

Abstract
Brexpiprazole is currently approved in the United States for the treatment of schizophrenia and as adjunctive treatment of major depressive disorder. In Canada, it is approved for the treatment of schizophrenia. This study evaluated the pharmacokinetics (PK) and safety of brexpiprazole in Japanese patients with schizophrenia. This phase 1 study comprised a 14‐day multiple‐dose administration of brexpiprazole 1, 4, and 6 mg/day (n = 7, 8, and 6, respectively). Plasma concentrations and PK parameters and the influence of CYP2D6 polymorphisms (intermediate metabolizers [IMs] and extensive metabolizers [EMs]) on PK were evaluated. Adverse events (AEs) were recorded. The Cmax and AUC24h of brexpiprazole and its metabolite, DM‐3411, showed dose‐proportionality. The Cmax and AUC24h of brexpiprazole showed accumulation of about 2.5‐ to 5.5‐fold on day 14, compared with those on day 1. The median tmax and the mean elimination half‐life of brexpiprazole were 4–5 and 52–92 hours, respectively, across all doses on day 14. The C24h of brexpiprazole reached steady state after day 10 in all dose groups. The dose‐normalized Cmax and AUC24h of brexpiprazole on day 14 were higher in IM patients than in EM patients. AEs were generally mild to moderate, with transient serum prolactin increase being the most common event. No clinically significant changes were observed for other clinical laboratory values. Brexpiprazole was safe and well tolerated in the studied Japanese patients with schizophrenia.
Keywords: brexpiprazole, phase 1, multiple dose, schizophrenia, pharmacokinetics
Antipsychotics have become the cornerstone in the treatment of patients diagnosed with a number of severe mental disorders, including schizophrenia, bipolar mania, and irritability associated with autistic spectrum disorder (ASD).1 Most currently available antipsychotics are functional dopamine D2 receptor antagonists.1 Aripiprazole was the first dopamine D2 partial agonist to be approved for the treatment of schizophrenia, bipolar mania, irritability associated with ASD, and Tourette's disorder and for adjunctive treatment in major depressive disorder (MDD).2 However, successful treatment outcomes are still challenged by the heterogeneity of therapeutic and adverse responses. Therefore, new therapeutic options with better efficacy and better tolerability are needed for patients with psychiatric disorders.
Brexpiprazole was approved in the United States for the treatment of schizophrenia and as adjunctive treatment of MDD in July 2015.3 In Canada, it is approved for the treatment of schizophrenia.4 Brexpiprazole is a serotonin‐dopamine activity modulator that acts as a partial agonist at the serotonin 5‐HT1A and dopamine D2 receptors and an antagonist at the serotonin 5‐HT2A and noradrenaline α1B/2C receptors, all at similar potencies.5 In the United States and Canada, the recommended starting dose for the treatment of schizophrenia is 1 mg/day, followed by up‐titration to the target dose range of 2 to 4 mg/day, and the maximum dose is 4 mg/day.3, 4 Cytochrome P450 (CYP) 2D6 and CYP3A4 are involved in the metabolism of brexpiprazole, and the major metabolite (DM‐3411) does not appear to contribute to the therapeutic effects of brexpiprazole.6 Brexpiprazole is not a substrate of efflux transporters (eg, P‐glycoprotein or breast cancer resistant protein [BCRP]) and shows little to no inhibition of CYP enzymes, according to in vitro studies.6 No brexpiprazole dosage adjustment is required with concomitant use of CYP2B6 inhibitors, gastric pH modifiers, CYP2D6, CYP3A4, or CYP2B6 substrates, or BCRP or P‐glycoprotein substrates.6 Patients known to be poor CYP2D6 metabolizers have higher brexpiprazole plasma concentrations than normal CYP2D6 metabolizers.3, 4, 6 Dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in patients taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers.3, 4, 6 To our knowledge, there are no reports showing the relationship between plasma levels of brexpiprazole and the occurrence of various adverse effects.
The aim of this study was to evaluate the pharmacokinetics (PK) including the influence of the CYP2D6 polymorphism and the safety profile of brexpiprazole in Japanese patients with schizophrenia after a 14‐day once‐daily repeated oral administration.
