

Abstract
Background
Functional similarity between molecules results in similar phenotypes, such as diseases. Therefore, it is an effective way to reveal the function of molecules based on their induced diseases. However, the lack of a tool for obtaining the similarity score of pair-wise disease sets (SSDS) limits this type of application.
Results
Here, we introduce DisSetSim, an online system to solve this problem in this article. Five state-of-the-art methods involving Resnik’s, Lin’s, Wang’s, PSB, and SemFunSim methods were implemented to measure the similarity score of pair-wise diseases (SSD) first. And then “pair-wise-best pairs-average” (PWBPA) method was implemented to calculated the SSDS by the SSD. The system was applied for calculating the functional similarity of miRNAs based on their induced disease sets. The results were further used to predict potential disease-miRNA relationships.
Conclusions
The high area under the receiver operating characteristic curve AUC (0.9296) based on leave-one-out cross validation shows that the PWBPA method achieves a high true positive rate and a low false positive rate. The system can be accessed from http://www.bio-annotation.cn:8080/DisSetSim/.
Keywords: Functional similarity, Similarity score, Disease sets, Disease-miRNA relationships
Background
The similarity of pair-wise disease sets (SDS) has drawn more and more attention in identifying functional similarity of the disease-caused molecules [1], predicting potential relationships between diseases and molecules [2–8], and so on. In previous studies, Wang et al. utilized the SDS to construct a human miRNA functional similarity network (MFSN) [1]. And Sun et al. used the SDS to predict novel disease lncRNA relationships [9].
The performance of calculating the SDS is mainly based on the method for computing the similarity of pair-wise diseases (SD). Currently, seven state-of-art methods involving Resnik’s [10], Lin’s [11], Wang’s [12], process-similarity based (PSB) [13], SemFunSim [14], ILNCSIM [15], and FMLNCSIM [16] methods were frequently used for computing the SD. Among these methods, Resnik’s [10], Lin’s [11], and Wang’s methods [12] are designed earlier for Gene Ontology (GO) [8, 17]. And these methods were introduced for calculating the SD by DOSim [18] and DisSim [19]. Resnik’s and Lin’s methods [10, 11] are based on information content (IC) for computing similarity between terms of ontology. And Wang’s method [12] is based on the hierarchical structure of the ontology. PSB and SemFunSim methods are newly developed for Disease Ontology (DO) [20]. PSB method [13] utilized the association of biological process between genes to calculate disease similarity. In comparison, SemFunSim method [14] considered more types of the functional associations including protein-protein interaction [21], human mRNA co-expression [22], and so on.
Resources for calculating the similarity score of pair-wise diseases (SSD) mainly includes the vocabularies of diseases and disease-related genes. The frequently used disease vocabularies contain Online Mendelian Inheritance in Man (OMIM) [23], Medical Subject Headings (MeSH) [24], and DO [20]. OMIM records the names of genetic disorders without providing semantic associations between them. MeSH provides a hierarchy of terms in biomedical domain. It contains 16 categories, of which only C and F03 involve disease names. In comparison with OMIM and MeSH, DO has been established around the concept of disease, and it aims to provide a clear definition for each disease. The disease-related genes are scattered in the databases, such as Gene Reference into Function (GeneRIF) [25], OMIM [23], Genetic Association Database (GAD) [26] and Comparative Toxicogenomics Database (CTD) [27]. It is better to use relationships of all of these databases.
“pair-wise-all pairs-maximum” (PWAPM) method and “pair-wise-best pairs-average” (PWBPA) method are optional for calculating similarity of pair-wise term sets [28]. For comparing multiple aspects, the best measure is the PWBPA method, which is widely utilized in calculating similarity of DO and GO term sets [1, 7, 9, 12].
Although DOSim [18] and DisSim [19] implemented the disease similarity methods in R package and web interface, no tools provided the function to calculate the similarity score of pair-wise disease sets (SSDS) currently. In this article, we designed and implemented an online tool DisSetSim to calculate the SSDS. Five state-of-art disease similarity methods (Resnik’s, Lin’s, Wang’s, PSB, and SemFunSim) and the PWBPA method was implemented in the tool. The system is freely available at http://www.bio-annotation.cn:8080/DisSetSim/.
