

Abstract
Objectives
Cognitive decline is common in Parkinson's disease (PD), but the underlying mechanisms for this complication are incompletely understood. Genotypes affecting dopamine transmission may be of importance. This study investigates whether genotypes associated with reduced prefrontal dopaminergic tone and/or reduced dopamine D2‐receptor availability (Catechol‐O‐methyltransferase [COMT] Val158Met genotype and DRD2 C957T genotype) affect the development of cognitive deficits in PD.
Materials and methods
One hundred and 34 patients with idiopathic PD, participating in a regional, population‐based study of incident parkinsonism, underwent genotyping. After extensive baseline investigations (including imaging and biomarker analyses), the patients were followed prospectively during 6‐10 years with neuropsychological evaluations, covering six cognitive domains. Cognitive decline (defined as the incidence of either Parkinson's disease mild cognitive impairment [PD‐MCI] or dementia [PDD], diagnosed according to published criteria and blinded to genotype) was studied as the primary outcome.
Results
Both genotypes affected cognition, as shown by Cox proportional hazards models. While the COMT 158Val/Val genotype conferred an increased risk of mild cognitive impairment in patients with normal cognition at baseline (hazard ratio: 2.13, P = .023), the DRD2 957T/T genotype conferred an overall increased risk of PD dementia (hazard ratio: 3.22, P < .001). The poorer cognitive performance in DRD2 957T/T carriers with PD occurred mainly in episodic memory and attention.
Conclusions
The results favor the hypothesis that dopamine deficiency in PD not only relate to mild cognitive deficits in frontostriatal functions, but also to a decline in memory and attention. This could indicate that dopamine deficiency impairs a wide network of brain areas.
Keywords: COMT, dementia, DRD2, mild cognitive impairment, neurodegeneration, Parkinson's disease, Parkinson's disease genetics, population‐based
1. INTRODUCTION
Cognitive decline affects the majority of patients with idiopathic Parkinson's disease (PD) and has a major impact on quality of life1 and morbidity,2 but the phenotype, pathophysiology, and genetics of the condition are incompletely understood.
Genetic factors are likely to contribute to cognitive dysfunction in PD, as reported in a small number of studies.3, 4, 5, 6 A functional Catechol‐O‐methyltransferase (COMT) gene polymorphism (Val158Met; rs4680) results in a COMT enzyme with reduced dopamine degradation activity. Human carriers of two COMT 158Met‐alleles (158Met homozygotes) show about 40% lower enzymatic activity in the brain compared to 158Val homozygotes,7 and higher prefrontal dopamine activation as measured by PET.8 The COMT 158Val‐allele correlates with lower scores in tests of working memory in healthy volunteers (showing complex relations to the same functions in PD), and with gray matter atrophy in dopamine‐innervated brain areas in patients with dementia disorders.9
Also affecting dopamine transmission, the C957T polymorphism of the Dopamine Receptor D2 (DRD2) gene (rs6277) affects messenger RNA stability,10 thereby influencing the level of expression of D2 receptors in the brain. The 957C/C genotype correlates with higher D2 receptor densities in extrastriatal, thalamic and neocortical areas,11 and better episodic memory in healthy elders.12 Hypofunction of extrastriatal dopamine receptor D2 subsystems, including in the insula, was recently found to contribute to cognitive decline in PD.13, 14
Variability in genes related to dopamine transmission can potentially clarify the role of dopamine deficiency in relation to cognitive decline in PD. Therefore, we investigated the above‐mentioned polymorphisms and cognitive functions in prospectively followed patients with PD. As a measurement of structural integrity, we included a sensitive CSF biomarker for neurodegeneration of myelinated axons (neurofilament light chain; NFL).15
2. MATERIALS AND METHODS
2.1. Participants
All patients with PD participated in a prospective, population‐based incidence study of unselected cases of new‐onset idiopathic parkinsonism, from a geographically defined area in Sweden. The study design has been described previously in detail.16 In brief, inclusion lasted from January 1, 2004, through April 30, 2009. Patients were followed by a movement disorder team at a University hospital, representing the only neurology clinic in the region. Patients with secondary parkinsonism (eg, drug induced parkinsonism) or dementia at baseline were excluded. Of 182 enrolled in the study, 144 patients of European ancestry were diagnosed as having PD, and of these, samples for COMT Val158Met and DRD2 C957T genotyping were collected from 134 (Figure 1). The patients with PD who did not provide samples for genotyping (n = 10) were older than the others (82.3 vs 70.4 years) and had lower Mini‐Mental State Examination (MMSE) scores, but were otherwise comparable. All patients were followed with standardized clinical examinations, including MMSE and motor assessments at least yearly for 6‐10 years, except 14 non‐demented patients that died and were followed shorter. A diagnosis of PD required two examiners (neurologists specialized in movement disorders) to agree that the clinical criteria for the diagnosis were fulfilled according to the UK Parkinson's Disease Society Brain Bank criteria (UK PDSBB).17 Having a family member with PD was not, however, treated as an exclusion criterium. All except three of the 134 patients with PD were examined with FP‐CIT SPECT (DAT‐scan), and all had pathological uptake. In three of the PD cases, the diagnosis was confirmed by autopsy. For comparison, COMT Val158Met and DRD2 C957T genotype and cognitive function in 30 adults, demographically similar, neurologically healthy controls with normal FP‐CIT uptake on SPECT, were also determined. All participants provided written informed consent. The study was approved by the Regional Medical Ethics Board in Umeå, Sweden, and was performed in accordance with the Declaration of Helsinki.