Methods
The protocol of this phase 1 trial was approved by the institutional review board at each trial site. This trial was conducted at 11 sites in Japan between June and November 2010 in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with Good Clinical Practice and in compliance with local regulatory regulations. Prior to study entry, written informed consent was obtained from each patient. In case of a minor patient (between 18 and 19 years old) or a patient who was hospitalized for protection, the patient's legally acceptable representative also received an explanation about the trial and provided written informed consent.
Study Population
Male and female patients were within the age range of 18 to 64 years old and diagnosed with schizophrenia according to the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, Text Revision (DSM‐IV‐TR) criteria. Body mass index (BMI) was between 19.0 and 35.0 kg/m2, and the patients were capable of being hospitalized during the trial. Patients with any evidence of organ dysfunction or with medically significant abnormality of laboratory tests, physical examination, or electrocardiogram were excluded. Patients who did not complete the required washout period for use of the following medications prior to the start of study medication administration or who were expected to use of any of the following medications during the study period were excluded: 5 days of oral antipsychotics, 1 full cycle plus 1 week of long‐acting antipsychotics, 2 weeks of benzodiazepines (could be used as rescue therapy during the trial), 7 days of selective serotonin reuptake inhibitor (SSRI) and serotonin‐noradrenaline reuptake inhibitor (SNRI), 14 days of paroxetine, 14 days of CYP2B6, CYP2D6, and CYP3A4 inhibitors and CYP3A4 inducers. Patients who had any DSM‐IV‐TR diagnosis other than schizophrenia, complications or history of diabetes, or been judged by the investigators to be inappropriate for inclusion in this trial for any other reasons were excluded.
Study Design
This study consisted of a 60‐day screening period, a 14‐day treatment period, and a 30‐day follow‐up period. Those who were receiving a drug designated as a prohibited concomitant drug were required to undergo a washout for the specified length of time that was described above. Patients who were judged eligible for the trial were hospitalized from no later than the last day of the screening period (day ‐1). During the treatment period, brexpiprazole at 1, 4, or 6 mg was orally administered once daily after breakfast for 14 days. After the treatment period, patients were hospitalized for 7 more days (up to day 21) for the washout of brexpiprazole. Regular blood sampling was performed up to day 21 for the evaluation of PK and safety. After the completion of the examinations on day 21, patients were discharged. Concomitant medications such as antipsychotics, anxiolytics benzodiazepines (could be used as rescue therapy during the trial), SSRI, SNRI, inhibitors/inducers of CYP3A4, and inhibitors of CYP2B6 and CYP2D6 were prohibited between the start of the treatment period and the completion of examinations on day 21 or discontinuation.
Sample Collection
Blood samples for the measurement of plasma concentrations of brexpiprazole and DM‐3411 were collected predose (on days 1, 10, 11, 12, 13, and 14) and postdose after the first and 14th (final) administrations. After the first dose, blood samples were collected at 1, 2, 4, 6, 8, 12, and 24 hours. After the final dose, blood samples were collected at 1, 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144, and 168 hours.
Bioanalysis
Plasma concentrations of brexpiprazole and its metabolite, DM‐3411, were determined using the validated liquid chromatography–tandem mass spectrometry (LC‐MS/MS) method at a bioanalytical laboratory, LSI Medience Corporation (Tokyo, Japan). Brexpiprazole, DM‐3411, and internal standards (brexpiprazole‐d8 for brexpiprazole and OPC‐14714 for DM‐3411) were extracted by protein precipitation and analyzed by LC‐MS/MS using TurboIonSpray with positive ionization (unpublished method). Intrabatch and interbatch precision (coefficient of variation) was less than 6.0%, accuracy (relative error) ranged from ‐14.2% to 11.6%, and the lower limit of quantitation of brexpiprazole and DM‐3411 in plasma was 0.5000 ng/mL. Stability studies showed that brexpiprazole and DM‐3411 were stable during the preparation and analytical processes.
