene, OR).
In Vivo siSOD Delivery and Gene Silencing
BALB/c mice (6 weeks old, female) were given a single intravenous injection of scL-siRNA complexes (180 μg of siRNA/injection). Twenty-four hours later, brains were harvested and total RNA isolated using RNeasy Plus mini kit (QIAGEN, Valencia, CA). Total RNA was reverse transcribed with Superscript VILO cDNA synthesis kit (Invitrogen, Carlsbad, CA). RT-PCR was performed using TaqMan gene expression assays for mouse SOD1 (Life Technologies, Carlsbad, CA) with a StepOnePlus RT-PCR system (Applied Biosystems, Foster City, CA). Relative SOD1 mRNA expression was normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and calculated using the ΔΔCt method. The level of SOD1 enzyme activity in the brain was measured with the SOD Assay Kit (Cayman Chemical, Ann Arbor, MI). The enzyme activity is denoted as units per milligram of total protein in brain tissue.
Western Blotting
To determine the expression level of the SOD1, western blot analysis was performed. Protein was isolated from mouse brain, and 40 μg of total cellular protein was separated on an 8% SDS-polyacrylamide gel, transferred to nylon membrane, and hybridized with antibodies against mouse SOD1 (Santa Cruz Biotechnology, Dallas, TX). An antibody recognizing GAPDH (Trevigen, Gaithersburg, MD) was utilized as an internal control for protein loading. Chemiluminescent detection was carried out using Supersignal west dura substrate (Pierce, Rockford, lL). Quantification of protein bands was carried out using ImageJ software (http://rsb.info.nih.gov/ij/).
TNF-α siRNA Transfection and Gene Silencing In Vitro
Silencing of TNF-α expression was monitored in vitro using Raw 264.7 murine macrophage cell line (ATCC, Manassas, VA). Cells were maintained at 37°C in a 5% CO2 atmosphere in DMEM supplemented with 10% fetal bovine serum (Sigma, St Louis, MO). Cells were plated in 6-well plates at 2.0 × 105 cells per well for 24 hr before transfection. Either control siRNA or TNF-α siRNA encapsulated in scL nanocomplex, prepared as above in serum-free media, was added to the wells at a final siRNA concentration of 100 nM. After incubation for 4 hr at 37°C, the medium was replaced with 2 mL of fresh complete medium, and the cells were further incubated for 24 hr before treated with 1 μg/mL of LPS from E. coli 0111:B4 (Sigma). Four hours after LPS treatment, cell pellet and culture media were collected.
Detection of TNF-α siRNA in Mouse Brain Cells
BALB/c mice (6 weeks old, female) were given a single intravenous injection of scL-siRNA complexes (180 μg siRNA/injection). TNF-α siRNA were encapsulated in scL nanocomplex or in Lip without a targeting moiety. Twenty-four hours after injection, brain cells were isolated using Neural Tissue Dissociation kit (Miltenyi Biotec, Auburn, CA) and Adult Brain Dissociation kit (Miltenyi Biotec). Isolated brain cells were magnetically sorted for astrocyte using anti-ACSA-2 microbead kit (Miltenyi Biotec), for microglia using CD11b microbeads (Miltenyi Biotec), and for neuron using Neuron Isolation kit (Miltenyi Biotec). Small RNAs were extracted using mirVana miRNA isolation kit (Ambion). Extracted small RNA was reverse transcribed with TaqMan microRNA reverse transcription kit (Life Technologies) using siTNF- and U6 snRNA-specific RT primers (Life Technologies). cDNA samples were subjected to RT-PCR using TaqMan small RNA assays for siTNF and U6 snRNA (Life Technologies) with a StepOnePlus RT-PCR system. Relative small RNA expression was normalized with U6 snRNA.
In Vivo TNF-α siRNA Delivery
BALB/c mice (7∼8 weeks old, female) were given daily injections for 3 consecutive days (total three injections) of scL-siRNA complexes (180 μg siRNA/injection, intravenous) as shown in Figure 6A. Either control siRNA or TNF-α siRNA was encapsulated in scL nanocomplex. siTNF was also encapsulated in Lip without a targeting moiety. Twenty-four hours after the last siRNA injection, mice were intraperitoneally injected with E. coli 0111:B4 LPS (30 μg/mouse), and organs were harvested 1 hr and 24 hr later. Animal survival was assessed 24 hr after LPS treatment.