Figure 1.
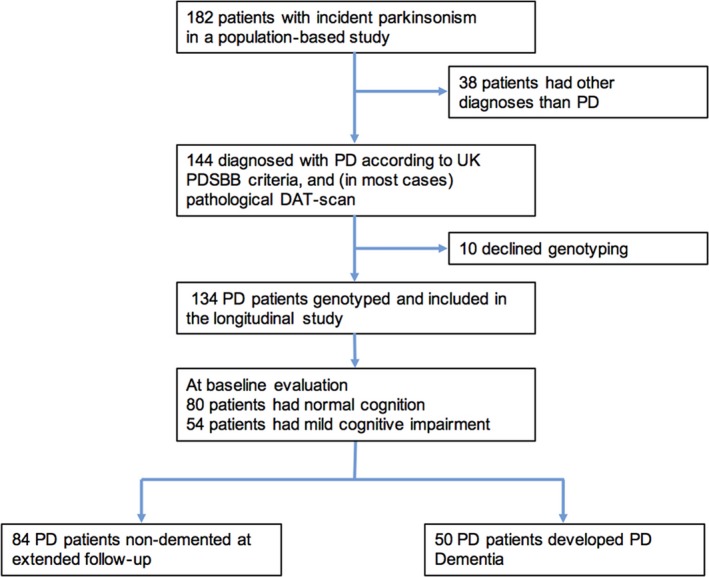
Flow of Parkinson's disease patients through the 10‐year study period. PD, Parkinson's disease; UK PDSBB, UK Parkinson's Disease Society Brain Bank
2.2. Genotyping and cerebrospinal fluid analysis
DNA was isolated from peripheral blood, using standard procedures. The polymorphisms of interest in the COMT and DRD2 genes, rs4680 and rs6277, were genotyped using TaqMan Assay‐by‐design (Applied Biosystems, Foster City, CA, USA). The assay was performed according to manufacturer's instructions, and genotypes were analyzed using the allelic discrimination function of the TaqMan 7900 HT Fast Real‐Time PCR system (Applied Biosystems, Foster City, CA, USA); 10% of the samples were run in duplicate, with 100% concordance. Genotype success rates of 98.3% (rs4680) and 100.0% (rs6277) were obtained. 103 of the patients with PD agreed to cerebrospinal fluid (CSF) collection by lumbar puncture at baseline, 1 year and/or 3 years. NFL concentration in CSF was measured with an ELISA method (NF‐light® ELISA kit, UmanDiagnostics AB, Umeå, Sweden) as described by the manufacturer by board‐certified laboratory technicians. The laboratory analyses were performed blinded from clinical data.
2.3. Cognitive impairment
Extensive neuropsychological testing, covering specific cognitive domains and used for PD‐mild cognitive impairment (MCI) and PD dementia (PDD) diagnostics, was performed at 0, 1, 3, 5, and 8 years.18 MCI was diagnosed according to Movement Disorder Society criteria.19 As the protocol included two tests of all cognitive domains but language, MCI was diagnosed on modified level 2 criteria, in keeping with previous research.20 See Table 1 for tests used for MCI classification, the partition of the tests and criteria for PDD diagnoses. Structural MRI and routine laboratory tests were performed in all patients to exclude non‐PDD causes of dementia. Diagnoses of PDD were determined by neurologists experienced in neurodegenerative disorders, blinded to the genotype of the patient. No patient with PD had the onset of dementia ≤1 year following motor onset (as in Lewy body dementia). All demented patients had PDD, according to published criteria.21
Table 1.
Tests used for Parkinson's disease cognitive impairment classification
| Neuropsychological function | Test |
|---|---|
| Episodic memory |
Free and Cued Selective Reminding Test (FCSRT)a
Logical memory and Paired associative learning from Wechsler memory scale (WMS)a Brief visuospatial memory test (BVMT) total recalla |
| Working memory |
Digit span forward, from Wechsler Adult Intelligence Scale (WAIS) IIIb
Digit span backwards, from Wechsler Adult Intelligence Scale (WAIS) IIIb |
| Attention | Trail Making Test (TMT) Ac and Bc |
| Verbal function |
Controlled Oral Word Association (COWA) Boston Naming Test (BNT) |
| Visuospatial function |
The Benton Judgement of Line Orientation testd
Pentagon copying from Mini‐Mental State Examination (MMSE) |
| Executive function |
Wisconsin card sorting test (WCST) – computer version 2e
Mental Control from Wechsler memory scale (WMS) Category fluency (animals in 60 seconds)e |
A domain score was calculated by the mean of standardized scores (Z‐scores) in the tests of episodic memorya, working memoryb, attentionc, visuospatial functiond, and executive functione.