CYP2D6 Genotyping
Using a QIAamp DNA Blood Mini Kit (Qiagen, Tokyo, Japan), DNA was extracted from 2 mL of venous blood that was collected in a blood collection tube containing ethylenediaminetetraacetic acid–2Na. To detect CYP2D6‐mutated alleles, direct sequencing and polymerase chain reaction–restriction fragment length polymorphism (PCR‐RFLP), for *2, *4, *10, *14A, *14B, *18, *21, and *41, and long‐PCR methods (for *5 gene‐deleted) were performed. An allele without any variants was determined as *1. Metabolic activity was defined as the normal alleles *1 and *2, the decreased‐activity alleles *10 and *41, the inactive alleles *4, *5, and *14A, and the unknown‐activity alleles *14B, *18, and *21 (www.cypalleles.ki.se).7 The CYP2D6 genotypes of subjects were classified into 3 categories, namely, extensive metabolizers (EMs; genotypes including at least 1 active allele), intermediate metabolizers (IMs; 2 decreased‐activity alleles or 1 decreased‐activity allele and 1 inactive allele or 1 decreased‐activity allele and 1 unknown‐activity allele), and poor metabolizers (PMs; 2 inactive alleles).
Pharmacokinetic Analysis
PK parameters were determined from the plasma concentrations of brexpiprazole and DM‐3411 using noncompartmental analysis in WinNonlin Enterprise version 5.3 (Pharsight Corporation). Descriptive statistics for the PK data were determined using SAS version 9.1.3 (SAS Institute Japan Inc.). Plasma concentrations and PK parameters, that is, maximum plasma concentration (Cmax), area under the plasma concentration–time curve (AUC) from time zero to 24 hours (AUC24h), plasma concentration measured 24 hours postdose (C24h), time to peak plasma concentration (tmax), terminal‐phase elimination half‐life (t1/2), apparent clearance of drug from plasma after extravascular administration (CL/F), and apparent volume of distribution during the terminal phase after extravascular administration (V/F) of brexpiprazole and/or DM‐3411 were estimated for each dose. The linearity of brexpiprazole pharmacokinetics was assessed by examining the log‐transformed Cmax and AUC24h on day 14 as a function of the administered log dose. Dose‐normalized PK parameters (Cmax/D and AUC24h/D) were calculated by dividing Cmax and AUC24h, respectively, by the dose; PK based on CYP2D6 polymorphism was also evaluated. Tmax was expressed by median, maximum, and minimum, and the other values were expressed by mean and standard deviation (SD).
Safety Assessment
Adverse events were recorded throughout the trial. Physical examination, body weight, vital signs, clinical laboratory measurements (hematology, biochemistry including serum prolactin levels, urinalysis), 12‐lead electrocardiogram (ECG), and extrapyramidal symptoms evaluation using the Drug‐Induced Extrapyramidal Symptoms Scale (DIEPSS),8 Abnormal Involuntary Movement Scale (AIMS),9 and Barnes Akathisia Rating Scale (BARS)10 were performed.
Results
In total, 29 patients were screened, of whom 21 were eligible to enter this study and received repeated administrations of brexpiprazole: 7, 8, and 6 patients received 1, 4, and 6 mg of brexpiprazole, respectively. The patients had a mean age of 50.8 ± 11.0 years and were predominantly male (67% male, n = 14). They had a mean body weight of 66.0 ± 15.7 kg, height of 163.0 ± 10.1 cm, and BMI of 24.6 ± 4.6 kg/m2. All patients enrolled in this study were included in the safety analysis and PK analysis on day 1, and 2 were excluded from the PK analysis on day 14 because of discontinuation of the study.
Pharmacokinetic Evaluation
The time‐course plots of mean plasma concentrations of brexpiprazole and DM‐3411 for multiple administrations of 1, 4, and 6 mg of brexpiprazole on day 1 and days 14 to 21 are shown in Figure 1. The PK parameters are summarized in Table 1. Following a multiple oral administration, the Cmax and AUC24h of brexpiprazole and DM‐3411 increased in a dose‐dependent manner. Regression of the log‐transformed Cmax and AUC24h on day 14 as a function of log dose showed that the 95% confidence interval (CI) of the slope included 1. These results showed dose‐proportionality for Cmax and AUC24h of brexpiprazole. Steady state, based on mean plasma concentrations of brexpiprazole in predosing samples, was estimated to have been attained after day 10. The Cmax and AUC24h of brexpiprazole following multiple administrations of 1, 4, and 6 mg of brexpiprazole were 2.5 to 5.5 times higher on day 14 compared with day 1 based on the accumulation index. Brexpiprazole showed higher plasma concentrations than DM‐3411 in all doses. The ratio of AUC24h of DM‐3411 to that of brexpiprazole on day 14 at each dose ranged from 0.5 to 0.8, indicating no significant differences among the dose groups.