TaqMan Analysis of TNF-α
Total RNAs were extracted from brains using PureLink RNA mini kit (Ambion). Extracted total RNA was reverse transcribed with Superscript VILO cDNA synthesis kit, and RT-PCR was performed using TaqMan gene expression assay for mouse TNF-α (Life Technologies) with a StepOnePlus RT-PCR system. Relative mRNA expression was normalized with GAPDH mRNA and calculated using the ΔΔCt method.
Measurement of TNF-α Production
Tissue samples were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher, Waltham, MA), and supernatants were collected by centrifugation at 10,000 rpm for 10 min at 4°C. Cell culture supernatants or serum samples were also collected. Each sample was assayed by TNF-α ELISA kit (eBioscience, San Diego, CA) according to the manufacturer’s protocol. In brief, samples were added to a 96-well plate pre-coated with capture antibodies specific for mouse TNF-α. Enzyme-linked detection antibodies were added to the wells. The intensity of color detected at 450 nm (correction wavelength 570 nm) was measured after addition of a substrate solution.
TUNEL Staining
Formalin-fixed mouse brains were paraffin embedded and sectioned at 5 μm. TUNEL staining was performed using the ApopTag peroxidase in situ apoptosis detection kit (Millipore) with minor modifications. In brief, sections were de-paraffinized with xylenes and rehydrated through a graded alcohol series. Heat induced epitope retrieval (HIER) was performed by immersing the tissue sections at 98°C for 20 min in 10 mM citrate buffer (pH 6.0) with 0.05% Tween. Slides were pretreated with 3% hydrogen peroxide at room temperature, 10 mM sodium citrate at 65°C, and equilibration buffer at room temperature. Slides were exposed to terminal transferase and digoxigenin labeled deoxyuridine triphosphate (dUTP) in reaction buffer for 2 hr at 37°C, stopped in wash buffer, and blocked with 10% normal goat serum at room temperature. Slides were exposed to HRP-conjugated anti-digoxigenin antibody (Roche) and DAB chromagen (Dako). Slides were counterstained with hematoxylin (Fisher, Harris Modified Hematoxylin) at a 1:17 dilution for 2 min, blued in 1% ammonium hydroxide for 1 min, dehydrated, and mounted with Acrymount. Images were captured using Olympus DP70 camera on Olympus BX61 microscope at 20× magnification.
Statistical Analysis
Results were presented as the mean ± standard deviation. All statistical significance was determined by the analysis of variance (ANOVA) (SigmaPlot, Systat Software, Chicago, IL). p values of < 0.05 were considered significant.
Author Contributions
S.-S.K., K.F.P., J.B.H., and E.H.C. directed the research, designed the experiments, and composed the manuscript. S.-S.K., A.R., and E.R.G.-S. performed the experiments. S.-S.K., A.R., and E.H.C. analyzed and interpreted the data.
Conflicts of Interest
E.H.C. and K.F.P. are two of the inventors of the described technology, for which several patents owned by Georgetown University have been issued. The patents have been licensed to SynerGene Therapeutics for commercial development. E.H.C. owns an equity interest in SynerGene Therapeutics and serves as a non-paid scientific consultant to SynerGene Therapeutics. S.-S.K. is salaried employee of SynerGene Therapeutics and owns stock in same. J.B.H. serves as salaried President & CEO of SynerGene Therapeutics and owns stock in same.
Acknowledgments
This study was supported in part by NCI grant 5R01CA132012-02 (E.H.C.) and a grant from SynerGene Therapeutics Inc. (K.F.P.). This study was conducted in part at the Lombardi Comprehensive Cancer Center Preclinical Imaging Research Laboratory, Microscopy & Imaging Shared Resource, Genomics & Epigenomics Shared Resource, Tissue Culture Shared Resource, Animal Core Facilities, and Histopathology & Tissue Shared Resource, which are supported by NCI grant P30-CA051008 and US Public Health Service grant 1S10RR15768-01. This study was performed in part in a facility constructed with support from Research Facilities Improvement grant C06RR14567 from the National Center for Research Resources, NIH.