Subjects were classified as having mild cognitive impairment (PD‐MCI) if: (i) impaired in a minimum of two tests in one domain (single domain MCI) or in a minimum of one test in two different domains (multiple domain MCI), (ii) impairments were ≥1.5 standard deviations below mean of normative data, (iii) self‐perceived cognitive decline was reported by Questionnaire and/or directly by patient and/or family member, and (iv) no functional impairment in basic activities of living.
Parkinson's disease dementia (PDD) diagnoses were based on neuropsychological test results, objective and subjective cognitive decline, and by the occurrence of functional impairment in basic activities of living (ie, driving a car, social or personal care, medication management) due to cognitive decline.
2.4. Statistical analyses
Baseline differences in demographic, cognitive, and CSF variables between different COMT and DRD2 genotype groups were tested by Kruskal‐Wallis tests, ANOVA, and Fisher's exact test, as appropriate. Based on previous research, a priori hypothesized risk genotypes (COMT 158Val/Val and DRD2 957T/T) were compared with other genotypes, using Kaplan‐Meier plots to investigate PD‐MCI and PDD incidence. To allow for correction for age, sex, disease duration, and cognitive status at baseline, hazard ratios were estimated by Cox proportional hazards regression models. Proportionality of hazards was evaluated by log‐log methods. To investigate potential domain‐specific effects, neuropsychological test scores were merged into six domains (episodic memory, working memory, attention, executive function, visuospatial function, and language), following the partition suggested by the Movement Disorder Society (see Table 1).19 The domain scores (measured by average standard deviations above or below the score mean at baseline; ie, Z‐score) in five domains, throughout the study period, and CSF NFL concentrations (tested at 0, 1, and 3 years), were compared in the genotype groups by linear mixed models, run with and without adjustment (see Table 4). The use of mixed‐effects models accounts for variability in length of follow‐up and is flexible with missing data. Although used in MCI diagnostics, language was excluded from mixed model analyses because of non‐normal distribution of scores. Differences in episodic memory (if found) were further analyzed in free‐ and cued recall conditions. P < .05 was considered significant. Holm‐Bonferroni correction for multiple comparisons was made in the Cox regression models and the mixed model analyses (see Tables 3 and 4). All statistical analyses were performed using a software program (SPSS 23.0; SPSS Inc).
Table 4.
Longitudinal cognitive function and cerebrospinal fluid data in 134 Parkinson's disease patients, in relation to COMT Val158Met‐ and DRD2 C957T genotype
| Cognitive domain | Mean difference, measured in SDs (95% CI) | P‐value |
|---|---|---|
| COMT 158Val/Val genotype vs. any Met allele | ||
| Episodic memory | −0.05 (−0.25‐0.15) | .598 |
| Working memory | −0.14 (−0.53‐0.25) | .482 |
| Attention | −0.18 (−0.20‐0.56) | .350 |
| Executive function | −0.19 (−0.51‐0.13) | .245 |
| Visuospatial function | −0.20 (−0.62‐0.22) | .343 |
| DRD2 957T/T genotype vs. any C allele | ||
| Episodic memory | −0.25 (−0.43 to −0.07) | .007a |
| Free recall | −0.42 (−0.78 to −0.07) | .021a |
| Cued recall | 0.36 (0.04‐0.67) | .026a |
| Working memory | −0.33 (−0.69‐0.01) | .060 |
| Attention | −0.58 (−0.91 to −0.26) | <.001a |
| Executive function | −0.31 (−0.61 to −0.02) | .034 |
| Visuospatial function | −0.38 (−0.75 to −0.01) | .044 |
| Cerebrospinal fluid NFL concentration | Difference, ng/L (95% CI) | P‐value |
|---|---|---|
| COMT 158Val/Val genotype vs. any Met allele | 3.8 (−433.0‐440.7) | .986 |
| DRD2 957T/T genotype vs. any C allele | 265.2 (−125.7‐656.1) | .181 |
Fixed effects estimates for differences at 0, 1, 3, 5, and 8 years (for NFL concentration at 0, 1, and 3 years), after correction for age, sex, disease duration, time of testing, and years of education.
Significant (P < 0.05) after Holm‐Bonferroni correction.
COMT, catechol‐O‐methyltransferase; DRD2, dopamine receptor D2; SD, standard deviation; NFL, neurofilament light chain protein.
Table 3.