Methods
Date sources
Data sets of DisSetSim are from open source databases, and they are listed in Table 1. DO [20] records disease names. It provides terms for calculating disease similarity. GeneRIF [25], OMIM [23], GAD [26] and CTD [27] are manually curated databases of disease-related genes. All of diseases in these databases are mapped to terms in DO based on SIDD [29]. GO annotation (GOA) [30] includes functional annotation of genes. HumanNet is the gene functional network of human. In addition, HMDD v2.0 [31] contains disease-related miRNAs, diseases of which were manually mapped to terms in DO by OAHG [32].
Table 1.
Data sources
| Data source | Web site |
|---|---|
| DO | http://disease-ontology.org/ |
| CTD | http://ctdbase.org/ |
| GeneRIF | http://www.ncbi.nlm.nih.gov/gene/about-generif |
| GAD | https://geneticassociationdb.nih.gov/ |
| OMIM | http://www.omim.org/ |
| GO & GOA | http://www.geneontology.org |
| HumanNet | http://www.functionalnet.org/humannet/ |
| OAHG | bio-annotation.cn/OAHG/ |
Methods for calculating similarity score of pair-wise diseases
Five state-of-art methods involving Resnik’s [10], Lin’s [11], Wang’s [12], PSB [13], and SemFunSim methods [14] have been implemented for calculating the SSD.
Resnik’s and Lin’s methods are based on IC. The IC of a disease t is described as Eq. 1:
| 1 |
where N is the total number of genes annotated by diseases, and n t is the number of genes annotated by t. Assuming t 1 and t 2 are two diseases, the similarity of them is defined by Resnik as following [10]:
| 2 |
where t MICA is the most informative common ancestor (MICA) of t 1 and t 2. Lin defines the similarity of t 1 and t 2 as Eq. 3 [11]:
| 3 |
Assuming T 1 is the set involving t 1 and all of its ancestor terms of ontology. Semantic contribution of term t to t 1 is represented as following:
| 4 |
where w is the contribution factor of each semantic relationship. According to Wang et al. [1], w is defined as 0.5 for ‘IS_A’ relationship of DO [20]. Then, all the semantic contributions of T 1 to t 1 is SV(t 1 ), which is defined as following:
| 5 |
Assuming T 2 is the set involving t 2 and all of its ancestor terms, the similarity between t 1 and t 2 is defined as following by Wang’s method [12]:
| 6 |
Assuming t 1 and t 2 can be related with m and n biological processes of GO based on hypergeometric test, respectively, the similarity of t 1 and t 2 is defined by the PSB method as following:
| 7 |
where p 1i and p 2j is the ith and jth significant related biological process terms of t 1 and t 2, respectively. Sim(p 1i, p 2j) represents similarity between two processes p 1i and p 2j, which is defined as Eq. 8:
| 8 |
where IC GO and IC DO represent IC based on GO and DO, respectively. n(p 1 ∩ p 2) and n(p 1 ∪ p 2) denote the number of common genes of p 1 and p 2, and the number of total genes of p 1 and p 2, respectively.
Assuming G 1 and G 2 represent related gene sets of t 1 and t 2, respectively. Then, the similarity of t 1 and t 2 by the SemFunSim method can be described as following:
| 9 |
where |GMICA| represents the number of genes in GMICA. m and n denote the number of genes in G 1 and G 2, respectively. Sim(g 1i, g 2j) is the functional similarity score between genes g 1i and g 2j, which could be obtained from HumanNet [33].
Method for calculating similarity score of pair-wise disease sets
The PWBPA method was utilized for calculating the SSDS. The similarity of two disease sets T 1 and T 2 is defined as following:
| 10 |
where T 1 and T 2 contains N and M diseases, respectively. t i and t j represents ith and jth terms of T 1 and T 2, respectively.
Predicting potential association between diseases and miRNAs
Functional similarity between miRNAs could be calculated based on their related disease sets. Similarities of each pair-wise miRNAs are utilized to establish a MFSN. Node of the network represents miRNA. Weight of edge is the functional similarity score. Then, disease-related miRNAs were prioritized using the network ranking algorithm named random walk with restart (RWR) [7].
