

Abstract
Our previous studies have raised the possibility that altered blood glucose levels may influence and/or be predictive of methamphetamine (METH) neurotoxicity. This study evaluated the effects of exogenous glucose and corticosterone (CORT) pretreatment alone or in combination with METH on blood glucose levels and the neural and vascular toxicity produced. METH exposure consisted of four sequential injections of 5, 7.5, 10, and 10 mg/kg (2 h between injections) D‐METH. The three groups given METH in combination with saline, glucose (METH+Glucose), or CORT (METH+CORT) had significantly higher glucose levels compared to the corresponding treatment groups without METH except at 3 h after the last injection. At this last time point, the METH and METH+Glucose groups had lower levels than the non‐METH groups, while the METH+CORT group did not. CORT alone or glucose alone did not significantly increase blood glucose. Mortality rates for the METH+CORT (40%) and METH+Glucose (44%) groups were substantially higher than the METH (< 10%) group. Additionally, METH+CORT significantly increased neurodegeneration above the other three METH treatment groups (≈ 2.5‐fold in the parietal cortex). Thus, maintaining elevated levels of glucose during METH exposure increases lethality and may exacerbate neurodegeneration. Neuroinflammation, specifically microglial activation, was associated with degenerating neurons in the parietal cortex and thalamus after METH exposure. The activated microglia in the parietal cortex were surrounding vasculature in most cases and the extent of microglial activation was exacerbated by CORT pretreatment. Our findings show that acute CORT exposure and elevated blood glucose levels can exacerbate METH‐induced vascular damage, neuroinflammation, neurodegeneration and lethality.
Cover Image for this issue: doi. 10.1111/jnc.13819.
Keywords: amphetamine, blood glucose, corticosterone, methamphetamine, neurotoxicity, vascular damage
Abbreviations used
- AMPH
amphetamine
- Ccl2
C‐C motif chemokine ligand 2
- CORT
corticosterone
- FJc
Fluoro‐Jade C
- Iba1
allograft inflammatory factor 1 (Aif1)
- METH
methamphetamine
- RECA‐1
rat endothelial cell antigen 1
The neurobiological effects of substituted amphetamines [e.g., methamphetamine (METH) and amphetamine (AMPH)] are well‐known (e.g., dopamine release/depletion, damage to dopaminergic neurons, neuroinflammation, etc.). A number of published reports document the ability of METH and AMPH to cause neurotoxicity in humans (Rothrock et al. 1988; Wilson et al. 1996; McCann et al. 1998; Ernst et al. 2000; Volkow et al. 2001a,b; McCann and Ricaurte 2004; Chang et al. 2007; Sekine et al. 2008; Ho et al. 2009), with even more reviews on research in laboratory animals relating to the neurotoxicity that METH and AMPH caused in the striatum (Bowyer and Holson 1995; Seiden and Sabol 1995; Fleckenstein et al. 2000, 2007; O'Callaghan and Miller 2001; Cadet et al. 2005, 2007; Bowyer et al. 2008; Bowyer and Hanig 2014; Yamamoto et al. 2010). There have also been a few reports on the in vivo vascular toxicity of METH and AMPH [see reviews: (Bowyer et al. 2014; Northrop et al. 2016)]. Furthermore, neuroinflammation is associated with a variety of neurotoxicant exposures, which induce damage to the CNS (Thomas et al. 2004; Thomas and Kuhn 2005; Block et al. 2007; Sriram and O'Callaghan 2007; Kraft and Harry 2011), and a number of neurodegenerative diseases [see review: (Prinz and Priller 2014]. More specifically, AMPH and METH can produce dopaminergic axonal and terminal damage as well as neuroinflammation in the striatum when concomitant non‐life threatening hyperthermia (40.0–41.0°C lasting from 2 to 5 h) occurs in the rodent (Bowyer et al. 1994, 1998, 2008, 2014; O'Callaghan and Miller 1994). However, when life‐threatening hyperthermic events occur during exposure, substantial neuroinflammation or neurodegeneration in the parietal cortex, limbic cortex, hippocampus, and thalamus also is observed (Bowyer et al. 1998, 2016). In some instances, ‘cerebrovascular inflammation’ (induced primarily due to vascular damage) can occur in the brain, thereby resulting in prominent microglial activation and some moderate astrocytic morphological changes in the absence of neurodegeneration (Bowyer et al. 2016). While these scenarios involve concomitant neuroinflammation and neural damage, it remains unclear whether these proinflammatory responses are independent, the cause, or the consequence of toxicant‐induced neural damage (Graeber and Streit 2010; Streit 2010).
Previously, we found that exogenous corticosterone (CORT), which mimicked a high physiologic stress episode, over the course of 1 week prior to METH exposure, significantly exacerbated the neuroimmune response and dopamine terminal damage produced by METH in the striatum (Kelly et al. 2012). These findings suggested that chronic exogenous CORT and, perhaps, persistent high physiological stress levels of endogenous CORT can sensitize the CNS to the neuroinflammatory and neurotoxic effects of METH. The neurodegeneration and neuroinflammation that can occur in the parietal cortex and thalamus after METH exposure were not evaluated in that study. Recent studies evaluating the effects of CORT pretreatment on other toxicant exposures has confirmed that enhanced neuroinflammatory responses can occur in other areas of the brain, such as the cortex and hippocampus (O'Callaghan et al. 2015; Locker et al. 2017).
While we have established the impact of METH on neuronal damage and inflammation, systemically delivered METH has the potential to disrupt non‐neuronal targets as well. We have anecdotally observed that blood glucose levels often plummet at the end of acute METH or AMPH exposures (Bowyer et al., unpublished data), indicating the possibility that blood glucose levels may influence and/or be predictive of METH‐induced neurotoxicity. In support of this hypothesis, other studies have indicated that METH has the potential to inhibit glucose uptake by neural cells and instigate blood–brain barrier (BBB) disruption through glucose transporter disruption (Abdul Muneer et al. 2011a, b). This occurs despite the fact that some stimulants such as cocaine are reported to elevate brain extracellular levels of glucose (Wakabayashi and Kiyatkin 2015). Interestingly, frequent hyperthermic events resulting from METH or AMPH exposure result in a greater number of transient breakdown/leakage events in the BBB that can occur each time body temperature rises above 41.6°C (Kiyatkin et al. 2007). As might be expected, this BBB leakage occurs more often in regions where neurodegeneration and neuroinflammation are detected (Bowyer and Ali 2006; Kiyatkin et al. 2007; Bowyer et al. 2014), arguing for a connection between blood glucose levels, BBB disruption, and METH neurotoxicity. While the mechanism by which CORT exerts its enhancing effects on toxicant‐induced neural damage and neuroinflammation is unknown, CORT pretreatment may affect blood glucose levels directly leading to damage to the brain or indirectly through enhanced METH‐induced neurotoxicity/neuroinflammation.
Thus, this study was designed to evaluate the effects of exogenous glucose and CORT pretreatment on blood glucose levels during METH exposure and determine whether they influence the subsequent neurodegeneration, neuroinflammation, and vascular toxicity that are seen in the parietal cortex and thalamus after METH exposure under certain conditions. The histological evaluation of these three adverse effects was determined at 3 days post‐METH exposure, which is the time point where they are readily observed [see reviews; (Bowyer et al. 2008, 2014)]. The level of exogenous glucose exposure was such that it was at the threshold of increasing blood glucose levels. This was done so that additive or synergistic effects of METH in combination with exogenous glucose on blood glucose levels might most readily be observed.
Materials and methods
Animals
This study was carried out in accordance with the declaration of Helsinki and the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. The use of animal testing in this study was done under protocols E7295 and E7519 (issued to John Bowyer) that were approved by the NCTR institutional animal care and use committee (IACUC), which is fully accredited (Food and Drug Administration ‐ National Center for Toxicological Research Accreditation #A4310‐01) by NIH‐OLAW. Sixty‐five day‐old (300–350 g) male Sprague‐Dawley rats, ninety five in total, were obtained from the Charles River Laboratories Inc., Wilmington, MA, USA Laboratories [Crl:CD(SD)] (RRID:RGD_734476). Upon arrival at NCTR, they received tail tattoos for identification. Prior to testing, rats were housed 2 per cage with food and water available ad libitum. Rats were housed on a daily 12 h light cycle with lights on at 6:00 am and off at 6:00 pm. During housing, the temperature (23°C) and humidity (53%) were controlled. The rats were tested between 85 and 90 days (12 weeks) of age. Prior to testing animals were randomly split into dosing groups with the exception that litter mates (as shipped by Charles River) could not be in the same dosing group. Since the experiments were carried out with five ‘replicates’ (see below), this further ensured that differences in shipment conditions and breeding selection of animals from source (Charles River) would have lesser effects on study results.
Administration of METH, glucose, and CORT
General considerations
We made a considerable effort to ensure that the exposed rats endured multiple (two to five) events of body temperature exceeding 41.7°C in order to facilitate METH toxicity and increase the likelihood of BBB disruption. The dose of exogenous glucose to be administered was one that was at the threshold of significantly increasing blood glucose so that, in combination with METH, the two treatments might act additively or synergistically to increase blood glucose levels.
Glucose levels were determined during METH administration to provide a relationship between hyperthermia, striatal dopamine depletions, and duration of METH exposure with CORT or exogenous glucose treatment. Determination of neurodegeneration and neuroinflammation and vascular changes were determined at 3 days after METH since this is the time‐point at which we observed that neurotoxicities were maximal in rats after amphetamine exposure (Bowyer et al. 2008, 2014).
An overview of the dosing design
The overall dosing design for the seven treatment groups is shown in Table 1. The seven treatments include Saline, Glucose, CORT, METH, METH+Glucose, METH+CORT, and METH+Veh. This part of the study was conducted over a 6‐month period and required that the data be collected in five ‘runs’ in order to monitor the animals closely and ensure low mortality. Each run contained 12–15 animals; at least six treatment groups were represented in each run. One day prior to testing, 20 mg/kg CORT or vehicle was injected s.c. (injection volume of 2 mL/kg in a 25% ethanol/75% sesame seed oil vehicle) at 8:00 am and 5:30 pm in the CORT, Meth+Veh, and METH+CORT groups. On the day of testing, all dosing commenced at 8:00 am and ended at 2:00 pm. Injections with the test drugs/articles occurred at 8 am, 10 am, 12 pm, and 2 pm. All groups, except Glucose and METH+Glucose, received four 2 mL/kg i.p. injections of normal saline with each injection spaced 2 h apart. The Saline group received four injections of 1 mL/kg normal saline s.c. following each injection of the 2 mL/kg normal saline i.p. The Glucose and METH+Glucose groups received four injections of 1 g/kg of D‐glucose (0.5 g glucose/mL in normal saline, 2 mL/kg volume).