Cognitive decline in 134 Parkinson's disease patients, in relation to COMT Val158Met‐ and DRD2 C957T genotype
| H.R. (95% CI) | P‐value | |
|---|---|---|
| Any cognitive decline (MCI or PDD) | ||
| ↑No. of COMT 158Val‐alleles | 1.51 (1.11‐2.06) | .009a |
| ↑No. of DRD2 957T‐alleles | 1.64 (1.17‐2.31) | .004a |
| COMT 158Val/Val vs. any Met allele | 1.79 (1.06‐3.03) | .030a |
| DRD2 957T/T vs. any C allele | 2.31 (1.41‐3.79) | <.001a |
| COMT 158Val/Val or DRD2 957T/T | 2.46 (1.54‐3.91) | <.001a |
| MCI development b | ||
| COMT 158Val/Val vs. any Met allele | 2.13 (1.11‐4.08) | .023a |
| DRD2 957T/T vs. any C allele | 2.12 (1.14‐3.94) | .018a |
| PDD development | ||
| COMT 158Val/Val vs. any Met allele | 1.11 (0.51‐2.45) | .791 |
| DRD2 957T/T vs. any C allele | 3.22 (1.64‐6.30) | <.001a |
Hazard Ratios (H.R.) are for risks within 6‐10 years, after correction for age, disease duration, sex, and baseline cognitive status (normal or MCI).
Significant (P < 0.05) after Holm‐Bonferroni correction.
Risk of PD‐MCI was analyzed in the 80 patients who had normal cognition at baseline, while the other outcomes were analyzed in all patients.
COMT, catechol‐O‐methyltransferase; DRD2, dopamine receptor D2; MCI, mild cognitive impairment; PDD, Parkinson's disease dementia; ↑, higher number (0, 1, or 2).
3. RESULTS
COMT Val158Met genotype was successfully determined in 132 of the patients with PD who donated samples and DRD2 C957T genotype in 134 (all) of the patients with PD who donated samples. The genotypes had relatively even distributions among the participants. At baseline, there were no major differences in clinical characteristics or NFL concentrations in the different COMT and DRD2 genotype groups (Table 2); 54 (40.3%) of the patients with PD had MCI at baseline. During follow‐up, 50 patients (37.3%) developed PDD, and 28 patients (20,9%) converted from PD‐normal to MCI. The genotyped patients had a median follow‐up of 7.0 years (interquartile range: 6.0‐9.0 years). There were no significant differences in medication dosages between the genotype groups during follow‐up, as measured by mean levodopa equivalent dose scores. In the healthy control group, 25 participants carried COMT 158Met‐alleles and 19 participants carried DRD2 957C‐alleles.
Table 2.
Baseline characteristics of 134 Parkinson's disease patients in relation to COMT Val158Met‐ and DRD2 C957T genotype
| Variable | COMT 158Met/Met genotype (n = 42) | COMT 158Val/Met genotype (n = 64) | COMT 158Val/Val genotype (n = 26) | P‐value |
|---|---|---|---|---|
| Age | 70.3 (9.0) | 69.3 (10.3) | 72.9 (9.1) | .273 |
| Disease duration in months, median (IQR) | 25.4 (12.6‐36.1) | 13.4 (8.1‐24.4) | 16.9 (10.0‐35.3) | .042 |
| Sex, M:F (% male) | 23:19 (54.8%) | 41:23 (64.1%) | 15:11 (57.7%) | .614 |
| UPDRS III, median (IQR) | 26 (13‐36) | 25 (18‐34) | 29 (24‐37) | .340 |
| MMSE | 28.6 (1.4) | 28.7 (1.4) | 28.7 (1.3) | .959 |
| CSF NFL concentration, ng/L, [number with baseline sample] | 1247 (760) [32] | 1499 (1601) [45] | 1508 (738) [18] | .638 |
| Variable | DRD2 957C/C genotype (n = 31) | DRD2 957T/C genotype (n = 66) | DRD2 957T/T genotype (n = 37) | P‐value |
|---|---|---|---|---|
| Age | 71.9 (9.7) | 69.1 (10.2) | 71.5 (9.3) | .286 |
| Disease duration in months, median (IQR) | 24.0 (10.2‐41.0) | 12.7 (8.0‐30.0) | 20.6 (12.2‐31.0) | .138 |
| Sex, M:F (% male) | 18:13 (58.1%) | 40:26 (60.6%) | 22:15 (59.5%) | .971 |
| UPDRS III, median (IQR) | 28 (21‐35) | 23 (16‐31) | 26 (19‐39) | .166 |
| MMSE | 28.5 (1.6) | 28.8 (1.3) | 28.4 (1.6) | .377 |
| CSF NFL concentration, ng/L, [number with baseline sample] | 1465 (762) [23] | 1269 (848) [50] | 1762 (1928) [27] | .247 |
Values are expressed as means (standard deviation) unless otherwise stated. Disease duration was defined by time from onset of first motor symptom to diagnosis of PD. COMT, catechol‐O‐methyltransferase; DRD2, dopamine receptor D2; PD, Parkinson's disease; IQR, interquartile range; UPDRS III, Unified Parkinson's Disease Rating Scale, part III (motor function); MMSE, Mini‐Mental State Examination; CSF, cerebrospinal fluid; NFL, neurofilament light chain protein.