Figure 1.
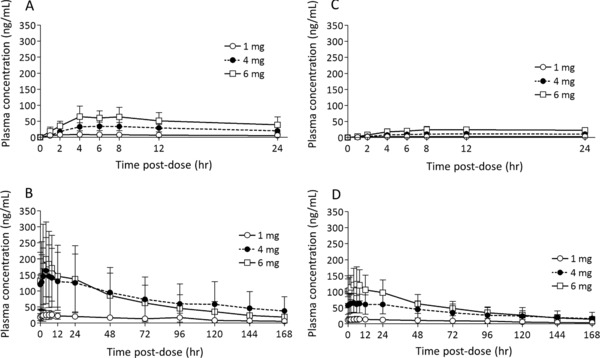
Mean plasma concentration–time profile of brexpiprazole (A, B) and DM‐3411 (C, D) on days 1 (A, C) and 14 (B, D) following multiple once‐daily oral dosing of brexpiprazole 1 mg (day 1, n = 7; day 14, n = 6), 4 mg (day 1, n = 8; day 14, n = 7), and 6 mg (n = 6). Error bars represent standard deviations.
Table 1.
Pharmacokinetic Parameters of Brexpiprazole and DM‐3411 Following Multiple Once‐Daily Administration of Brexpiprazole
| 1 mg | 4 mg | 6 mg | ||||
|---|---|---|---|---|---|---|
| Day 1 | Day 14 | Day 1 | Day 14 | Day 1 | Day 14 | |
| Parameter | (n = 7) | (n = 6) | (n = 8) | (n = 7) | (n = 6) | (n = 6) |
| Brexpiprazole | ||||||
| Cmax (ng/mL) | 10.2 (5.0) | 29.3 (15.1) | 37.0 (13.5) | 165 (102) | 69.9 (29.1) | 206 (123) |
| AUC24h (ng·h/mL) | 160 (67) | 537 (264) | 601 (197) | 3238 (2184) | 1151 (551) | 3738 (2474) |
| tmax (h)a | 4.1 (1.3–8.0) | 5.0 (2.0–7.9) | 6.0 (4.0–8.3) | 4.0 (1.8–4.3) | 4.1 (2.2–8.0) | 4.2 (2.0–8.0) |
| t1/2 (h) | — | 91.9 (47.6) | — | 70.6 (26.9) | — | 51.9 (6.5) |
| CL/F (L/h) | — | 2.17 (0.82) | — | 1.96 (1.46) | — | 2.31 (1.41) |
| V/F (L) | — | 307.8 (282.7) | — | 178.4 (122.8) | — | 175.7 (119.7) |
| DM‐3411 | ||||||
| Cmax (ng/mL) | 3.04 (1.30) | 15.3 (9.3) | 11.7 (6.7) | 66.9 (29.2) | 25.8 (8.4) | 128 (52) |
| AUC24h (ng·h/mL) | 60.6 (28.9) | 319 (190) | 218 (125) | 1467 (697) | 489 (185) | 2605 (1056) |
| tmax (h)a | 12.0 (8.0–23.8) | 6.0 (3.9–24.0) | 11.7 (8.0–23.5) | 3.9 (1.0‐23.9) | 10.1 (8.0–23.9) | 6.0 (2.0–8.0) |
| t1/2 (h) | — | 81.6 (28.7) | — | 95.3 (64.5) | — | 54.2 (8.5) |
Values are expressed as mean (standard deviation) unless specified otherwise.
Cmax, maximum plasma concentration; AUC, area under the concentration–time curve; AUC24h, AUC from time zero to 24 hours postdose; tmax, time to Cmax; t1/2, elimination half‐life; CL/F, apparent total plasma clearance.
Results are expressed as median (range).