Table 1.
Overview of animal dosing groups in study
| Treatment group | 2 mL/kg salinea | 1 g/kg D‐glucoseb | CORT vehiclec | CORT pretreatmentd | METH | Number of animals tested | Mortality |
|---|---|---|---|---|---|---|---|
| Salinee | Yes | No | No | No | No | 5 | 0 |
| Glucosee | No | Yes | No | No | No | 5 | 0 |
| CORTe | Yes | No | No | Yes | No | 5 | 0 |
| METH | Yes | No | No | No | Yes | 11f | 1 |
| METH+Glucose | No | Yes | No | No | Yes | 16f | 7 |
| METH+CORT | Yes | No | No | Yes | Yes | 15 f | 6 |
| METH+Veh | Yes | No | Yes | No | Yes | 9 | 2 |
Four injections of 2 mL/kg i.p. normal saline (injections spaced by 2 h intervals).
Four injections of solutions containing 0.5 g/mL D‐glucose in normal saline at 2 mL/kg i.p.
One day prior to testing, 2 mL/kg of 25% ethanol and 65% sesame seed oil (CORT vehicle) was injected s.c. at 8:00 am and 5:30 pm.
One day prior to testing, 20 mg/kg CORT (dissolved in vehicle described above) in a 2 mL/kg volume was injected s.c. at 8:00 am and 5:30 pm.
Group also received an 4 injection of 1 mL/kg normal saline s.c. in addition to the four injections of 2 mL/kg normal saline i.p.
Glucose levels were omitted from three animals in these groups. This is primarily due to either the difficulty in obtaining sufficient blood from the toe pads or death prior to the 4th dose of METH.
This glucose dose was determined by a preliminary study (n = 3 per group) to determine the threshold that demonstrated a marked increase blood glucose levels. We observed that 0.5 mg/kg glucose had no effect on blood glucose, 1 g/kg increased the blood glucose levels 25–50% above control, and a 2 g/kg dose of glucose increased blood glucose levels in two animals to 60% and 75% above control. Thus, we chose 1 g/kg D‐glucose/ dextrose (RRID:PubChem CID Bethesda, MD, USA: 107526) to explore the additive effects of METH and exogenous glucose on blood glucose. Four groups (see Table 1) received four injections of D‐METH (5, 7.5, 10, and 10 mg/kg s.c. spaced at 2 h intervals); the D‐METH (D‐methamphetamine HCl; Sigma‐Aldrich, St. Louis, MO, USA (RRID: PubChem CID: 10836) dose was dissolved in normal saline.
It was assumed that blood glucose levels would be decreased at the end of neurotoxic METH exposures, as we have previously observed with neurotoxic exposures to amphetamine. However, we had no previous data indicating exactly what would occur with blood glucose levels during the earlier time points of METH exposure or data on how CORT would interact with METH. Therefore, this study was designed as a pilot study, where it was assumed that a sample size of n = 5 per group would be necessary and sufficient to provide meaningful results. Additional animals were included in the METH treatment groups to account for potential data problems such as mortality. Moreover, post hoc power analysis was performed, using the SAS procedure PROC POWER. Based on a two‐sample t‐test with a specified mean difference of 20 mg/dL in blood glucose levels between control and treated group, an estimated standard deviation, and α = 0.05, power > 0.8 for a sample size of n = 5 per group.
When body temperatures exceeded 41.6°C during dosing in any of the four groups receiving METH, the lethal effects of hyperthermia were prevented by placing the animals unrestrained on crushed ice for 15–25 min in a clean, wood chip free cage to allow their temperatures to drop below 40.5°C. Animals were behaviorally observed for 6 h after the last injection of METH and body temperature was monitored until it dropped below 39.5°C. Animals that exhibited moribund signs, primarily hind limb paralysis or respiratory distress for over 15 min, were given an overdose of Euthasol® and then perfused for histological analysis. Perfusion < 6 h after the last METH injection had to be performed in moribund animals in the METH+Veh, METH+Glucose, and METH+CORT groups (n = 2, n = 6, and n = 4, respectively). Two animals in the METH+Glucose and one in the METH+CORT group died overnight and were not perfused. The rest of the animals were perfused 3 days after dosing.
Determination of blood glucose levels
Multiple blood samples were drawn linearly over time. Blood was obtained initially from both the hindfoot toe pad and lateral tail vein for determining glucose levels. Glucose levels obtained from the tail vein were lower (usually 30–50%) than those from the hindfoot toe pad, particularly so at the later time points in animals receiving METH. Thus, blood draws from the tail vein were discontinued early during the experiments. For obtaining hindfoot blood, the animals were restrained by hand by one of the investigators while another first wiped the hindfoot with 70% ethanol in water and then inserted a 25 gauge needle deep into one of the toe pads to obtain 5–10 μL of blood. The hindfoot was then again wiped with 70% ethanol in water. Glucose levels were not determined after the 9 h time point (3 h after last injection) since we have previously observed that glucose levels rebound after that time point.
One drop (5–10 μL) of blood was immediately applied to blood glucose test strips to determine glucose levels. Levels were determined, using a Contour®Next blood glucose monitoring system and blood glucose test strips from Bayer (Bayer Health Care LLC; Mishawaka, IN, USA). The monitoring system accuracy was validated by testing with known standard solutions of glucose supplied by Bayer. Recorded levels were 90–95% of that expected from the test solutions.
Animal sacrifice and preparation for histological analysis
The perfusion process necessary for histological processing was initiated by giving a lethal dose of approximately 150–300 mg/kg i.p. of pentobarbital and 20–40 mg/kg sodium phenytoin (0.25–0.5 mL of Euthasol® from Virbac AH Inc., Fort Worth, TX, USA) containing in 390 mg/mL pentobarbital and 50 mg/mL sodium phenytoin). When the rat's respiration waned to barely detectable and there was no response to tail pinch (pain), it was perfused with 50 mL of saline followed by 200 mL of 4% formaldehyde in NaPB. Details of the perfusion process have been previously reported (Bowyer et al. 1998). Brains were postfixed for 36 h in 4% formaldehyde in NaPB and then transferred to a 20% sucrose and 0.1 M sodium phosphate buffer for at least 3 days. Sectioning of the brain to produce 25–30 μm sections was performed either, using unfrozen fixed brain as previously described (Bowyer et al. 1998) or using a cryostat. For cryostat sectioning, the brain was removed from the sucrose and rapidly frozen in crushed/powdered dry ice. It was then embedded in OCT compound (Electron Microscope Science, Hatfield, PA, USA) and mounted on a Leica Cryostat for cutting coronal sections 25 μm thick from +1.2 to −6.0 relative to the Bregma (Paxinos and Watson, 1995) and collected in 0.1% formaldehyde with NaPB. While half of the sections were stored at 4°C in NaPB containing 4% formalin until histological processing, half of the sections were transferred to an anti‐freezing solution and stored at −20°C until further processing.
Histochemical and immunohistological labeling
Fluoro‐Jade C labeling
Methods similar to Schmued et al. (2005) were used to detect degenerating neurons, dendrites, axons, and terminals in the forebrain, using Fluoro‐Jade C (FJc). Briefly, sections were mounted on gelatin (Sigma; St. Louis, MO, USA, 300Bloom) coated slides and dried at 50°C. They were then transferred sequentially through solutions of 100% ethanol [Decon Labs. Inc., King of Prussia, PA, USA; RRID:PubChem CID: 702 (ethanol)] for 8 min, 95% ethanol (2 min), 70% ethanol (2 min), and 2X double deionized water (2 min). They were then immersed in double distilled water containing 0.06% potassium permanganate for 8 to 10 min followed by 2X double deionized water (2 min). Labeling was performed in a 0.0001% FJc (AG325; Millipore Corporation, Bedford, MA, USA; in 0.1% acetic acid and distilled water solution followed by rinsing in double distilled water 3X (1 min per wash) to remove excess label. Slides were then rapidly air dried, xylene‐cleared, and coverslipped with DPX (P‐Xylene‐bis(N‐pyridinium bromide); N,N'‐4‐Xylylenebis(pyridinium); 1,1'‐(p‐Phenylenedimethylene)bis(pyridinium bromide); Fluka/Sigma; RRID:Pubchem CID: 3036637) mounting media.
Counting FJc labeled cells in the parietal cortex and thalamus
Three independent investigators (JB, KT, and LM) counted the number of FJc+ neurons in the histologically fixed sections. Two of the investigators (KT and LM) were totally blind to the animal's treatment group. Investigators counted FJc‐labeled neurons from the following sections: somatosensory parietal cortex (between −0.5 to −1.6 mm from bregma); thalamic paraventricular region (between −1.6 to −2.8); and the combined regions of the ventrolateral (VL), ventromedial (VM), and intralaminar (IL) thalamus (between −2.3 to −3.3 from bregma). The VL, VM, and IL counts were combined because the boundaries, the VM, and VL and the IL (central medial and paracentral) thalamic nuclei were difficult to determine in the medial thalamus. It is important to note that automated counting of FJc+ cells was not feasible for the following reasons: (i) multiple criteria were necessary to determine whether the FJc‐labeled cells were actually neurons, (ii) the region over which cells needed to be detected was expansive e.g., the entire somatosensory parietal cortex, (iii) the intensity of FJc‐labeled neurons within the same animal was variable and more so between animals, and (iv) there was a relatively few number of FJc‐labeled neurons (normally ≤ 30/section/hemisphere). In order for an FJc‐labeled structure to be deemed a neuron, the structure must have had a labeled soma and at least one dendritic process. Furthermore, FJc puncta surrounding the apparent soma and dendrite(s) had to be present.