References
- 1.Ali I.U., Chen X. Penetrating the blood-brain barrier: promise of novel nanoplatforms and delivery vehicles. ACS Nano. 2015;9:9470–9474. doi: 10.1021/acsnano.5b05341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldsmith M., Abramovitz L., Peer D. Precision nanomedicine in neurodegenerative diseases. ACS Nano. 2014;8:1958–1965. doi: 10.1021/nn501292z. [DOI] [PubMed] [Google Scholar]
- 3.Pardridge W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012;32:1959–1972. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones A.R., Shusta E.V. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm. Res. 2007;24:1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang M., Hu M., Song Q., Song H., Huang J., Gu X., Wang X., Chen J., Kang T., Feng X. GM1-modified lipoprotein-like nanoparticle: multifunctional nanoplatform for the combination therapy of Alzheimer’s disease. ACS Nano. 2015;9:10801–10816. doi: 10.1021/acsnano.5b03124. [DOI] [PubMed] [Google Scholar]
- 6.Xiao C., Davis F.J., Chauhan B.C., Viola K.L., Lacor P.N., Velasco P.T., Klein W.L., Chauhan N.B. Brain transit and ameliorative effects of intranasally delivered anti-amyloid-β oligomer antibody in 5XFAD mice. J. Alzheimers Dis. 2013;35:777–788. doi: 10.3233/JAD-122419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDannold N., Arvanitis C.D., Vykhodtseva N., Livingstone M.S. Temporary disruption of the blood-brain barrier by use of ultrasound and microbubbles: safety and efficacy evaluation in rhesus macaques. Cancer Res. 2012;72:3652–3663. doi: 10.1158/0008-5472.CAN-12-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allard E., Passirani C., Benoit J.P. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30:2302–2318. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 9.Miyagami M., Tsubokawa T., Tazoe M., Kagawa Y. Intra-arterial ACNU chemotherapy employing 20% mannitol osmotic blood-brain barrier disruption for malignant brain tumors. Neurol. Med. Chir. (Tokyo) 1990;30:582–590. doi: 10.2176/nmc.30.582. [DOI] [PubMed] [Google Scholar]
- 10.Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001;47:65–81. doi: 10.1016/s0169-409x(00)00122-8. [DOI] [PubMed] [Google Scholar]
- 11.Abbott N.J., Rönnbäck L., Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 12.Chen X., Gawryluk J.W., Wagener J.F., Ghribi O., Geiger J.D. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer’s disease. J. Neuroinflammation. 2008;5:12. doi: 10.1186/1742-2094-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zlokovic B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Kumar P., Wu H., McBride J.L., Jung K.E., Kim M.H., Davidson B.L., Lee S.K., Shankar P., Manjunath N. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 15.Kwon E.J., Skalak M., Lo Bu R., Bhatia S.N. Neuron-targeted nanoparticle for siRNA delivery to traumatic brain injuries. ACS Nano. 2016;10:7926–7933. doi: 10.1021/acsnano.6b03858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian M., Jones M.F., Lal A. Long non-coding RNAs embedded in the Rb and p53 pathways. Cancers (Basel) 2013;5:1655–1675. doi: 10.3390/cancers5041655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S.S., Harford J.B., Pirollo K.F., Chang E.H. Effective treatment of glioblastoma requires crossing the blood-brain barrier and targeting tumors including cancer stem cells: the promise of nanomedicine. Biochem. Biophys. Res. Commun. 2015;468:485–489. doi: 10.1016/j.bbrc.2015.06.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendricks B.K., Cohen-Gadol A.A., Miller J.C. Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg. Focus. 2015;38:E10. doi: 10.3171/2015.1.FOCUS14767. [DOI] [PubMed] [Google Scholar]
- 19.Farrington G.K., Caram-Salas N., Haqqani A.S., Brunette E., Eldredge J., Pepinsky B., Antognetti G., Baumann E., Ding W., Garber E. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014;28:4764–4778. doi: 10.1096/fj.14-253369. [DOI] [PubMed] [Google Scholar]
- 20.Dennis M.S., Watts R.J. Transferrin antibodies into the brain. Neuropsychopharmacology. 2012;37:302–303. doi: 10.1038/npp.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friden P.M., Walus L.R., Musso G.F., Taylor M.A., Malfroy B., Starzyk R.M. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 1991;88:4771–4775. doi: 10.1073/pnas.88.11.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCoy M.K., Tansey M.G. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pirollo K.F., Chang E.H. Targeted delivery of small interfering RNA: approaching effective cancer therapies. Cancer Res. 2008;68:1247–1250. doi: 10.1158/0008-5472.CAN-07-5810. [DOI] [PubMed] [Google Scholar]