The random walker starts on one or several seed nodes and then randomly transits to neighboring nodes considering the probabilities of the edges between the two nodes. And the probability to return to the seed nodes is supposed as γ. Then, RWR algorithm can be defined as following:
| 11 |
where P 0 denotes the initial probability vector, P t is a vector in which the ith element represents the probability of finding the walker at node i and step t, A is the column-normalized adjacency matrix of the network. The algorithm was performed until the difference between P t and P t+1 falling below 10−10, which means all the nodes become stable.
In this study, the known miRNAs of a disease were considered as seed nodes. The unknown miRNAs of it could be scored based on RWR on the MFSN. After ranking the miRNAs based on the scores, disease-related miRNAs could be prioritized.
Implementation
DisSetSim has been implemented on a JavaEE framework and run on the web server (2-core (2.26 GHz) processors) of Ucloud [34]. The four-layer architecture involving DATABASE, ALGORITHM, TOOLS, and VIEW layer is shown in Fig. 1 The detailed description of the architecture is fixed as following.
Fig. 1.
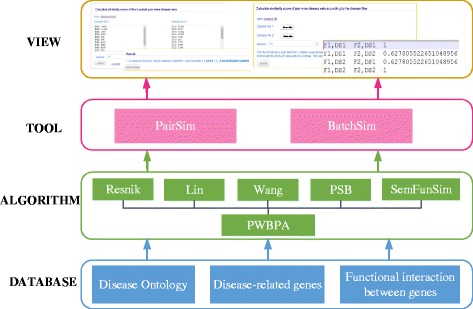
System overview of DisSetSim
(1) DATABASE layer. This layer stores DO, disease-related genes, and functional associations between genes. These are exploited by ALGORITHM layer for calculating the similarity between disease sets.
(2) ALGORITHM layer. Five algorithms of measuring the similarity between DO terms have been implemented, which include Resnik’s, Lin’s, Wang’s, PSB, and SemFunSim methods. And the method named PWBPA for calculating the SSDS were also implemented.
(3) TOOL layer. Two tools including PairSim and BatchSim have been provided for exploring the SSDS. PairSim calculates the similarity for a given pair of disease sets, and BatchSim computes similarity between each pair of multiple disease sets.
(4) VIEW layer. Web pages are provided for viewing the results. It shows the similarity of pair-wise disease sets.
Results
Web interface
DisSetSim provides two tools PairSim and BatchSim for querying the SSDS. The details about the usage of these two tools are described as follows.
The usage of PairSim
Figure 2a shows a case for searching the similarity score of a given pair of disease sets. The web page for inputting disease sets is http://www.bio-annotation.cn:8080/DisSetSim/ basic-init. Each of these disease sets could be inputted in a textbox. A disease set is comprised by several diseases. And each disease is represented by the identifier of term in DO. All the term identifiers could be downloaded from the hyperlink ‘disease terms’ in the inputting page. Here, we click the ‘example’ button to use our example. Then, we choose one of the five methods (Resnik’s, Lin’s, Wang’s, PSB, and SemFunSim) for calculating the SSD. After submitting this pair of disease sets, the system could return the similarity score based on the PWBPA method.
Fig. 2.