Iba1 and RECA‐1 immunohistochemistry
For immuno‐reactive labeling of brain sections with antibody to allograft inflammatory factor 1 (Aif1) (Iba1) (a.k.a, Aif1), the following procedures were used with free‐floating sections agitated on an orbital shaker. Sections were first washed 15 min in NaPB and then incubated in phosphate buffer containing 0.3% H2O2 for 30 min to destroy the endogenous peroxidases. From this point on, except with the last step of 3,3'‐diaminobenzidine (DAB) processing, the incubation and washing solutions consisted of NaPB containing 0.4% Triton X‐100 in double deionized water. The sections were subsequently washed 3X for 5 min. After a 30 min preincubation in 4% normal goat serum, the sections were incubated in serum and rabbit anti‐Iba1 (1 : 1000 dilution, Wako Inc., Osaka, Japan; RRID:AB_2665520) antibody for 1 to 2 h followed 18 to 24 h at 5°C. Sections were then washed 3X for 10 min and incubated in secondary antibody for 1–2 h. In cases where only single labeling for Iba1 was performed, the signal was amplified, using the avidin and biotinylated horseradish peroxidase macromolecular complex (ABC, Vector Laboratories, Burlingame, CA, USA) and visualized with 0.4 mg/mL of DAB in 50 mM Tris buffer. When dual labeling of sections with rat endothelial cell antigen 1 (RECA‐1) and IBA1 antibodies was performed, the sections were incubated in a streptavidin‐TRITC (1 : 250, Jackson Immunoresearch, Philadelphia, PA, USA; RRID:AB_2337237) for 2 h at 23°C . The sections were then washed 3X with buffer and mounted on slides. Sections on the slides were dried, xylene‐cleared, and cover slipped with DPX.
Immunolabeling of RECA‐1 was accomplished using methods similar to those used for Iba1 immunoreactivity. After the 0.3% H2O2 treatment, the sections were treated with 10% normal horse serum (GIBCO, Rockville, MD, USA) in NaPB for 30 min prior to incubation in a mouse anti‐rat RECA‐1 antibody solution (Abcam; Cambridge, MA, USA; RRID:AB_296613) at 1 : 1000 dilution for 2 h at 23°C and then at 4°C overnight. The sections were then washed 3X in NaPB and 0.5% Triton‐X‐100 in PBS and incubated in secondary biotinylated donkey anti‐mouse IgG (1 : 200; Jackson Immunoresearch; RRID:AB_2307438) for 2 h on a shaker at 23°C. The signal was amplified using the avidin and biotinylated horseradish peroxidase macromolecular complex (ABC, Vector Laboratories) and visualized with 0.4 mg/mL of DAB in 50 mM Tris buffer. Sections were then washed thoroughly in NaPB four times to remove excess antibody, rinsed in distilled water, dried, cleared in xylene, and cover slipped with DPX mounting medium.
Microscopy visualization
Histological examination of the slide‐mounted tissue was examined, using a Nikon epifluorescent microscope 80i (Nikon Instruments Inc., Melville, NY, USA) and an X‐Cite 120 LED light source (Excelitas Technologies®, Waltham, MA, USA). The following sets of filters were used for visualizing fluorescent labeling in conjunction with the histochemical or immuno‐histological labeling: TRITC (for Iba1) excitation of 533–553 nm and an emission of 573–613 nm; FITC (for FJc) excitation of 464.5–499.5 and an emission of 516–556 nm. Visualization of DAB‐labeled sections was done with incandescent illumination. All photomicrographs of the images were taken using a Nikon DS‐Ri1 using the NIS software from Nikon USA.
Quantification of microglial morphology/activation in the parietal cortex
Similar to the approach of counting the FJc labeled neurons in the parietal cortex, we examined both hemispheres from three sections of the parietal cortex to characterize microglia in Saline, CORT, METH, METH+Veh, and METH+CORT animals. Photomicrographs (4X) of each hemisphere were taken to capture microglia in cortical layers II through Layer V of the vibrissae somatosensory S1 barrel fields and a portion of the more ventral S2 barrel fields. NIS Elements Software (Nikon) was used to detect and measure microglia within the analyzed region of interest (ROI). The ROI was standardized and kept constant (see Figure S6) in Results for more details of ROI and morphology of objects captured). The intensity range selection points for detecting the microglia were set to eliminate (as much as possible) as many of the minor processes not connected to somas. However, many of the darkly stained isolated distal process were still captured. Setting intensity capture parameters to exclude all of the darkly stained distal processes was not an option since it resulted in exclusion of smaller or more lightly stained microglia soma and adjoining processes. The sphericity parameter was not utilized during object selection since many of the microglia were not ovate or spheroid in nature. The unconnected distal processes, which were smaller with an area of < 16 μm2, were removed through data filtering (see Statistical analysis section).
Statistical analysis
Blood glucose and body temperature
In this study, blood glucose levels and body temperature measurements were analyzed similarly. First, the six treatment groups formed by the two crossed factors Glucose Modulator (levels: saline, glucose, corticosterone) and METH (levels: no‐methamphetamine, methamphetamine) were analyzed with a repeated measures analysis of variance (anova) model. The anova model included the fixed main effects of Glucose Modulator and METH and the interaction term Glucose Modulator × METH. The factor Time (blood glucose levels: 0, 1, 3, 5, 7, 9 h; body temperature levels: 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 h) was considered a within‐subjects factor in the model. Mauchly's sphericity test was used to evaluate whether there was significant departure from sphericity. If the data violated this assumption, the Greenhouse‐Geisser epsilon corrected p‐values were utilized to evaluate significance.
In addition, a separate repeated measures anova model was used to assess the effect of METH+CORT versus METH+Veh over time. This anova model included the fixed main effect of Group (levels: METH+CORT, METH+Veh) and the within‐subjects factor Time. As stated previously, the Greenhouse‐Geisser epsilon correction was applied when data violated the assumption of sphericity (as determined by Mauchly's sphericity test). Differences between METH+CORT and METH+Veh were evaluated at each time point.
All analyses were carried out in SAS (version 9.3; SAS Institute, Inc., Cary, NC, USA) and evaluated at a significance level of 0.05.
FJc+ cells in the parietal cortex and thalamus
The intraclass coefficient (ICC) (Shrout and Fleiss 1979) was used to assess inter‐rater reliability for the numbers of FJc+ cells counted by all three independent investigators. A two‐way random effects model (based on the absolute agreement definition and average measures) was used to estimate the ICC, which was computed, using the irr package in R (http://r-project.org). Measures of the ICC exceeded 0.95 for all three regions of the brain. Thus, counts were averaged across investigators. The resulting numbers of FJc+ cells were analyzed using a linear mixed effects model, where the effect due to treatment was modeled as a fixed effect and the effect due to animal was a nested random effect. Furthermore, pairwise comparisons were performed for the following tests: METH+Veh versus METH, METH+Glucose versus METH, METH+CORT versus METH, METH+Glucose versus METH+CORT, and METH+CORT versus METH+Veh. Adjusted p < 0.05 were deemed statistically significant. Descriptive statistics for the number of FJc+ cells for all seven treatment groups are presented as mean ± standard error of the mean (SEM) in Table 2.
Table 2.
Effects of exogenous glucose and corticosterone on METH‐induced FJc+ labeling/neurodegeneration
| Treatment group | Parietal cortex | PV nucleus thalamus | VM, VL & IL nuclei thalamus | Sample sizea |
|---|---|---|---|---|
| Saline | 0.39 ± 0.13 | 0.21 ± 0.07 | 0.29 ± 0.09 | 5 |
| Glucose | 0.18 ± 0.06 | 0.09 ± 0.06 | 0.01 ± 0.01 | 5 |
| CORT | 0.25 ± 0.09 | 0.10 ± 0.07 | 0.02 ± 0.01 | 5 |
| METH | 6.40 ± 0.62 | 5.03 ± 0.72 | 53.31 ± 11.18 | 6 |
| METH+glucose | 8.22 ± 1.01 | 16.11 ± 4.66 | 85.00 ± 24.03 | 6 |
| METH+CORT | 18.14 ± 1.39b | 6.54 ± 1.18 | 69.59 ± 13.26 | 7 |
| METH+Veh | 8.58 ± 1.25 | 2.67 ± 0.39 | 14.87 ± 2.88 | 6 |
Mean ± SEM of FJc+/section/hemisphere are shown.
The number of animals per group evaluated is less than expected by 2–3 animals per treatment group from the mortality loss shown in Table 1. This is because animals that did not reach 41.7°C body temperatures were excluded from analysis.
Indicates neurodegeneration was significantly different than each of the other METH groups.
Microglia morphology in the parietal cortex
The Sholl analysis (Sholl 1956) as it has been applied recently [as an example (Kongsui et al. 2014)] is a more complex and sophisticate type of analysis looking at microglia process morphology and length as well as volume. It has the capability of differentiating different types of microglia even in resting or non‐activated states. However, in our experiments, activated microglia were clearly visible due to the obvious increase in their size, and we feel the method we have implemented adequately detects the activated microglia in an unbiased but simplified method. Microglia area (soma + adjoining proximal processes) greater than 50 μm2 were considered activated; measurements less than 50 μm2 were removed from analysis. We evaluated the number of activated microglia based on several area size categories. Categories were largely defined by equally spaced intervals of size 100 μm2; however, there was one exception. The last interval was categorized as (550, 1443] μm2 to avoid having many intervals with small observed counts. Data from each animal was summarized by counting the number of microglia in each size category. Counts were then averaged across the six replicates (each hemisphere from three sections of the parietal cortex) for each animal. The mean number of microglia (±SEM) per treatment group is summarized in a bar graph (see Results). For each size category, significant difference was determined by the Kruskal–Wallis test followed by Dunn's pairwise multiple comparisons method with Bonferroni adjustment. However, p‐values < 0.05 were considered statistically significant.
Results
METH effects on blood glucose levels
Time‐specific estimates of mean blood glucose levels and SEM are presented in Fig. 1. Analysis of the six treatment groups resulting from the two crossed factors Glucose Modulator and METH (METH+Veh was not included in this portion of the analysis) was adjusted for the Greenhouse‐Geisser epsilon correction since Mauchly's test indicated a violation of the sphericity assumption (p < 0.0001). Results from the repeated measures anova indicate that: (i) blood glucose levels changed with time (p < 0.0001); (ii) change in mean blood glucose levels over time did not depend on Glucose Modulator levels (i.e. Saline, Glucose, and CORT) (p = 0.4962); (iii) the change in mean blood glucose level over time was affected by METH levels (i.e. no‐METH and METH) (p < 0.0001); and (iv) the change in mean blood glucose levels over time did not vary by the interaction between Glucose Modulator and METH levels (p = 0.2581). Thus, the data demonstrates that mean blood glucose levels change over time and are significantly affected by METH. Furthermore, trend analysis indicated that there was a significant quadratic trend over time (p < 0.0001) and a significant interaction with the presence of methamphetamine (p = 0.0004). Figure S1 presents mean blood glucose levels for the METH treatment groups versus the no‐METH treatment groups over time. Following anova, analysis was performed at each time point to assess significant difference between the METH and no‐METH treatment groups. Using a significance level of 0.05, significance was attained at time points 1, 3, 5, and 9 h.