- 24.Xu L., Frederik P., Pirollo K.F., Tang W.H., Rait A., Xiang L.M., Huang W., Cruz I., Yin Y., Chang E.H. Self-assembly of a virus-mimicking nanostructure system for efficient tumor-targeted gene delivery. Hum. Gene Ther. 2002;13:469–481. doi: 10.1089/10430340252792594. [DOI] [PubMed] [Google Scholar]
- 25.Sade H., Baumgartner C., Hugenmatter A., Moessner E., Freskgård P.O., Niewoehner J. A human blood-brain barrier transcytosis assay reveals antibody transcytosis influenced by pH-dependent receptor binding. PLoS ONE. 2014;9:e96340. doi: 10.1371/journal.pone.0096340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanodia J.S., Gadkar K., Bumbaca D., Zhang Y., Tong R.K., Luk W., Hoyte K., Lu Y., Wildsmith K.R., Couch J.A. Prospective design of anti-transferrin receptor bispecific antibodies for optimal delivery into the human brain. CPT Pharmacometrics Syst. Pharmacol. 2016;5:283–291. doi: 10.1002/psp4.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Y.J., Atwal J.K., Zhang Y., Tong R.K., Wildsmith K.R., Tan C., Bien-Ly N., Hersom M., Maloney J.A., Meilandt W.J. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci. Transl. Med. 2014;6:261ra154. doi: 10.1126/scitranslmed.3009835. [DOI] [PubMed] [Google Scholar]
- 28.Wiley D.T., Webster P., Gale A., Davis M.E. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc. Natl. Acad. Sci. USA. 2013;110:8662–8668C667. doi: 10.1073/pnas.1307152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S.S., Ye C., Kumar P., Chiu I., Subramanya S., Wu H., Shankar P., Manjuath N. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol. Ther. 2010;18:993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baratz R., Tweedie D., Wang J.Y., Rubovitch V., Luo W., Hoffer B.J., Greig N.H., Pick C.G. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflammation. 2015;12:45. doi: 10.1186/s12974-015-0237-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rait A.S., Pirollo K.F., Xiang L., Ulick D., Chang E.H. Tumor-targeting, systemically delivered antisense HER-2 chemosensitizes human breast cancer xenografts irrespective of HER-2 levels. Mol. Med. 2002;8:475–486. [PMC free article] [PubMed] [Google Scholar]
- 32.Pirollo K.F., Nemunaitis J., Leung P.K., Nunan R., Adams J., Chang E.H. Safety and efficacy in advanced solid tumors of a targeted nanocomplex carrying the p53 gene used in combination with docetaxel: a phase 1b study. Mol. Ther. 2016;24:1697–1706. doi: 10.1038/mt.2016.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siefker-Radtke A., Zhang X.Q., Guo C.C., Shen Y., Pirollo K.F., Sabir S., Leung C., Leong-Wu C., Ling C.M., Chang E.H. A phase l study of a tumor-targeted systemic nanodelivery system, SGT-94, in genitourinary cancers. Mol. Ther. 2016;24:1484–1491. doi: 10.1038/mt.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S.S., Rait A., Rubab F., Rao A.K., Kiritsy M.C., Pirollo K.F., Wang S., Weiner L.M., Chang E.H. The clinical potential of targeted nanomedicine: delivering to cancer stem-like cells. Mol. Ther. 2014;22:278–291. doi: 10.1038/mt.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S.S., Rait A., Kim E., Pirollo K.F., Nishida M., Farkas N., Dagata J.A., Chang E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano. 2014;8:5494–5514. doi: 10.1021/nn5014484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S.S., Rait A., Kim E., Pirollo K.F., Chang E.H. A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine (Lond.) 2015;11:301–311. doi: 10.1016/j.nano.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han I., You Y., Kordower J.H., Brady S.T., Morfini G.A. Differential vulnerability of neurons in Huntington’s disease: the role of cell type-specific features. J. Neurochem. 2010;113:1073–1091. doi: 10.1111/j.1471-4159.2010.06672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L., Huang C.C., Huang W., Tang W.H., Rait A., Yin Y.Z., Cruz I., Xiang L.M., Pirollo K.F., Chang E.H. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol. Cancer Ther. 2002;1:337–346. [PubMed] [Google Scholar]
- 39.Yu W., Pirollo K.F., Yu B., Rait A., Xiang L., Huang W., Zhou Q., Ertem G., Chang E.H. Enhanced transfection efficiency of a systemically delivered tumor-targeting immunolipoplex by inclusion of a pH-sensitive histidylated oligolysine peptide. Nucleic Acids Res. 2004;32:e48. doi: 10.1093/nar/gnh049. [DOI] [PMC free article] [PubMed] [Google Scholar]