By CYP2D6 genotype, 15 and 6 patients were classified as EMs and IMs, respectively. No patients were classified as PMs or other. The number of EM patients was 6, 5, and 4 in the 1‐, 4‐, and 6‐mg groups, respectively, and the number of IM patients was 1, 3, and 2 in the 1‐, 4‐, and 6‐mg groups, respectively. The mean dose‐normalized plasma concentrations of brexpiprazole and DM‐3411 by CYP2D6 genotype are shown in Figure 2. The PK parameters by CYP2D6 genotype are summarized in Table 2. For PK parameters of brexpiprazole, both Cmax/D and AUC24h/D were higher in IM patients than in EM patients. The t1/2 was also longer in IM patients; however, except for 1 IM patient with quite a long t1/2, no significant difference was observed between the groups. CL/F was lower in IM patients than in EM patients. Steady state, based on mean plasma concentrations per dose of brexpiprazole in predosing samples, was estimated to have been attained after day 10 in both the EM and IM groups. The Cmax and AUC24h in EM and IM patients of brexpiprazole following multiple administrations of brexpiprazole were 2.6–2.8 and 4.5–5.8 times higher, respectively, on day 14 compared with day 1 based on the accumulation index. The ratio of the AUC24h of DM‐3411 to that of brexpiprazole on day 14 in EM and IM patients was 0.8 and 0.3, respectively. Cmax/D and AUC24h/D of DM‐3411 on day 1 were lower in IM patients than in EM patients, and those on day 14 were similar for EM and IM patients. The Cmax and AUC24h in EM and IM patients of DM‐3411 following multiple administrations of brexpiprazole were 3.9–5.1 and 7.4–9.5 times higher, respectively, on day 14 compared with day 1 based on the accumulation index.
Figure 2.
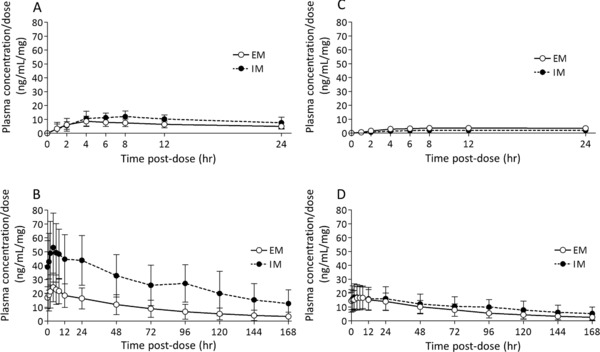
Mean dose‐normalized plasma concentration/dose–time profile of brexpiprazole (A, B) and DM‐3411 (C, D) by CYP2D6 genotype on days 1 (A, C) and 14 (B, D) following multiple once‐daily oral dosing of 1, 4, and 6 mg brexpiprazole. Error bars represent standard deviations. EM, CYP2D6 extensive metabolizer (day 1, n = 15; day 14, n = 13); IM, CYP2D6 intermediate metabolizer (n = 6).
Table 2.
Pharmacokinetic Parameters of Brexpiprazole and DM‐3411 by CYP2D6 Genotype Following Multiple Once‐Daily Administration of Brexpiprazole
| EM | IM | ||||
|---|---|---|---|---|---|
| Parameter | Day 1 (n = 15) | Day 14 (n = 13) | Day 1 (n = 6) | Day 14 (n = 6) | |
| Brexpiprazole | |||||
| Cmax/D (ng/mL/mg) | 9.20 (4.12) | 26.0 (11.7) | 12.9 (3.6) | 55.3 (22.1) | |
| AUC24h/D (ng·h/mL/mg) | 146 (60) | 462 (211) | 214 (68) | 1103 (451) | |
| t1/2 (h) | — | 62.9 (23.5) | — | 89.9 (47.1) | |
| CL/F (L/h) | — | 2.55 (1.08) | — | 1.25 (1.07) | |
| DM‐3411 | |||||
| Cmax/D (ng/mL/mg) | 3.78 (1.57) | 17.7 (8.4) | 2.31 (0.84) | 17.9 (9.0) | |
| AUC24h/D (ng·h/mL/mg) | 73.5 (31.3) | 366 (170) | 40.9 (14.8) | 389 (205) | |
| t1/2 (h) | — | 65.7 (36.4) | — | 105 (51) | |
EM, CYP2D6 extensive metabolizer; IM, CYP2D6 intermediate metabolizer; Cmax/D, dose‐normalized maximum plasma concentration; AUC, area under the concentration–time curve; AUC24h/D, dose‐normalized AUC from time zero to 24 hours postdose; t1/2, elimination half‐life; CL/F, apparent total plasma clearance.
Values are expressed as mean (standard deviation).