Figure 1.
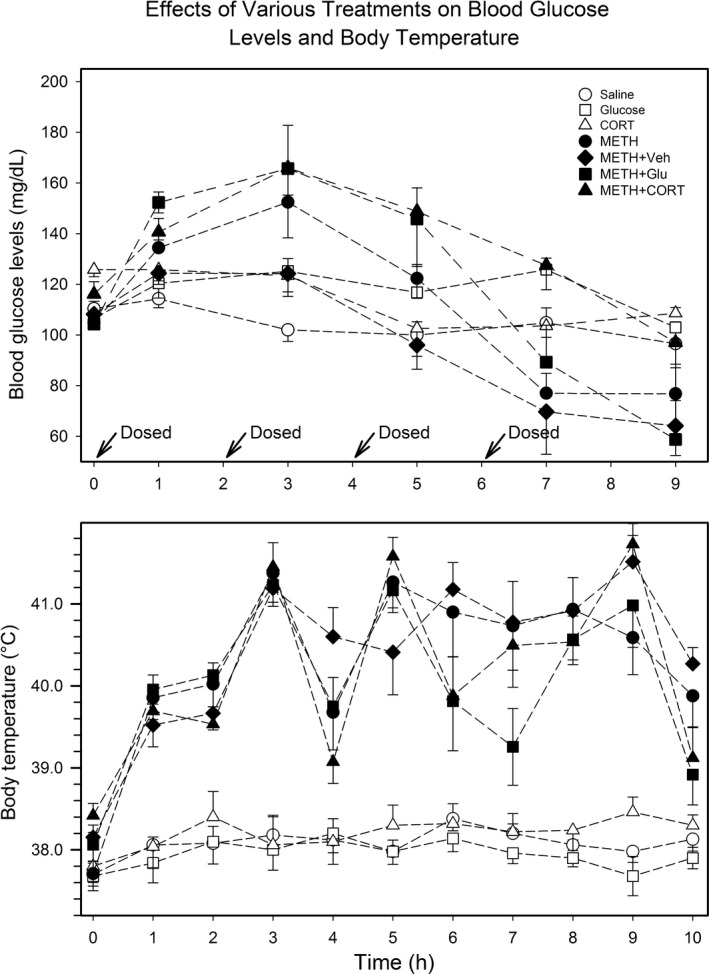
Blood glucose levels and body temperature at 3 h post‐ dosing. The blood glucose levels (top panel) and body temperature (bottom panel) are shown for all seven treatment groups. Blood glucose was determined 1 h after each of injection of METH and also at 3 h after the last set injection from blood obtained from the hindfoot toe pad. Upper colonic temperature was determined every hour.
In our subsequent analysis of blood glucose levels comparing METH+CORT and METH+Veh over time, Mauchly's test indicated a significant departure from sphericity (p < 0.0001). Thus, the Greenhouse‐Geisser corrected results of the repeated measures anova indicate:
(i) blood glucose levels changed as a function of time (p < 0.0001) and (ii) the change in mean blood glucose across time did not depend on Group levels (i.e. METH+CORT and METH+Veh) (p = 0.0938). Trend analysis indicated a significant quadratic trend over time (p < 0.0001) that did not vary by Group level (p = 0.0595). Subsequent analyses were carried out to compare METH+CORT and METH+Veh at each time point; there was a significant difference at 5 h and 7 h (Figure S2).
METH effects on body temperature
Results from the repeated measures anova adjusted for the Greenhouse‐Geisser epsilon correction indicated that: (i) mean body temperature changed with time (p < 0.0001); (ii) the change in mean body temperature over time did not depend on Glucose Modulator levels (i.e. Saline, Glucose, and CORT) (p = 0.5673); (iii) the change in mean body temperature over time was affected by METH levels (i.e., no‐METH and METH) (p < 0.0001); and (iv) the METH effect on the change in mean body temperature over time did not vary significantly by Glucose Modulator level (p = 0.8835). Trend analysis demonstrated a significant quartic trend over time (p < 0.0001), which varied by the presence of METH (p = 0.0003). Mean body temperature (and SEM) is plotted in Fig. 1 . Lastly, subsequent analysis indicated that mean body temperature for the METH treatment groups was significantly different than the no‐METH treatment groups at all 11 time points (Figure S3).
The Greenhouse‐Geisser epsilon corrected results of the anova model comparing METH+CORT to METH+Veh showed that the change in body temperature over time was significant (p < 0.0001) and differed by Group (p = 0.0100). However, the significant quartic trend in the data (p < 0.0001) did not differ by treatment group (p = 0.1626). There was a significant difference between METH+CORT and METH+Veh at time points 4, 5, 6, and 10 h (Figure S4). However, there was no consistent pattern of expression over time.
METH effects on neurodegeneration
A summary of the number of FJc+ neurons for all seven treatment groups is provided in Table 2. The raw data amassed from all three investigators is presented in File S1. The number of animals per group is less than the numbers presented earlier in the paper due to mortality loss (as indicated in Table 1). We also excluded a few animals with body temperatures that did not reach 41.7°C more than one time during METH exposure. These animals would be expected to have significantly lower levels of neurodegeneration and their inclusion may have biased statistical analysis of the treatment groups. There were no significant differences in the average number of times an animal had a body temperatures above 41.7°C between the four METH groups (METH: 3.6 ± 0.5; METH+Veh: 3.7 ± 0.5; METH+Glucose: 3.5 ± 0.6; METH+CORT: 3.5 ± 0.3).
Representative images of histological sections labeled with FJc for animals in the METH, METH+Glucose, and METH+CORT groups are presented in Figure S5. There was a significant difference in neurodegeneration in the parietal cortex when comparing METH+CORT to METH, METH+Glucose, and METH+Veh (Table 2). There were no differences in neurodegeneration observed in the CORT or Glucose groups when compared to the Saline group. There was a trend toward increased neurodegeneration in the thalamus when comparing METH+CORT to METH+Veh and METH+Glucose to METH. However, none of the comparisons were significant in the two thalamic regions because of the considerable amount of variability in neurodegeneration. This was not due to differences in counting between the three investigators but to the wide variability in the neurodegeneration present in the thalamus compared to the parietal cortex.
Microglial‐types associated with vasculature changes after METH
Activated microglia in the parietal cortex (layer III and boundary of layer IV) detected by Iba1‐DAB immunostaining were observed in the same regions of FJc labeling in all four METH treated groups (Fig. 2). Microglia in the parietal cortex of saline‐, glucose‐, and CORT‐treated animals are shown in panels a, b, and c, respectively. In panels d–f, the darker arrows identify activated microglia that are often associated with degenerating neurons (Streit 1990, 2002) and neurotoxicity produced by amphetamines (Bowyer et al. 1998). Such activated microglia were seen with METH, METH+Glucose, and METH+CORT (panels d, e and f, respectively). As well, there are some activated microglia in cortical layer II all the way to boundary of layer I, which are identified with the lighter gray arrows.
Figure 2.
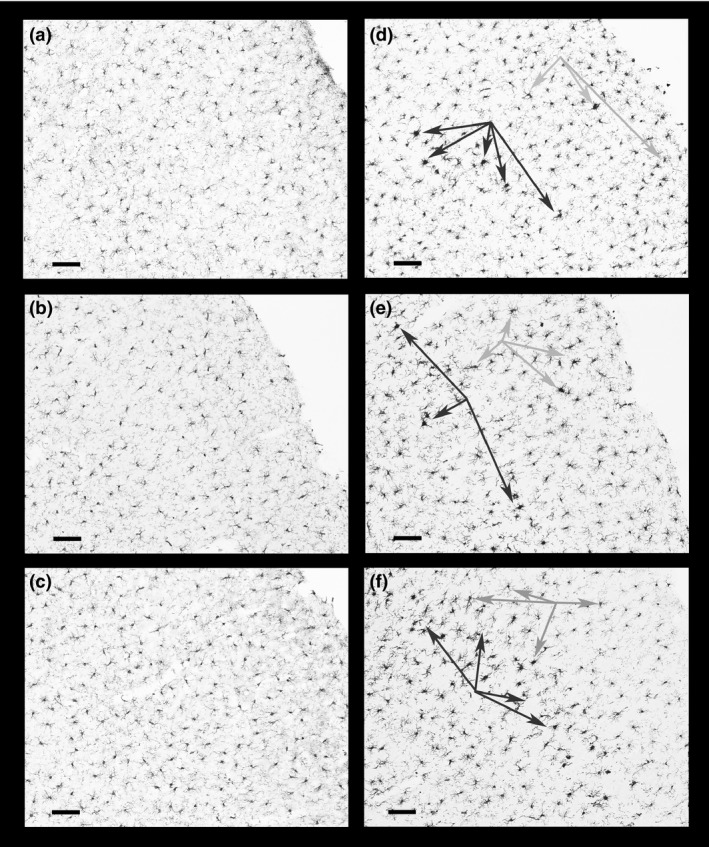
Microglial activation in the parietal cortex. Activated microglia often associated with degenerating neurons, located in the barrel fields of primary somatosensory cortex layer III, are identified by the dark arrows in panels (d) (METH), (e) (METH+Glucose) and (f) (METH+CORT) and can be compared to the absence of activated microglia in panels (a) (Saline) and (b) (Glucose). There is a hint of slight microglial activation in layer III of the CORT group (panel c). Fewer numbers of such activated microglia can also be seen in cortical layers II and IV as indicated by the gray arrows. Magnification bar (bottom left on panels) = 100 μm.
Figure 3 shows the co‐localization of vasculature with activated microglia in the barrel fields of the primary somatosensory cortex. The photomicrographs in this figure are formed by merging the visible light images with the fluorescent image of sections dual labeled with DAB‐Iba1 antibody and TRITC‐RECA‐1‐antibody. In the upper two panels, the ruby labeled microglia in layers I through V of the barrel fields of the parietal cortex can be seen at two magnifications in a control animal (FG307). Microglia in the same region of an animal in the CORT group (FG308) appear somewhat larger but no clear activation is present. However, in some of the animals in the three METH groups, activated microglia could be seen in most layers of the cortex, not just layers III and IV where neurodegeneration occurs. In the bottom two panels, activated microglia are shown in a METH+Glucose animal (FG314). At the higher magnification, activated microglia surrounding the vasculature are seen from the bottom of layer I through II, which are cortical where there is little or no FJc+/degenerating neurons. The instances of such microglia vasculature associations were less in the METH and METH+Veh groups and prominent in the METH+CORT animals as seen in panels b through f in Fig. 4 .