3.1. COMT genotype in PD
As shown in Figure 2A, the COMT 158Val/Val genotype increased the risk of any cognitive decline in PD during follow‐up (decline from PD‐normal to PD‐MCI or from PD‐MCI to PDD, P = .006). However, as shown by Cox regression (Table 3) and in Figure 2B, the increased risk of cognitive decline was driven by a higher incidence of PD‐MCI among carriers of two COMT 158Val alleles (COMT 158Val/Val homozygotes). The COMT 158Val/Val homozygotes had 2.1 times higher hazard for developing PD‐MCI compared to PD patients with other COMT 158 genotypes, after correction for age, sex, disease duration, and baseline cognitive status (P = .023), but the COMT 158Val/Val genotype was not a risk factor for PDD. Analyses of survival times with no cognitive impairment showed that the increased risk of cognitive decline in COMT 158Val/Val carriers occurred between baseline and the fifth year of follow‐up. There was no measurable effect related to COMT genotype in the analysis of single cognitive domains (estimated by linear Mixed Models). The concentration of NFL in cerebrospinal fluid (CSF) did not significantly differ between the different COMT genotype groups. The COMT genotype had no effects on cognitive functions in the healthy controls (data not shown).
Figure 2.
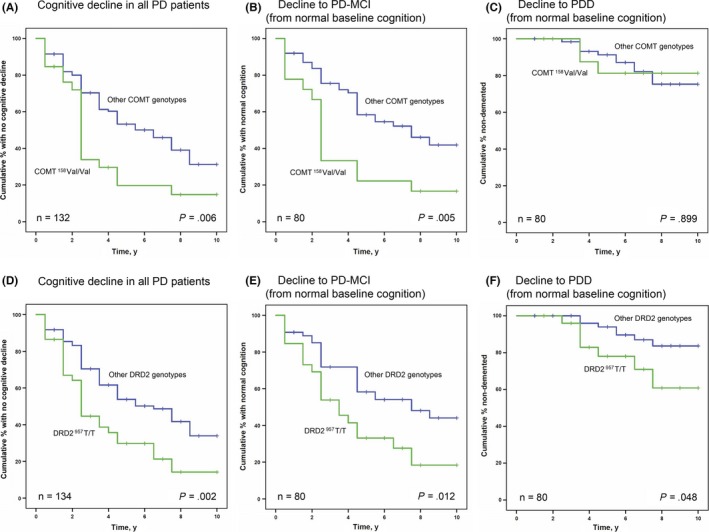
Effect of the COMT Val158Met and DRD2 C957T genotypes on cognitive decline in Parkinson's disease. Kaplan‐Meier plots showing the incidence of cognitive decline in relation to COMT (A–C) and DRD2 (D–F) genotype. The specific incidence of PD‐MCI and PDD (B, C, E, and F) is shown here only for the patients who had a normal cognitive function at baseline (n = 80). PD, Parkinson's disease; COMT, catechol‐O‐methyltransferase; DRD2, dopamine receptor D2; MCI, mild cognitive impairment; PDD, Parkinson's disease dementia
3.2. DRD2 genotype in PD
As shown in Figure 2D, the DRD2 957T/T genotype increased the risk of any cognitive decline in PD, measured by incidence of either PD‐MCI or PDD (P = .002). More specifically, carrying a DRD2 957T/T genotype increased the hazard for developing PDD 3.2 times, in comparison with PD patients with other DRD2 genotypes (P < .001; Table 3), after correction for age, sex, disease duration, and baseline cognitive status. The 957T/T genotype was also a risk factor for PD‐MCI development among patients with normal cognition at baseline (n = 80, P = .018, Table 3). Conversely, carriers of any DRD2 957C alleles had a lower risk of all types of cognitive decline (Figure 2FD‐; Table 3). Analysis of test scores in specific cognitive domains showed that carriers of the DRD2 957T/T genotype had significantly poorer performances in episodic memory (on average 0.25 standard deviations lower scores, P = .007) and in attention (on average 0.58 standard deviations lower scores, P < .001) after correction for confounding factors (Table 4). Patients with PD with a DRD2 957T/T genotype tended to perform slightly worse in all domains, but apart from the differences in episodic memory and attention, effects were non‐significant after correction for multiple comparisons. Detailed analysis of episodic memory showed opposite effects of DRD2 genotype in free versus cued recall. While PD patients with the DRD2 957T/T genotype had a significantly poorer performance in free recall (P = .021), they performed better in cued recall (P = .026). NFL concentrations in CSF did not differ between the DRD2 genotypes.
In the group of healthy control participants, DRD2 957T/T carriers performed poorer in tests of attention at an uncorrected statistical threshold (on average 0.47 standard deviations lower scores, P = .031, after correction for age, sex, time of testing, and years of education), similar to the performance in patients with PD. The difference was, however, non‐significant corrected for multiple comparisons.
4. DISCUSSION
By prospectively following a population‐representative cohort of patients with PD with neuropsychological evaluations for up to 10 years, we found that two common, functional polymorphisms in genes related to dopamine transmission (the COMT and DRD2 genes), affect the evolution of cognitive deficits, and alter the risks of MCI and PDD.