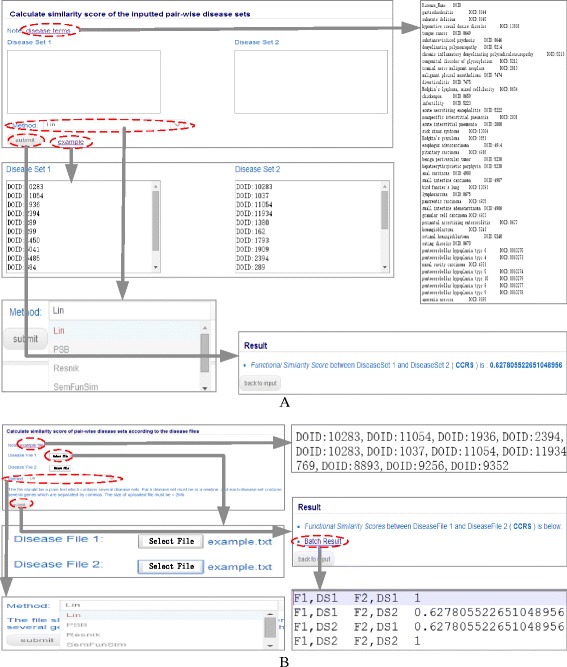
Schematic workflow of DisSetSim. a Schematic workflow of PairSim. b Schematic workflow of BatchSim
The usage of BatchSim
Figure 2b shows a case for searching the similarity score of all the pairs based on the selected files. The web page for inputting disease sets is http://www.bio-annotation.cn:8080/DisSetSim/batch-init. Two files including disease sets should be selected before submitting. The file should be a plain text which contains several disease sets. Each disease set must be in a newline, and each disease set contains several disease IDs which are separated by commas. The size of uploaded file must be <2 Mb. Here, we selected our example file in this page. Then, we choose one of the five methods for calculating the SSD. After clicking the ‘submit’ button, the system could return the similarity score of all the pairs of the selected files based on the PWBPA method.
miRNA functional similarity network
By applying DisSetSim to the inputted disease sets of miRNAs, the similarity score of each miRNA pair could be obtained. Using miRNA as node and similar miRNAs as edge, the MFSN was constructed based on various similarity cutoffs. As shown in Fig. 3a, the number of links dramatically decreases when the cutoff increases from low value to high value. When the cutoff is equal to or bigger than 0.7, the link numbers remain relatively stable. Therefore, we use 0.7 as cutoff for the MFSN. In total, 1042 miRNA-miRNA functional associations between 346 miRNAs were obtained as MFSN (Fig. 3c). Similar to the most of the reported biological networks, the degree of this MFSN also shows a scale-free distribution [5, 9, 35–37]. It means that most of the miRNAs only have a few functionally similar miRNAs, and a few of miRNAs have a numerous functional similar miRNA (Fig. 3b).
Fig. 3.

Construction and characteristics of the miRNA functional similarity network. a Cumulative distribution of the edges between miRNAs when using various similarity cutoffs. b Degree distribution for miRNA in the miRNA functional similarity network. c The miRNA functional similarity network
Here, the PWBPA method was utilized for calculating similarity between disease sets, and SemFunSim was used as computing the similarity of pair-wise diseases. This is because that the SemFunSim method was proven to obtain the best performance [14]. Alternatively, other state-of-art methods could also be chosen to construct MFSN.
Disease-related miRNAs
By applying the above similarity scores of miRNAs, novel disease-related miRNAs were predicted based on RWR algorithm (See ‘Methods’ section). To evaluate the performance of the similarity scores of miRNAs, leave-one-out cross validation of 5710 known experimentally confirmed miRNA-disease associations, including 265 diseases with at least two miRNAs, were used for this assessment. For a disease of interest, each known miRNA of this disease was left out as the testing case, and the remaining miRNAs of this disease were used as seed nodes. All the miRNAs except the miRNAs of this disease were considered as candidate miRNAs. We then examined how well the testing miRNA ranked relative to the candidate miRNAs. If the ranking of this testing miRNA exceeded a given cutoff, we regarded this miRNA-disease association as successfully predicted. As a result, an area under the ROC curve (AUC) of 0.9296 was achieved (Fig. 4), which demonstrated that our miRNA functional similarity was effective in recovering known experimentally confirmed disease-related miRNAs.
Fig. 4.
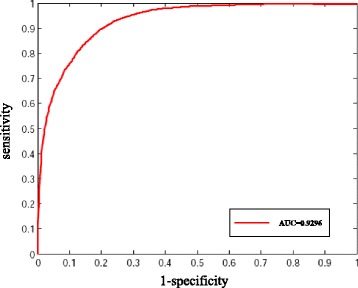
ROC curve of the PWBPA method based on leave-one-out cross validation on known experimentally verified miRNA-disease associations
Discussion
As the best of our knowledge, non-coding RNAs (ncRNAs) attract more and more attentions because of their important regulation roles in molecular level. However, the lack of protein limits the identification of their function. Here the application of our tool in constructing MFSN and predicting miRNA-disease associations provides a novel way to help for exploring the function of miRNAs especially for prioritizing miRNA-disease associations. This application can be extended to other ncRNAs, such as lncRNAs and circRNAs. Although methods for calculating the SDS have been implemented by previous methods, it is not easy to calculate the SSDS. Therefore, DisSetSim benefits researchers for exploring the function of disease-related molecular.