Figure 3.
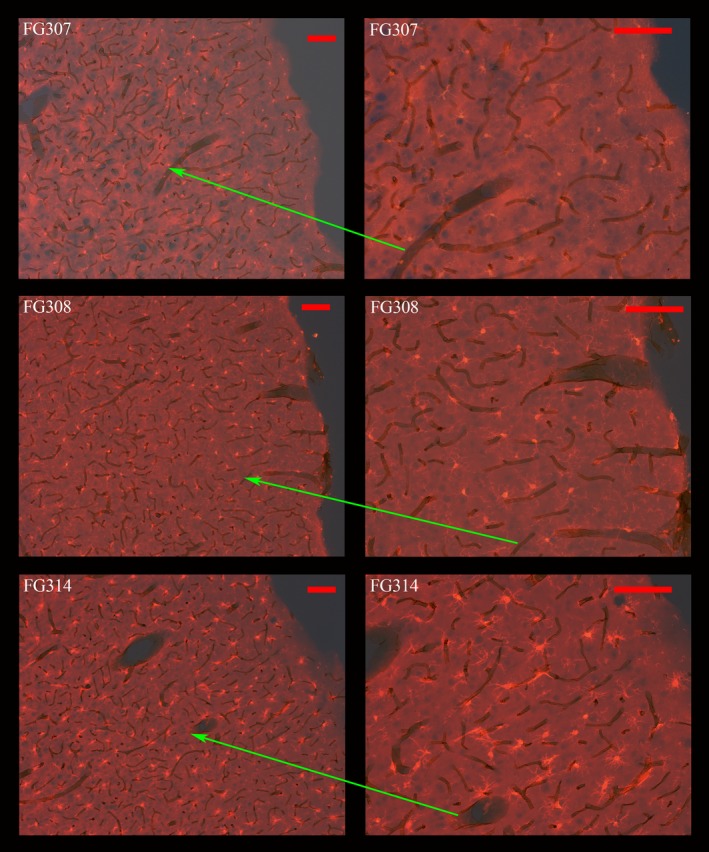
Co‐localization of activated microglia and vasculature in the parietal cortex. The photomicrographs panels were formed by merging the visible light images with the fluorescent images of sections dual labeled with 3,3'‐diaminobenzidine (DAB)‐RECA1 antibody and TRITC‐Iba1‐antibody. Bright red TRITC labeled microglia in layers I through III can be seen in a Saline animal (FG307) at two magnifications (top two panels). In the middle two panels, slightly larger microglia can be seen in the same cortical regions in an animal from the CORT group (FG308). Finally, in the bottom two panels, activated microglia in an animal from the METH+Glucose group (FG314) can be seen from cortical layer III all the way into the lower portions of layer I. The ends of the red arrows identify the identical region at higher magnification. Magnification bar = 100 μm.
Figure 4.
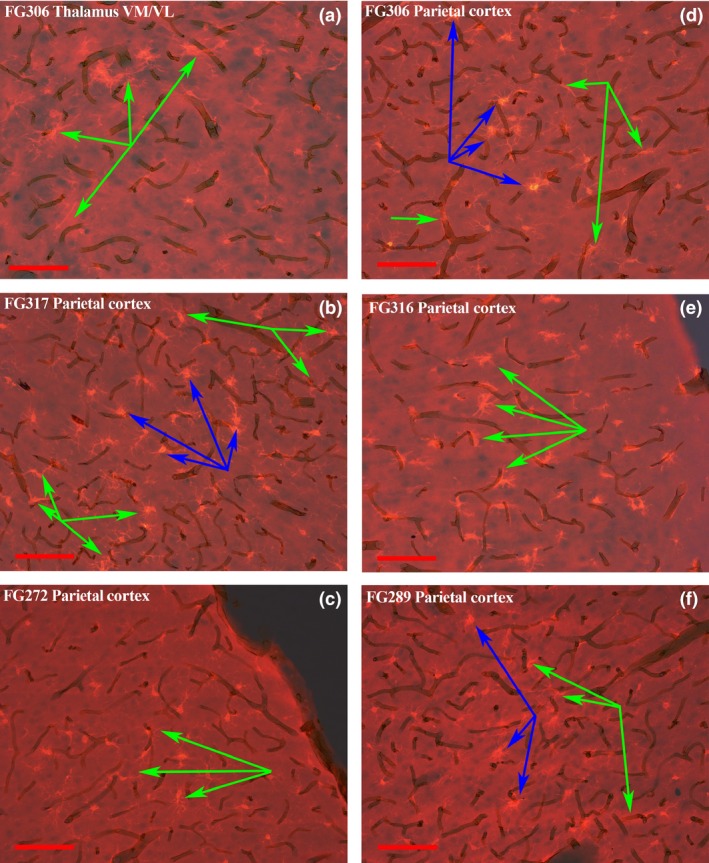
Co‐localization of activated microglia and vasculature in the parietal cortex after METH+CORT. The photomicrographs panels were formed in the same manner as those in Fig. 3. Activated microglia associated with vasculature, identified by the green arrows, are present at the border of the ventrolateral and ventromedial thalamus in panel (a). This panel was from a micrograph of an animal from the METH+CORT group, as are all the other panels in this figure. The micrographs in panels (b) through (f) show activated microglia, identified by the blue arrows, in association with vasculature in layers II and IV of the parietal cortex in regions where there is minimal or no FJc labeling/neurodegeneration. Magnification bar = 100 μm.
A bar graph of the mean number of microglia for each treatment group is presented in Fig. 5. The METH+Glucose group was not evaluated because the neurodegeneration was no different than the METH and METH+Veh groups; also, neurodegeneration in the METH+Glucose group was significantly less than the METH+CORT group. A general idea of the various sizes of microglia areas captured can be seen in Figure S6. The mean number of microglia was not significantly different between treatment groups for the two smallest size categories: (50,150] and (150,250] μm2. In contrast, mean number of microglia sized 250–350 μm2 was significantly different between METH and Saline and METH+CORT and Saline. Similarly, the mean number of microglia for the groups METH and METH+CORT were significantly different from both Saline and CORT for microglia sized 350–450, 450–550, and 550–1433 μm2 (with the exception of the pairwise comparison between METH+CORT and Saline, which was not significant (p = 0.06) in the last category).
Figure 5.
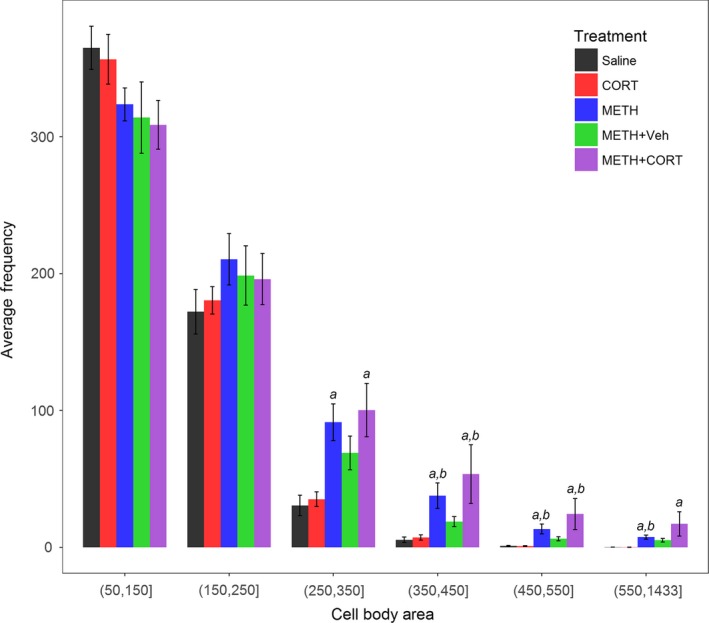
Bar plot of microglia in the parietal cortex after treatment. Average number and SEM of microglia categorized by microglial area (soma + adjoining proximal processes) in Saline, CORT, METH, METH+Veh and METH+CORT treatment groups. anumbers significantly greater than Saline at p < 0.05 as adjusted for multiple comparisons. bnumbers significantly greater than CORT at p < 0.05 as adjusted for multiple comparisons.
Microglia areas above 385 μm2 would normally be classified as ‘activated’ by visual inspection. There was, on average, five and eight microglia above this level for Saline and CORT, respectively. However, there was, on average, 95, 58, and 30 microglia of this magnitude in METH+CORT, METH, and METH+Veh, respectively. However, from the more classical visual inspection methods, the higher numbers of microglia above 385 μm2 in METH and METH+Veh, located primarily in cortical layers III and IV where neurodegeneration occurs, would be classified as regions of microglial activation. It should be noted that in the METH+CORT group, on average, approximately 10% of the counts in the (550,1433] category exceeded 1000 μm2. The counts with areas microglia areas > 1000 μm2 are likely not single activated microglia but two that merge into one due to their close proximity that could not be distinguished as separate by NIS or by visual inspection. Although the more sophisticated methods of Sholl methods were not used to analyze microglial activation, they were not imperative for analysis showing microglial activation of this great a magnitude.
There is also clear immunohistological evidence that METH exposure caused vascular changes and thinning in areas of the parietal cortex, particularly in the METH+CORT group at 3 days after treatment. The thinning of vasculature in the parietal cortex, as indicated by the decreased DAB‐collagen IV immunolabeling, was particularly evident in the METH+CORT group (Fig. 6). As well, there was a loss of DAB‐Glut‐1 immunolabeling of vasculature in the same region. However, there was no evidence of prominent loss of vasculature or obvious presence of degenerating vasculature in this region.
Figure 6.