Biomarkers predictive of cognitive dysfunction in idiopathic PD are of value because of the serious impact of such complications. Furthermore, common variations in genes with known biological effects have been favored as a mean to test causality of disease mechanisms in observational studies.22 The present study extends earlier observations of a relationship between frontostriatally based cognitive functions and genes associated with dopamine transmission and is, to our knowledge, the first to demonstrate the DRD2 957T/T genotype as a risk factor for PDD. The observed effects were not explained by differences in demographic factors such as age or by differences in medication.
Converging evidence points to a multifactorial etiology of cognitive dysfunction in PD. Patients with early PD underrecruit an extensive frontostriatal brain network as a consequence of dopaminergic hypofunction,23 contributing to cognitive impairments. In later disease phases, accumulating cortical Lewy bodies, Alzheimer disease‐type brain pathology and hypofunction in non‐dopaminergic (ie, acetylcholinergic) transmitter systems contribute to PD dementia (PDD).24 According to the “dual syndrome hypothesis,” the early, dopamine dysfunction‐related impairments of frontostriatal, executive functions in PD are stable and do not strongly relate to dementia, while the more “posterior” deficits in visuospatial function or memory relate to cortical pathology that is predictive of PDD.25 The present findings potentially modify this hypothesis. In particular, the finding of DRD2 957T/T genotype as a risk factor for dementia (with the strongest detrimental effects seen in attention and episodic memory) suggests that dopamine hypofunction in PD may have effects beyond “frontostriatal” brain functioning including, possibly, alterations of distributed, dopamine‐dependent attention‐ and/or memory networks.
In healthy subjects, the high‐activity COMT 158Val‐allele has generally been found to impair executive functions such as working memory and cognitive flexibility.26, 27 In PD, studies by Williams‐Gray et al. showed that in later disease phases (>1.6 years after diagnosis), the Val‐allele correlated with impairments in executive functioning (as measured by the “Tower of London” test), similar to findings in controls, while in early disease (<1.6 years after diagnosis), the effect was opposite.4, 28 This was suggested to result from an inverted U‐shaped relation between cortical dopamine levels and prefrontal function, where a transient up‐regulation of prefrontal dopamine would cause “overdosing” in Met‐carriers in early PD, while the same patients would have an advantage in later disease phases, when progressive degeneration of dopaminergic systems produce more marked dopamine deficits. The present finding of mild cognitive impairments (MCI, as diagnosed according to the Movement Disorder Society) in COMT 158Val/Val carriers with PD could relate to the fact that the mean age at baseline was higher than in the CamPaIGN cohort, possibly making the studied population more vulnerable to a high prefrontal dopamine clearance. The invariably detrimental effect of the 158Val/Val genotype on cognition in the present study is also compatible with structural effects in the brain, for example an increased gray matter atrophy in dopamine‐innervated areas, such as reported in previous studies.9
In relation to DRD2 function, patients with PD often experience memory problems characterized by an inability to recall information. This may partly relate to dopamine deficiency. The hippocampus is strongly interconnected with dopaminergic neurons in the midbrain,29 and several lines of evidence have established the importance of D2 receptor function in attention and memory.30 Specifically, the DRD2 system is linked to transient memory updating processes.31, 32 A reduction in D2 receptors has been found in PD13 and in imaging studies, memory deficits in PD are correlated with reduced extrastriatal D2 receptor binding in the medial temporal lobe, including the insula, in parts of the so‐called salience network.14 The present findings of poorer performances in attention and episodic memory and an increased risk of dementia in DRD2 957T/T carriers with PD could, potentially, relate to reduced function or increased vulnerability in D2‐dependent “salience networks” of the brain (eg, in medial, temporal structures). In this respect, the opposite effect on episodic memory in free recall compared to cued recall, related to the DRD2 957T/T genotype, is intriguing. This might suggest that the typical memory impairment in PD (memory deficits that improve with external cues) is a hypodopaminergic trait, specifically related to reduced function in D2 receptor signaling. This would be consistent with the proposed role of D2 receptors in flexible memory updating, and the corresponding memory deficit could gradually become more detrimental in PD compared to effects induced by COMT enzyme heterogeneity (related to the COMT Val158Met polymorphism).
Interestingly, the DRD2 957C/C genotype (and the DRD2 gene locus) has previously been found to be associated to susceptibility to schizophrenia in younger populations.33, 34 The seemingly opposite effects of the DRD2 957C allele in different populations (increasing the risk of schizophrenia in young populations, while protecting against cognitive decline in PD) may be an example of genetic pleiotropism. The precise mechanisms behind the DRD2‐genotype‐induced effects on cognition were not determined by this study but merits further investigation.