Safety Assessment
Overall, all doses of brexpiprazole were well tolerated. There were no deaths. Treatment‐emergent adverse events (TEAEs) were reported in 5 of 7 patients (71.4%) in the 1‐mg dose group and in all patients in the 4‐ and 6‐mg dose groups (Table 3). The incidence of TEAEs following administration of brexpiprazole is shown in Table 3. Overall, the severity of TEAEs was generally mild to moderate. However, 2 patients, 1 each in the 1‐ and 4‐mg groups, experienced serious TEAEs; hence,they prematurely withdrew from the study. Both were because of worsening of the underlying condition (schizophrenia): severe in the 1‐mg group and moderate in the 4‐mg group. Both events were resolved.
Table 3.
Treatment‐Emergent Adverse Events
| Dose | 1 mg (n = 7) | 4 mg (n = 8) | 6 mg (n = 6) |
|---|---|---|---|
| Incidence of TEAEs, n (%) | |||
| At least 1 TEAE | 5 (71.4) | 8 (100) | 6 (100) |
| Serious TEAE | 1 (14.3) | 1 (12.5) | 0 |
| Severe TEAE | 1 (14.3) | 0 | 0 |
| Discontinuation because of TEAE | 1 (14.3) | 1 (12.5) | 0 |
| TEAEs occurring in patients in any group, n (%) | |||
| Blood prolactin increased | 2 (28.6) | 3 (37.5) | 3 (50.0) |
| Blood creatine phosphokinase increased | 1 (14.3) | 2 (25.0) | 0 |
| Blood prolactin decreased | 0 | 1 (12.5) | 1 (16.7) |
| Blood pressure increased | 1 (14.3) | 0 | 1 (16.7) |
| Nausea | 0 | 2 (25.0) | 0 |
| Insomnia | 0 | 1 (12.5) | 1 (16.7) |
| Schizophrenia | 1 (14.3) | 1 (12.5) | 0 |
| Nasopharyngitis | 0 | 2 (25.0) | 0 |
| Blood pressure decreased | 0 | 0 | 1 (16.7) |
| Blood corticotrophin increased | 0 | 0 | 1 (16.7) |
| Blood cortisol increased | 0 | 0 | 1 (16.7) |
| Back pain | 0 | 0 | 1 (16.7) |
| Muscle rigidity | 0 | 0 | 1 (16.7) |
| Musculoskeletal pain | 1 (14.3) | 0 | 0 |
| Convulsion | 0 | 0 | 1 (16.7) |
| Dizziness | 0 | 0 | 1 (16.7) |
| Somnolence | 0 | 1 (12.5) | 0 |
| Salivary hypersecretion | 0 | 0 | 1 (16.7) |
| Constipation | 0 | 1 (12.5) | 0 |
| Diarrhea | 0 | 1 (12.5) | 0 |
| Stomatitis | 0 | 1 (12.5) | 0 |
| Vomiting | 0 | 1 (12.5) | 0 |
| Skin laceration | 0 | 0 | 1 (16.7) |
| Foot fracture | 1 (14.3) | 0 | 0 |
| Joint sprain | 1 (14.3) | 0 | 0 |
| Erythema | 0 | 1 (12.5) | 0 |
| Skin exfoliation | 1 (14.3) | 0 | 0 |
| Decreased appetite | 0 | 1 (12.5) | 0 |
TEAE, treatment‐emergent adverse event.
The most common TEAE was increase in serum prolactin level. Eight patients (2 male patients in the 1‐mg group, 2 male and 1 female patients in the 4‐mg group, and 1 male and 2 female patients in the 6‐mg group) showed increased prolactin level, and 2 patients (1 female patient in the 4‐mg group and 1 male patient in the 6‐mg group) showed decreased prolactin level as TEAEs. All events were judged to be mild in severity. The time course for serum prolactin levels is shown in Figure 3. Temporary increases in serum prolactin were observed in 5 of 6 patients in the 6‐mg group 6 hours postdose on day 1.
Figure 3.