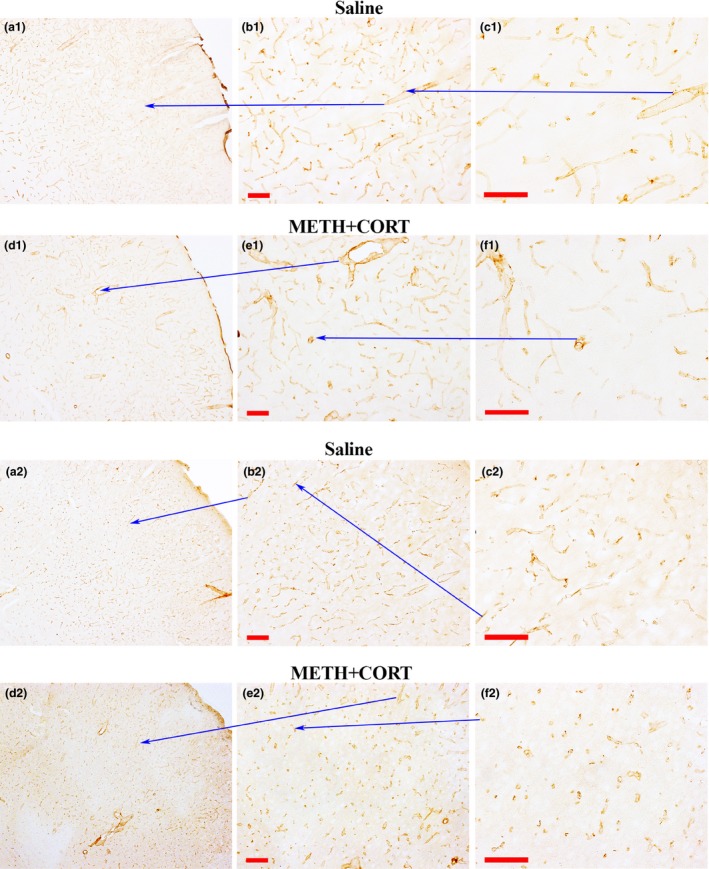
Collagen IV and Glut‐1 depletions in the vasculature of the parietal cortex after METH+CORT. Collagen IV immune‐labeling in the S1/S2 somatosensory barrel fields of the cortex are shown at three different magnifications (a1, b1 and c1) at 3 days post‐saline. METH+CORT cause decreased labeling in this region (d1, e1, and f1) as shown at the same magnification levels. Likewise, compared to Saline at several magnification (a2, b2, and c2), the METH+CORT exposure resulted in some depletion of the Glut‐1 transporter (d2, e2, and f2) and some moderate changes in vascular distribution in the same region of the parietal cortex. Magnification bar = 100 μm.
Discussion
The current study found that normally: (i) blood glucose levels are initially increased at the start of METH exposure but decrease below normal at the very end of exposure; (ii) CORT pretreatment exacerbates the early increase in blood glucose by METH and prevents the late drop in levels; and (iii) microglia are activated in the parietal cortex and are associated with vasculature outside of the regions of neurodegeneration. Thus, these results addressed the research questions/objectives of this study. However, they raise many questions about the mechanisms that are involved in the physiological and vascular effects produced by exogenous CORT and glucose as well as the interactions of these treatments with the effects of high‐dose METH exposure on neurotoxic and neuroimmune outcomes.
A previous study showed that CORT pretreatment can enhance striatal cytokine levels due to METH exposure and also can exacerbate dopamine depletions/terminal neurotoxicity in the striatum (Kelly et al. 2012). The present study shows that CORT pretreatment, and possibly exogenous glucose, exacerbate METH neurodegeneration and neuroinflammation in laboratory animals. These results have potential translational relevance with respect to METH abusers. Stress has been demonstrated to produce a physiological CORT response that is capable of modulating the response to METH (Halpin et al. 2014) and neuroinflammatory (Loram et al., 2011, Gibb et al., 2013) exposures. Studies in humans have shown cross sensitization of dopamine release between stress and AMPH or METH use (Yui et al., 2000; Booij et al., 2016). Thus, it is possible that individuals undergoing stress prior to the start of METH abuse may be more likely to suffer adverse neurotoxic or neuroimmune responses.
METH would be expected to raise glucose levels in the blood through release of norepinephrine from liver noradrenergic innervation and possibly through effects on the adrenal gland (Exton and Park 1968; Sugano et al. 1980). However, increased tissue metabolism of glucose by METH, particularly in brain, would tend to decrease blood glucose levels (Abreu et al. 1949; Berntman et al. 1978). The early increases we observed in blood glucose produced by METH alone may be due to liver increases in gluconeogenesis while the prominent drops in glucose levels toward the end of METH exposure, when severe hyperthermia occurs, are likely due to the documented depletions of liver glycogen stores (Levi et al. 2013). As such, varying degrees of liver damage can be produced by AMPH exposure (Halpin and Yamamoto 2012; Levi et al. 2013). Environmentally induced heat stroke in the absence of amphetamines also can lead to glycogen depletion and liver damage in rodents (Levi et al. 2013) but, interestingly, this does not lead to the large decreases in blood glucose levels [(Bowyer et al. 2014); Bowyer, unpublished data] that are seen with AMPH (see Fig. 1). CORT was able to block the prominent drop in blood glucose levels produced by METH in the current study. It is not clear why this occurred, but because levels of glucose remained higher than in the other METH groups, it could partially explain the observed increase in neurotoxicity.
There are reports showing that METH abuse has the potential to interact adversely with elevated blood glucose and diabetes (Treweek et al. 2009) and chronic high blood glucose levels are associated with vascular damage evidenced by BBB permeability (Huber et al. 2006; Hawkins et al. 2007; VanGilder et al. 2009). While acute rescue from hyperglycemia does not reverse vascular damage (Hawkins et al. 2007), chronic rescue with exogenous insulin is reported to reduce BBB permeability (Huber et al. 2006). Additionally, antioxidant/anti‐inflammatory treatments can rescue vascular damage without affecting blood glucose (VanGilder et al. 2009).
One mechanism by which high glucose can damage vasculature is through the generation of Maillard reaction products and other subsequent chemical reactions taking place in vivo, outcomes known to contribute to advanced glycation end products (AGEs) (Brownlee et al. 1988; Treweek et al. 2009). Such reactions leading to AGEs have been implicated in disease states, in particular diabetes (Monnier and Cerami 1981; Monnier et al. 1984; Treweek et al. 2009). The amine functionality of METH and AMPH allow them to take part in the Maillard reaction. The hyperthermia produced during acute METH or AMPH exposure in laboratory animals is well documented to be necessary for vascular damage, neurotoxicity, and a robust neuroimmune response (Bowyer et al. 2014). Thus, the increases in blood glucose, severe hyperthermia, and hypertension produced by METH and the potentiation of glucose levels by CORT seen in the present experiments indicate that AGEs may play a role in vascular damage. Although glycation products may be unaffected in the face of evidence of striatal damage from known dopaminergic neurotoxicants such as MPTP (Viana et al. 2016), this is not necessarily the case for METH toxicity which involves pronounced hyperthermia and hypertension (unlike MPTP). Also, the vascular damage and neurodegeneration resulting in the parietal cortex and thalamus due to METH exposure may not involve exactly the same mechanisms as neurodegeneration in the other brain regions in which MPTP produces neurotoxicity. Thus far, we have not succeeded in our attempts to histologically identify increases in carboxymethyl lysine linked proteins present on the luminal side of vascular endothelium or the presence of High mobility group B1 (HMGB1) in the nuclei of vascular endothelium after acute METH exposures. These would be expected findings based on our vascular data, but actual changes may have been below the detection range for the antibodies we used.
Elevated blood glucose has been reported to activate the inflammasome in brain and vasculature, but the mechanisms are still not fully understood (Lenart et al. 2016; Song et al. 2017). ROS genes linked to inflammatory responses, such as Gpx1, Hmox1, and Sod1, increase from 2‐ to 20‐fold after AMPH (Bowyer et al. 2013). However, mRNA expression of the inflammasome genes Aim2, Nlrc4, Nlrp1, and Nlrp3 are reduced (≤ 50%) in the blood and brain of AMPH exposed rats at 3 h and 1 days post‐exposure, which is a time they might be expected to show elevation. While AMPH exposure alone may not result in expression changes in the inflammasome, several studies have found that prior exposure to glucocorticoids or stress activates this signaling cascade (Frank et al., 2014; Frank et al., 2015; Weber et al., 2015; Zhang et al., 2015; Sobesky et al., 2016). Further work is necessary to evaluate whether activation of these inflammasome genes occurs when CORT or glucose levels are elevated in combination with METH or AMPH exposure.
Nonetheless, there is clear evidence from our previous studies examining gene expression in the parietal cortex, and even more so in the meninges and cerebral surface vasculature, that exposures to AMPH that produce multiple acute episodes of severe hyperthermia result in pronounced ROS production, vascular dysregulation, and damage (Thomas et al. 2009, 2010). In a recent study, we found that METH results in pronounced microglia activation directed at vasculature, even in areas of the anterior medial dorsal hippocampus and lateral septum where there was no evidence of neurodegeneration or nerve terminal damage (Bowyer et al. 2016). We take these findings as further evidence of high glucose levels correlating with increased vascular damage.
A possible explanation for how CORT alone may produce or exacerbate METH‐induced microglial responses to cerebral vasculature is through increases in C‐C motif chemokine ligand 2 (Ccl2), Ccl7, and Ccl20. Support vasculature for the brain present in the meninges and cerebral surface vasculature as well as the choroid plexus have significant levels of Ccl2 mRNA under control conditions, but these levels are increased 6‐fold by AMPH exposures that induce severe hyperthermia (Thomas et al. 2009; Bowyer et al. 2013). The levels of these three cytokines are much lower in striatum and cortex under control conditions relative to choroid plexus and the meninges and cerebral surface vasculature, presumably due to the much lower levels of vasculature. However, the levels in the striatum (15‐fold) and parietal cortex (50‐fold) are also increased greatly by AMPH exposure. Pretreatment with CORT just prior to METH can increase Ccl2 levels in the range of 60–72 fold (in the cortex and striatum, respectively) over that seen with METH alone in a mouse model of METH toxicity (Kelly et al. 2012). We postulate that Ccl2, Ccl7, and Ccl20 may be the signals directing the microglia to respond to damaged vasculature in a manner similar to how these cytokines may be involved in damage to the kidney and pulmonary function related to diabetes (de Boer et al. 2007; Ichinose et al. 2007). The highest levels of Ccl2, Ccl7, and Ccl20 mRNA expression in human are in the smooth muscle related to vasculature (BioGPS website), which indirectly implies they may have a role in vascular endothelium repair.
It has been difficult to resolve to what extent vascular damage versus excitotoxicity plays in the sporadic neurodegeneration that occurs after exposure to amphetamines. Neurotoxic exposures to either AMPH or METH that result in severe hyperthermia produce the most damage and disruption of vasculature in the thalamus along the border between the ventral intralaminar, ventromedial and ventromedial thalamic nuclei (Bowyer et al. 1998, 2016). In this region, FJc labeling of degenerating neurons is only apparent at 48 h or more after AMPH or METH exposure, while the FJc+ neurons in the parietal cortex appear within 24 h. Thus, there is indirect evidence in the thalamus that vascular damage precedes neuronal death.