While the effects on cognition were different for the COMT and DRD2 genotypes, none of the genotypes affected cerebrospinal fluid NFL concentration (which has been found predictive of PDD).35 This suggests that the COMT and DRD2 genotypes did not affect cognition through gross effects on neurodegeneration, at least not as measured by injury of myelinated axons. This is consistent with the trend of a similarly poorer performance in tests of attention in the healthy control persons with a DRD2 957T/T genotype, which may indicate that the DRD2 genotype‐related effect on attention is not specific to degenerating dopaminergic systems, but rather is an effect on functions common to both disease‐affected and normal brains. The effect may, however, be amplified in PD. It could also be of interest to investigate the studied genotypes in Lewy Body Dementia.
A limitation was the small number of healthy controls and patients and the theoretical classification of cognitive tests in cognitive domains, which is, however, not standardized in clinical research settings. Our study also has strengths, namely a population‐based and longitudinal design, evaluation of incident cases, and the use of an extensive neuropsychological test battery at several times during the follow‐up period. The risk of incorrect PD diagnosis was minimized by the long follow‐up periods, the finding of pathologic uptake on DAT‐scan examination in all (100%) of the examined patients with PD, and by confirmation of the diagnosis by autopsy in a small number of cases.
In conclusion, although confirmation in independent, larger studies is needed, our results suggest that the COMT 158Val/Val genotype (which is known to produce a decrease in synaptic dopamine availability) increases the risk of MCI in PD and that the DRD2 957T/T genotype is a risk factor for PDD. These findings provide new insights into the mechanisms of cognitive dysfunction in PD. In the clinical setting, the DRD2 957T/T genotype could possibly be used as a marker for PDD risk.
CONFLICT OF INTERESTS
David Bäckström, Magdalena Eriksson, Gabriel Granåsen, Sofia Mayans, Eva Elgh, Henrik Zetterberg, Kaj Blennow, Lars Forsgren – Reports no disclosures. Jan Linder reports receiving honoraria for lectures from GSK, Lundbeck, Boehringer Ingelheim, Abbott, AbbVie, Solvay, Orion Pharma, UCB, Nordic InfuCare, Medtronic, and IPSEN and serving on advisory boards for Boehringer Ingelheim, Lundbeck, and GSK.
ACKNOWLEDGEMENTS
Mona Edström, RN, and Jörgen Andersson, laboratory technician (Department of Pharmacology and Clinical Neuroscience, Umeå University), provided valuable assistance with data collection. The authors are grateful to all the patients and healthy control subjects for their participation.
Bäckström D, Eriksson Domellöf M, Granåsen G, et al. Polymorphisms in dopamine‐associated genes and cognitive decline in Parkinson's disease. Acta Neurol Scand. 2018;137:91–98. https://doi.org/10.1111/ane.12812
Funding information
This study was supported by grants from the Swedish Medical Research Council, Erling Persson Foundation, Parkinson Foundation in Sweden, Umeå University, Västerbotten County Council, King Gustaf V and Queen Victoria Freemason Foundation, Swedish Parkinson Foundation, Kempe Foundation, and Swedish Parkinson's Disease Association
REFERENCES
- 1. Reginold W, Duff‐Canning S, Meaney C, et al. Impact of mild cognitive impairment on health‐related quality of life in Parkinson's disease. Dement Geriatr Cogn Disord. 2013;36:67‐75. [DOI] [PubMed] [Google Scholar]
- 2. Fletcher P, Leake A, Marion MH. Patients with Parkinson's disease dementia stay in the hospital twice as long as those without dementia. Mov Disord. 2011;26:919. [DOI] [PubMed] [Google Scholar]
- 3. Kalinderi K, Bostantjopoulou S, Fidani L. The genetic background of Parkinson's disease: current progress and future prospects. Acta Neurol Scand. 2016;134:314‐326. [DOI] [PubMed] [Google Scholar]
- 4. Williams‐Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow‐up of the CamPaIGN cohort. Brain. 2009;132:2958‐2969. [DOI] [PubMed] [Google Scholar]
- 5. Winder‐Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain. 2013;136:392‐399. [DOI] [PubMed] [Google Scholar]
- 6. Ferreira M, Massano J. An updated review of Parkinson's disease genetics and clinicopathological correlations. Acta Neurol Scand. 2017;135:273‐284. [DOI] [PubMed] [Google Scholar]
- 7. Chen J, Lipska BK, Halim N, et al. Functional analysis of genetic variation in catechol‐O‐methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu K, O'Keeffe D, Politis M, et al. The catechol‐O‐methyltransferase Val(158)Met polymorphism modulates fronto‐cortical dopamine turnover in early Parkinson's disease: a PET study. Brain. 2012;135:2449‐2457. [DOI] [PubMed] [Google Scholar]
- 9. Gennatas ED, Cholfin JA, Zhou J, et al. COMT Val158Met genotype influences neurodegeneration within dopamine‐innervated brain structures. Neurology. 2012;78:1663‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duan J, Wainwright MS, Comeron JM, et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12:205‐216. [DOI] [PubMed] [Google Scholar]