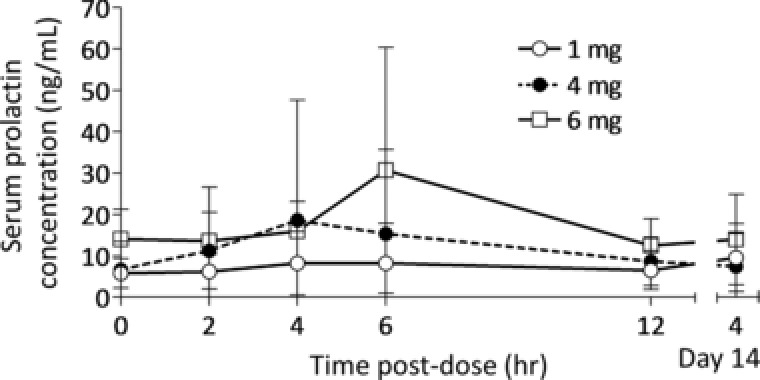
Mean serum prolactin concentration–time profile on days 1 and 14 following multiple once‐daily oral dosing of brexpiprazole 1 mg (day 1, n = 7; day 14, n = 6), 4 mg (day 1, n = 8; day 14, n = 7), and 6 mg (n = 6). Error bars represent standard deviations.
No clinically significant changes were observed for other clinical laboratory values, 12‐lead ECG, vital signs, body weight, DIEPSS, AIMS, and BARS.
Discussion
This phase 1 study was conducted in Japan to investigate the PK and safety of repeated administrations of brexpiprazole at 1, 4, and 6 mg in patients with schizophrenia. Brexpiprazole was safe and well tolerated, and TEAEs observed were generally mild to moderate. The results of PK analysis demonstrated that the Cmax and AUC24h of brexpiprazole showed dose‐proportionality. The plasma concentrations of brexpiprazole reached steady state after day 10 of administration for all dose groups, and the Cmax and AUC24h of brexpiprazole showed accumulation of about 2.5‐ to 5.5‐fold on day 14 compared with those on day 1. The t1/2 of brexpiprazole ranged from approximately 52 to 92 hours. This long t1/2 of brexpiprazole was similar to that in the US prescribing information and Canada Product Monograph (91 hours),3, 4 and it may account for the accumulation of brexpiprazole and supports the use of brexpiprazole for once‐daily dosing.
For brexpiprazole, IM patients showed higher Cmax/D and AUC24h/D than EM patients. CL/F was lower in IM patients than in EM patients. These findings were considered to be caused by high metabolism of brexpiprazole in EM patients with high CYP2D6 enzyme activity and low metabolism in IM patients with low CYP2D6 enzyme activity, suggesting that CYP2D6 contributes to the metabolism of brexpiprazole.
In addition, the Cmax and AUC24h of its main metabolite, DM‐3411, showed dose‐proportionality in a pattern similar to brexpiprazole. The Cmax and AUC24h of DM‐3411 showed a tendency for greater accumulation in IM patients than in EM patients. This finding may suggest the contribution of CYP2D6 to the metabolic process of DM‐3411. However, it does not appear to contribute to the therapeutic effects of brexpiprazole.6
In the study, 8 of 21 patients showed increased prolactin level and 2 of 21 patients showed decreased prolactin level as a TEAE. Antipsychotics have a blocking effect on the dopamine D2 receptor and therefore can elevate the secretion of prolactin.11, 12 Conversely, a dopamine partial agonist suppresses prolactin secretion.11, 13 In most studies with aripiprazole, prolactin levels were found to be decreased, even below those expected from placebo, or to remain unchanged across all dosages.11 Brexpiprazole has lower intrinsic activity at dopamine D2 receptors than does aripiprazole.5 The intrinsic activity level of brexpiprazole may be slightly lower than no influence level for inhibition of prolactin secretion in some patients who showed slightly increased prolactin levels. In this study, the mean serum prolactin levels of the 4‐ and 6‐mg dose groups were transiently increased 4 or 6 hours postdose on day 1, and they were higher than that 4 hours postdose on day 14, even though brexpiprazole mean plasma concentration on day 14 was higher than that on day 1. This discrepancy between brexpiprazole concentration elevation and prolactin level elevation by multiple doses of drugs did not correlate with the trend seen with other dopamine D2 receptor antagonist antipsychotics.12
There is a limitation of the present study from the small number of patients enrolled. All patients (9 EM patients and 5 IM patients) in the 4‐ and 6‐mg groups showed at least 1 TEAE. This study sample size was too small to determine the relationship between adverse effects of brexpiprazole and plasma concentration or CYP2D6 genotype. This study was designed because the PK and safety of repeated administration of brexpiprazole have not yet been investigated in Japan. Despite this limitation, the steady‐state plasma levels and predictable PK parameters demonstrated in this study suggested that brexpiprazole can be administered on a once‐daily basis, in line with approved dosing in the United States and Canada. For further investigations, studies involving larger sample sizes and longer study periods will be required to further evaluate the efficacy, safety, and tolerability of brexpiprazole for the treatment of Japanese patients with schizophrenia.