Conclusions
Overall, it is clear that blood glucose levels are initially elevated significantly by METH when pronounced hyperthermia accompanies exposure and that CORT pretreatment, and possibly exogenous glucose, during METH exposure potentiate hyperglycemia. Paradoxically, METH exposure often produces a precipitous drop in blood glucose at the end of exposure when the signs of neurotoxic effects are just becoming apparent. This drop may precede death if severe enough but it is not necessarily predictive of overall lethality. Episodes of hypoglycemia during METH exposure are not necessary for neurotoxicity since CORT pretreatment 1 days prior to METH actually increases neurodegeneration in specific brain regions but prevents hypoglycemia late in METH exposure. Exogenous glucose or CORT significantly increased METH lethality, and CORT significantly increased neurodegeneration produced by METH. CORT pretreatment alone may have minor to moderate adverse effects on vascular integrity, however, in combination with METH the disruption is clearly observed. The potentiation of neurodegeneration and vascular leakage by CORT during METH exposure is not due to higher body temperatures and may not be due solely to the elevation of blood glucose.
Disclaimer
The authors are solely employed by the U.S. government and are not affiliated or employed with any company or have any financial interests relating to the research presented or its implications. The contents of this manuscript do not necessarily reflect the views and policies of the U.S. Food and Drug Administration, nor does the mention of trade names or commercial products constitute endorsement or a recommendation for use. In addition, the findings In addition, the findings and conclusions in this report are those of the author(s) and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Supporting information
File S1. Raw counts of FJc‐ labeled neurons for each evaluator that that were used in determining treatment effects on degeneration.
Figure S1. METH versus noMETH blood glucose levels.
Figure S2. METH+CORT versus METH+Veh blood glucose levels.
Figure S3. METH versus noMETH body temperature levels.
Figure S4. METH+CORT versus METH+Veh body temperature levels.
Figure S5. FJc‐labeling of degenerating neurons in three of the METH treatment groups.
Figure S6. “Screen Shot” of Nikon NIS Display of an image of the parietal and setting an parameters used for image analysis.
Acknowledgments and conflict of interest disclosure
This study was supported by FDA protocol E7519.
All experiments were conducted in compliance with the ARRIVE guidelines. The authors have no conflict of interest to declare.
Cover Image for this issue: doi. 10.1111/jnc.13819.
The copyright line for this article was changed on 14 December 2017 after original online publication.
References
- Abdul Muneer P. M., Alikunju S., Szlachetka A. M. and Haorah J. (2011a) Methamphetamine inhibits the glucose uptake by human neurons and astrocytes: stabilization by acetyl‐L‐carnitine. PloS one 6, e19258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul Muneer P. M., Alikunju S., Szlachetka A. M., Murrin L. C. and Haorah J. (2011b) Impairment of brain endothelial glucose transporter by methamphetamine causes blood‐brain barrier dysfunction. Mol Neurodegener 6, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu B. E., Liddle G. W. et al (1949) Influence of amphetamine sulfate on cerebral metabolism and blood flow in man. J. Am. Pharm. Assoc. Am. Pharm. Assoc. 38, 186–188. [DOI] [PubMed] [Google Scholar]
- Berntman L., Carlsson C., Hagerdal M. and Siesjo B. K. (1978) Circulatory and metabolic effects in the brain induced by amphetamine sulphate. Acta Physiol. Scand. 102, 310–323. [DOI] [PubMed] [Google Scholar]
- Block M. L., Zecca L. and Hong J. S. (2007) Microglia‐mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8, 57–69. [DOI] [PubMed] [Google Scholar]
- de Boer W. I., Alagappan V. K. and Sharma H. S. (2007) Molecular mechanisms in chronic obstructive pulmonary disease: potential targets for therapy. Cell Biochem. Biophys. 47, 131–148. [DOI] [PubMed] [Google Scholar]
- Booij L., Welfeld K., Leyton M., et al. (2016) Dopamine crosssensitization between psychostimulant drugs and stress in healthy male volunteers. Transl Psychiatry 6, e740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer J. F. and Ali S. (2006) High doses of methamphetamine that cause disruption of the blood‐brain barrier in limbic regions produce extensive neuronal degeneration in mouse hippocampus. Synapse 60, 521–532. [DOI] [PubMed] [Google Scholar]
- Bowyer J. F. and Hanig J. P. (2014) Amphetamine‐ and methamphetamine‐induced hyperthermia: implications of the effects produced in brain vasculature and peripheral organs to forebrain neurotoxicity. Temperature 1, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer J. F. and Holson R. R. (1995) Methamphetamine and amphetamine neurotoxicity: characteristics, interactions with body temperature and possible mechanisms, in Handbook of Neurotoxicology, vol. 2, (Chang L. W. and Dyer R. S., eds.), pp. 845–870. Marcel Dekker Inc, New York. [Google Scholar]
- Bowyer J. F., Davies D. L., Schmued L., Broening H. W., Newport G. D., Slikker W., Jr and Holson R. R. (1994) Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J. Pharmacol. Exp. Ther. 268, 1571–1580. [PubMed] [Google Scholar]
- Bowyer J. F., Peterson S. L., Rountree R. L., Tor‐Agbidye J. and Wang G. J. (1998) Neuronal degeneration in rat forebrain resulting from D‐amphetamine‐induced convulsions is dependent on seizure severity and age. Brain Res. 809, 77–90. [DOI] [PubMed] [Google Scholar]
- Bowyer J. F., Thomas M. T., Schmued L. C. and Ali S. F. (2008) Brain region‐specific neurodegenerative profiles showing the relative importance of amphetamine dose, hyperthermia, seizures and the blood‐brain barrier. Ann. N.Y. Acad. Sci. 1139, 127–139. [DOI] [PubMed] [Google Scholar]
- Bowyer J. F., Patterson T. A., Saini U. T., Hanig J. P., Thomas M., Camacho L., George N. I. and Chen J. J. (2013) Comparison of the global gene expression of choroid plexus and meninges and associated vasculature under control conditions and after pronounced hyperthermia or amphetamine toxicity. BMC Genom. 14, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer J. F., Tranter K. M., Sarkar S., Raymick J., Hanig J. P. and Schmued L. C. (2014) Systemic administration of fluoro‐gold for the histological assessment of vascular structure, integrity and damage. Curr. Neurovasc. Res. 11, 31–47. [DOI] [PubMed] [Google Scholar]
- Bowyer J. F., Sarkar S., Tranter K. M., Hanig J. P., Miller D. B. and O'Callaghan J. P. (2016) Vascular‐directed responses of microglia produced by methamphetamine exposure: indirect evidence that microglia are involved in vascular repair? J. Neuroinflammation 13, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M., Cerami A. and Vlassara H. (1988) Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 318, 1315–1321. [DOI] [PubMed] [Google Scholar]
- Cadet J. L., Jayanthi S. and Deng X. (2005) Methamphetamine‐induced neuronal apoptosis involves the activation of multiple death pathways. Review. Neurotox. Res. 8, 199–206. [DOI] [PubMed] [Google Scholar]
- Cadet J. L., Krasnova I. N., Jayanthi S. and Lyles J. (2007) Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox. Res. 11, 183–202. [DOI] [PubMed] [Google Scholar]
- Chang L., Alicata D., Ernst T. and Volkow N. (2007) Structural and metabolic brain changes in the striatum associated with methamphetamine abuse. Addiction 102(Suppl 1), 16–32. [DOI] [PubMed] [Google Scholar]
- Ernst T., Chang L., Leonido‐Yee M. and Speck O. (2000) Evidence for long‐term neurotoxicity associated with methamphetamine abuse: a 1H MRS study. Neurology 54, 1344–1349. [DOI] [PubMed] [Google Scholar]
- Exton J. H. and Park C. R. (1968) Control of gluconeogenesis in liver. II. Effects of glucagon, catecholamines, and adenosine 3',5'‐monophosphate on gluconeogenesis in the perfused rat liver. J. Biol. Chem. 243, 4189–4196. [PubMed] [Google Scholar]
- Fleckenstein A. E., Gibb J. W. and Hanson G. R. (2000) Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur. J. Pharmacol. 406, 1–13. [DOI] [PubMed] [Google Scholar]
- Fleckenstein A. E., Volz T. J., Riddle E. L., Gibb J. W. and Hanson G. R. (2007) New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 47, 681–698. [DOI] [PubMed] [Google Scholar]
- Frank M. G., Hershman S. A., Weber M. D., Watkins L. R. and Maier S. F. (2014) Chronic exposure to exogenous glucocorticoids primes microglia to pro‐inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 40, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank M. G., Watkins L. R. and Maier S. F. (2015) The permissive role of glucocorticoids in neuroinflammatory priming: mechanisms and insights. Curr Opin Endocrinol Diabetes Obes 22, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb J., Al‐Yawer F. and Anisman H. (2013) Synergistic and antagonistic actions of acute or chronic social stressors and an endotoxin challenge vary over time following the challenge. Brain. Behav. Immun. 28, 149–58. [DOI] [PubMed] [Google Scholar]
- Graeber M. B. and Streit W. J. (2010) Microglia: biology and pathology. Acta Neuropathol. 119, 89–105. [DOI] [PubMed] [Google Scholar]
- Halpin L. E. and Yamamoto B. K. (2012) Peripheral ammonia as a mediator of methamphetamine neurotoxicity. J. Neurosci. 32, 13155–13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins B. T., Lundeen T. F., Norwood K. M., Brooks H. L. and Egleton R. D. (2007) Increased blood‐brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia 50, 202–211. [DOI] [PubMed] [Google Scholar]
- Ho E. L., Josephson S. A., Lee H. S. and Smith W. S. (2009) Cerebrovascular complications of methamphetamine abuse. Neurocrit. Care 10, 295–305. [DOI] [PubMed] [Google Scholar]
- Huber J. D., VanGilder R. L. and Houser K. A. (2006) Streptozotocin‐induced diabetes progressively increases blood‐brain barrier permeability in specific brain regions in rats. Am. J. Physiol. Heart Circ. Physiol. 291, H2660–H2668. [DOI] [PubMed] [Google Scholar]
- Ichinose K., Kawasaki E. and Eguchi K. (2007) Recent advancement of understanding pathogenesis of type 1 diabetes and potential relevance to diabetic nephropathy. Am. J. Nephrol. 27, 554–564. [DOI] [PubMed] [Google Scholar]