- 11. Hirvonen MM, Lumme V, Hirvonen J, et al. C957T polymorphism of the human dopamine D2 receptor gene predicts extrastriatal dopamine receptor availability in vivo. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:630‐636. [DOI] [PubMed] [Google Scholar]
- 12. Li SC, Papenberg G, Nagel IE, et al. Aging magnifies the effects of dopamine transporter and D2 receptor genes on backward serial memory. Neurobiol Aging. 2013;34:358. e1‐10. [DOI] [PubMed] [Google Scholar]
- 13. Christopher L, Duff‐Canning S, Koshimori Y, et al. Salience network and parahippocampal dopamine dysfunction in memory‐impaired Parkinson disease. Ann Neurol. 2015;77:269‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christopher L, Marras C, Duff‐Canning S, et al. Combined insular and striatal dopamine dysfunction are associated with executive deficits in Parkinson's disease with mild cognitive impairment. Brain. 2014;137:565‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bacioglu M, Maia LF, Preische O, et al. Neurofilament light chain in blood and csf as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron. 2016;91:494‐496. [DOI] [PubMed] [Google Scholar]
- 16. Linder J, Stenlund H, Forsgren L. Incidence of Parkinson's disease and parkinsonism in northern Sweden: a population‐based study. Mov Disord. 2010;25:341‐348. [DOI] [PubMed] [Google Scholar]
- 17. Gibb WR, Lees AJ. The significance of the Lewy body in the diagnosis of idiopathic Parkinson's disease. Neuropathol Appl Neurobiol. 1989;15:27‐44. [DOI] [PubMed] [Google Scholar]
- 18. Elgh E, Domellöf M, Linder J, Edström M, Stenlund H, Forsgren L. Cognitive function in early Parkinson's disease: a population‐based study. Eur J Neurol. 2009;16:1278‐1284. [DOI] [PubMed] [Google Scholar]
- 19. Litvan I, Goldman JG, Tröster AI, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord. 2012;27:349‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yarnall AJ, Breen DP, Duncan GW, et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE‐PD study. Neurology. 2014;82:308‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22:1689‐1707; quiz 837. [DOI] [PubMed] [Google Scholar]
- 22. Ebrahim S. DAVEY SMITH G. Mendelian randomization: can genetic epidemiology help redress the failures of observational epidemiology? Hum Genet. 2008;123:15‐33. [DOI] [PubMed] [Google Scholar]
- 23. Ekman U, Eriksson J, Forsgren L, Mo SJ, Riklund K, Nyberg L. Functional brain activity and presynaptic dopamine uptake in patients with Parkinson's disease and mild cognitive impairment: a cross‐sectional study. Lancet Neurol. 2012;11:679‐687. [DOI] [PubMed] [Google Scholar]
- 24. Svenningsson P, Westman E, Ballard C, Aarsland D. Cognitive impairment in patients with Parkinson's disease: diagnosis, biomarkers, and treatment. Lancet Neurol. 2012;11:697‐707. [DOI] [PubMed] [Google Scholar]
- 25. Kehagia AA, Barker RA, Robbins TW. Cognitive impairment in Parkinson's disease: the dual syndrome hypothesis. Neurodegener Dis. 2013;11:79‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917‐6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dickinson D, Elvevåg B. Genes, cognition and brain through a COMT lens. Neuroscience. 2009;164:72‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williams‐Gray CH, Hampshire A, Barker RA, Owen AM. Attentional control in Parkinson's disease is dependent on COMT val 158 met genotype. Brain. 2008;131:397‐408. [DOI] [PubMed] [Google Scholar]
- 29. Lisman JE, Grace AA. The hippocampal‐VTA loop: controlling the entry of information into long‐term memory. Neuron. 2005;46:703‐713. [DOI] [PubMed] [Google Scholar]
- 30. Floresco SB, Magyar O. Mesocortical dopamine modulation of executive functions: beyond working memory. Psychopharmacology. 2006;188:567‐585. [DOI] [PubMed] [Google Scholar]
- 31. Bilder RM, Volavka J, Lachman HM, Grace AA. The catechol‐O‐methyltransferase polymorphism: relations to the tonic‐phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology. 2004;29:1943‐1961. [DOI] [PubMed] [Google Scholar]
- 32. Nyberg L, Andersson M, Forsgren L, et al. Striatal dopamine D2 binding is related to frontal BOLD response during updating of long‐term memory representations. NeuroImage. 2009;46:1194‐1199. [DOI] [PubMed] [Google Scholar]
- 33. Liu L, Fan D, Ding N, et al. The relationship between DRD2 gene polymorphisms (C957T and C939T) and schizophrenia: a meta‐analysis. Neurosci Lett. 2014;583:43‐48. [DOI] [PubMed] [Google Scholar]
- 34. Consortium SWGOTPG . Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014;511:421‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bäckström DC, Eriksson Domellöf M, Linder J, et al. Cerebrospinal fluid patterns and the risk of future dementia in early, incident Parkinson disease. JAMA Neurol. 2015;72:1175‐1182. [DOI] [PubMed] [Google Scholar]