Conclusions
During repeated administrations of brexpiprazole at 1, 4, and 6 mg once daily for 14 days, the concentration of brexpiprazole reached steady state after day 10 in all dose groups. The Cmax and AUC24h of brexpiprazole and DM‐3411 showed dose‐proportionality. CYP2D6 contributed to the metabolism of brexpiprazole. Brexpiprazole was safe and well tolerated in the studied Japanese patients with schizophrenia.
Acknowledgments
We gratefully acknowledge the patients who have participated in this study. We thank the investigators at each study site.
Declaration of Conflicting Interests
Dr. Ishigooka has received research support or speakers' honoraria from, or has served as a consultant to Otsuka, Dainippon Sumitomo, Eli Lilly, Astellas, Mitsubishi Tanabe, MSD, Takeda, Shionogi, Novartis, Kyowa, Teva, and Meiji Seika Pharma. Mr. Iwashita, Mr. Higashi, Dr. Liew, and Dr. Tadori are employees of Otsuka Pharmaceutical Co., Ltd.
Funding
This study was supported by Otsuka Pharmaceutical Co., Ltd.
References
- 1. Correll CU. From receptor pharmacology to improved outcomes: individualising the selection, dosing, and switching of antipsychotics. Eur Psychiatry. 2010;25(suppl 2):S12–S21. [DOI] [PubMed] [Google Scholar]
- 2. Abilify Prescribing Information . Otsuka Pharmaceutical Co Ltd.; 2016.
- 3. Otsuka Pharmaceutical Co Ltd . Prescribing Information for Rexulti® (brexpiprazole) tablets. 2015. http://www.accessdata.fda.gov.
- 4. Otsuka Pharmaceutical Co Ltd . Product monograph for RexultiTM (brexpiprazole) tablets. 2017. http://www.healthcanada.gc.ca.
- 5. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin‐dopamine activity modulator. J Pharmacol Exp Ther. 2014;350:589–604. [DOI] [PubMed] [Google Scholar]
- 6. Garnock‐Jones KP. Brexpiprazole: a review in schizophrenia. CNS Drugs. 2016;30:335–342. [DOI] [PubMed] [Google Scholar]
- 7. Sakuyama K, Sasaki T, Ujiie S, et al. Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A‐B, 18, 27, 36, 39, 47–51, 53–55, and 57). Drug Metab Dispos. 2008;36:2460–2467. [DOI] [PubMed] [Google Scholar]
- 8. Inada T, Beasley CM Jr, Tanaka Y, Walker DJ. Extrapyramidal symptom profiles assessed with the Drug‐Induced Extrapyramidal Symptom Scale: comparison with Western scales in the clinical double‐blind studies of schizophrenia patients treated with either olanzapine or haloperidol. Int Clin Psychopharmacol. 2003;18:39–48. [DOI] [PubMed] [Google Scholar]
- 9. Guy W. ECDEU Assessment Manual for Psychopharmacology. US Department of Health Education and Welfare Publication (ADM) 76–338. Rockville, MD: National Institute of Mental Health; 1976. [Google Scholar]
- 10. Barnes TR. A rating scale for drug‐induced Akathisia. Br J Psychiatry. 1989;154:672–676. [DOI] [PubMed] [Google Scholar]
- 11. Peusken J, Pani L, Detraux J, De Hert M. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs. 2014;28:421–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berwaerts J, Cleton A, Rossenu S, et al. A comparison of serum prolactin concentrations after administration of paliperidone extended‐release and risperidone tablets in patients with schizophrenia. J Psychopharmacol. 2010;24:1011–1018. [DOI] [PubMed] [Google Scholar]
- 13. Tadori Y, Kitagawa H, Forbes RA, McQuade RD, Stark A, Kikuchi T. Differences in agonist/antagonist properties at human dopamine D2 receptors between aripiprazole, bifeprunox and SDZ 208–912. Eur J Pharmacol. 2007;574:103–111. [DOI] [PubMed] [Google Scholar]