- Kelly K. A., Miller D. B., Bowyer J. F. and O'Callaghan J. P. (2012) Chronic exposure to corticosterone enhances the neuroinflammatory and neurotoxic responses to methamphetamine. J. Neurochem. 122, 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyatkin E. A., Brown P. L. and Sharma H. S. (2007) Brain edema and breakdown of the blood‐brain barrier during methamphetamine intoxication: critical role of brain hyperthermia. Eur. J. Neurosci. 26, 1242–1253. [DOI] [PubMed] [Google Scholar]
- Kongsui R., Beynon S. B., Johnson S. J. and Walker F. R. (2014) Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J. Neuroinflammation 11, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft A. D. and Harry G. J. (2011) Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ Res. Public Health 8, 2980–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin L. E., Collins S. A. and Yamamoto B. K. (2014) Neurotoxicity of methamphetamine and 3,4‐methylenedioxymethamphetamine. Life Sci. 97, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenart N., Brough D. and Denes A. (2016) Inflammasomes link vascular disease with neuroinflammation and brain disorders. J. Cereb. Blood Flow Metab. 36, 1668–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M. S., Patton R. E., Hanig J. P., Tranter K. M., George N. I., James L. P., Davis K. J. and Bowyer J. F. (2013) Serum myoglobin, but not lipopolysaccharides, is predictive of AMPH‐induced striatal neurotoxicity. Neurotoxicology 37C, 40–50. [DOI] [PubMed] [Google Scholar]
- Locker A. R., Michalovicz L. T., Kelly K. A., Miller J. V., Miller D. B. and O'Callaghan J. P. (2017) Corticosterone primes the neuroinflammatory response to Gulf War‐relevant organophosphates independently of acetylcholinesterase inhibition. J. Neurochem. 142, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram L. C., Taylor F. R., Strand K. A., Frank M. G., Sholar P., Harrison J. A., Maier S. F. and Watkins L. R. (2011) Prior exposure to glucocorticoids potentiates lipopolysaccharide induced mechanical allodynia and spinal neuroinflammation. Brain Behav Immun 25, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann U. D. and Ricaurte G. A. (2004) Amphetamine neurotoxicity: accomplishments and remaining challenges. Neurosci. Biobehav. Rev. 27, 821–826. [DOI] [PubMed] [Google Scholar]
- McCann U. D., Wong D. F., Yokoi F., Villemagne V., Dannals R. F. and Ricaurte G. A. (1998) Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN‐35,428. J. Neurosci. 18, 8417–8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier V. M. and Cerami A. (1981) Nonenzymatic browning in vivo: possible process for aging of long‐lived proteins. Science 211, 491–493. [DOI] [PubMed] [Google Scholar]
- Monnier V. M., Kohn R. R. and Cerami A. (1984) Accelerated age‐related browning of human collagen in diabetes mellitus. Proc. Natl Acad. Sci. USA 81, 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop N. A., Halpin L. E. and Yamamoto B. K. (2016) Peripheral ammonia and blood brain barrier structure and function after methamphetamine. Neuropharmacology 107, 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Callaghan J. P. and Miller D. B. (1994) Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J. Pharmacol. Exp. Ther. 270, 741–751. [PubMed] [Google Scholar]
- O'Callaghan J. P. and Miller D. B. (2001) Neurotoxic effects of substituted amphetamines in rats and mice, in Handbook of Neurotoxicology, Vol. 2, (Massaro E. J., ed.), pp. 269–301. Humana Press Inc., Totowa, NJ. [Google Scholar]
- O'Callaghan J. P., Kelly K. A., Locker A. R., Miller D. B. and Lasley S. M. (2015) Corticosterone primes the neuroinflammatory response to DFP in mice: potential animal model of Gulf War Illness. J. Neurochem. 133, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G. and Watson C. (1995) The Rat Brain in Stereotaxic Coordinates. CA Academic Press, San Diego. [Google Scholar]
- Prinz M. and Priller J. (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. [DOI] [PubMed] [Google Scholar]
- Rothrock J. F., Rubenstein R. and Lyden P. D. (1988) Ischemic stroke associated with methamphetamine inhalation. Neurology 38, 589–592. [DOI] [PubMed] [Google Scholar]
- Schmued L. C., Stowers C. C., Scallet A. C. and Xu L. (2005) Fluoro‐Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 1035, 24–31. [DOI] [PubMed] [Google Scholar]
- Seiden L. S. and Sabol K. E. (1995) Neurotoxicity of methamphetamine‐related drugs and cocaine, in Handbook of Neurotoxicology (Chang L. W. and Dyer R. S., eds), Vol. 2, pp. 824–844. Marcel Dekker, Inc., New York, Basel, Hong Kong. [Google Scholar]
- Sekine Y., Ouchi Y., Sugihara G. et al (2008) Methamphetamine causes microglial activation in the brains of human abusers. J. Neurosci. 28, 5756–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl D. A. (1956) The measurable parameters of the cerebral cortex and their significance in its organization. Prog. Neurobiol., 1956, 324–333. [PubMed] [Google Scholar]
- Shrout P. E. and Fleiss J. L. (1979) Intraclass correlations: uses in assessing rater reliability. Psychol. Bull. 86, 420–428. [DOI] [PubMed] [Google Scholar]
- Sobesky J. L., D'Angelo H. M., Weber M. D., Anderson N. D., Frank M. G., Watkins L. R., Maier S. F. and Barrientos R. M. (2016) Glucocorticoids mediate short‐term high‐fat diet induction of neuroinflammatory priming, the NLRP3 Inflammasome, and the danger signal HMGB1. eNeuro, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Pei L., Yao S., Wu Y. and Shang Y. (2017) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci. 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K. and O'Callaghan J. P. (2007) Divergent roles for tumor necrosis factor‐alpha in the brain. J. Neuroimmune Pharmacol. 2, 140–153. [DOI] [PubMed] [Google Scholar]
- Streit W. J. (1990) An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I‐B4). J. Histochem. Cytochem. 38, 1683–1686. [DOI] [PubMed] [Google Scholar]
- Streit W. J. (2002) Microglia and the response to brain injury. Ernst Schering Res. Found Workshop, 39, 11–24. [DOI] [PubMed] [Google Scholar]
- Streit W. J. (2010) Microglial activation and neuroinflammation in Alzheimer's disease: a critical examination of recent history. Front. Aging Neurosci. 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano T., Shiota M., Tanaka T., Miyamae Y., Shimada M. and Oshino N. (1980) Intracellular redox state and stimulation of gluconeogenesis by glucagon and norepinephrine in the perfused rat liver. J. Biochem. 87, 153–166. [DOI] [PubMed] [Google Scholar]
- Thomas D. M. and Kuhn D. M. (2005) MK‐801 and dextromethorphan block microglial activation and protect against methamphetamine‐induced neurotoxicity. Brain Res. 1050, 190–198. [DOI] [PubMed] [Google Scholar]
- Thomas D. M., Francescutti‐Verbeem D. M., Liu X. and Kuhn D. M. (2004) Identification of differentially regulated transcripts in mouse striatum following methamphetamine treatment–an oligonucleotide microarray approach. J. Neurochem. 88, 380–393. [DOI] [PubMed] [Google Scholar]
- Thomas M., George N. I., Patterson T. A. and Bowyer J. F. (2009) Amphetamine and environmentally induced hyperthermia differentially alter the expression of genes regulating vascular tone and angiogenesis in the meninges and associated vasculature. Synapse 63, 881–894. [DOI] [PubMed] [Google Scholar]
- Thomas M., George N. I., Saini U. T., Patterson T. A., Hanig J. P. and Bowyer J. F. (2010) Endoplasmic reticulum stress responses differ in meninges and associated vasculature, striatum, and parietal cortex after a neurotoxic amphetamine exposure. Synapse 64, 579–593. [DOI] [PubMed] [Google Scholar]
- Treweek J. B., Dickerson T. J. and Janda K. D. (2009) Drugs of abuse that mediate advanced glycation end product formation: a chemical link to disease pathology. Acc. Chem. Res. 42, 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanGilder R. L., Kelly K. A., Chua M. D., Ptachcinski R. L. and Huber J. D. (2009) Administration of sesamol improved blood‐brain barrier function in streptozotocin‐induced diabetic rats. Exp. Brain Res. 197, 23–34. [DOI] [PubMed] [Google Scholar]
- Viana S. D., Valero J., Rodrigues‐Santos P., Couceiro P., Silva A. M., Carvalho F., Ali S. F., Fontes‐Ribeiro C. A. and Pereira F. C. (2016) Regulation of striatal astrocytic receptor for advanced glycation end‐products variants in an early stage of experimental Parkinson's disease. J. Neurochem. 138, 598–609. [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Chang L., Wang G. J. et al (2001a) Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am. J. Psychiatry 158, 383–389. [DOI] [PubMed] [Google Scholar]
- Volkow N. D., Chang L., Wang G. J. et al (2001b) Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 158, 377–382. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K. T. and Kiyatkin E. A. (2015) Central and peripheral contributions to dynamic changes in nucleus accumbens glucose induced by intravenous cocaine. Front. Neurosci. 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M. D., Frank M. G., Tracey K. J., Watkins L. R. and Maier S. F. (2015) Stress induces the danger‐associated molecular pattern HMGB‐1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 35, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. M., Kalasinsky K. S., Levey A. I. et al (1996) Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 2, 699–703. [DOI] [PubMed] [Google Scholar]
- Yamamoto B. K., Moszczynska A. and Gudelsky G. A. (2010) Amphetamine toxicities: classical and emerging mechanisms. Ann. N. Y. Acad. Sci. 1187, 101–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui K., Goto K., Ikemoto S., Ishiguro T. and Kamata Y. (2000) Increased sensitivity to stress in spontaneous recurrence of methamphetamine psychosis: noradrenergic hyperactivity with contribution from dopaminergic hyperactivity. J Clin Psychopharmacol. 20, 165–174. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu L., Liu Y.Z., Shen X.L., Wu T.Y., Zhang T., Wang W., Wang Y.X. and Jiang C.L. (2015) NLRP3 inflammasome mediates chronic mild stress‐induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol, 18, pii–pyv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. Raw counts of FJc‐ labeled neurons for each evaluator that that were used in determining treatment effects on degeneration.
Figure S1. METH versus noMETH blood glucose levels.
Figure S2. METH+CORT versus METH+Veh blood glucose levels.
Figure S3. METH versus noMETH body temperature levels.
Figure S4. METH+CORT versus METH+Veh body temperature levels.
Figure S5. FJc‐labeling of degenerating neurons in three of the METH treatment groups.
Figure S6. “Screen Shot” of Nikon NIS Display of an image of the parietal and setting an parameters used for image analysis.