Conclusions
In this article, we designed and developed a web system DisSetSim to calculate the SSDS. Five state-of-art methods were implemented (see ‘METHODS’ section) for calculating disease similarity. And the PWBPA method was implemented for calculating the SSDS. Two tools involving PairSim and BatchSim provide the function to obtain the SSDS by inputting a pair-wise disease sets and multiple disease sets, respectively.
The functional similarity of miRNAs could be calculated based on our system. Here, the similarity of each pair-wise miRNAs was calculated. And then a MFSN was constructed based on miRNA similarity. The network was further utilized to predicate disease-related miRNAs based on RWR. The high AUC (0.9296) shows the MFSN is very suitable for predicting potential relationships between diseases and miRNAs.
Acknowledgments
Yadong Wang and Liang Cheng are the corresponding authors. Lingling Zhao and Zhiyan Liu are the co-first authors.
Funding
This work was supported by the Major State Research Development Program of China [No. 2016YFC1202302], the National Natural Science Foundation of China (Grant No. 61502125, $2000), Heilongjiang Postdoctoral Fund (Grant No. LBH-Z15179, $800), and China Postdoctoral Science Foundation (Grant No. 2016 M590291, $1000).
Availability of data and materials
The system can be accessed from http://www.bio-annotation.cn:8080/DisSetSim/.
About this supplement
This article has been published as part of Journal of Biomedical Semantics Volume 8 Supplement 1, 2017: Selected articles from the Biological Ontologies and Knowledge bases workshop. The full contents of the supplement are available online at https://jbiomedsem.biomedcentral.com/articles/supplements/volume-8-supplement-1.
Abbreviations
- CTD
Comparative Toxicogenomics Database
- DO
Disease Ontology
- GAD
Genetic Association Database
- GeneRIF
Gene Reference into Function
- GO
Gene Ontology
- GOA
GO annotation
- IC
information content
- MFSN
miRNA functional similarity network
- MICA
the most informative common ancestor
- ncRNA
non-coding RNAs
- OMIM
Online Mendelian Inheritance in Man
- PSB
process-similarity based
- PWAPM
pair-wise-all pairs-maximum
- PWBPA
pair-wise-best pairs-average
- PWR
random walk with restart
- SD
pair-wise diseases
- SSD
the similarity score of pair-wise diseases
- SSDS
the similarity score of pair-wise disease sets.
Authors’ contributions
YH, LZ and ZL implemented the first version of the online system. HJ, HS, PX updated the system. YW and LC wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yang Hu, Email: huyang@hrbmu.edu.cn.
Lingling Zhao, Email: zhaoll@hit.edu.cn.
Zhiyan Liu, Email: liuzhy@nebulagene.com.
Hong Ju, Email: hongju.hit@hotmail.com.
Hongbo Shi, Email: shihongbo@ems.hrbmu.edu.cn.
Peigang Xu, Email: peigang.xu@hit.edu.cn.
Yadong Wang, Email: ydwang@hit.edu.cn.
Liang Cheng, Email: liangcheng@hrbmu.edu.cn.
References
- 1.Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26(13):1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, Yan CC, Luo C, Ji W, Zhang Y, Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci Rep. 2015;5:11338. doi: 10.1038/srep11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, Yan CC, Zhang X, You Z-H. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2016:bbw060. [DOI] [PMC free article] [PubMed]
- 4.Chen X, You Z, Yan G, Gong D. IRWRLDA: improved random walk with restart for LncRNA-disease association prediction. Oncotarget. 2016;7(36):57919–57931. doi: 10.18632/oncotarget.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng J, Wang T, Hu J, Wang Y, Chen J. Constructing networks of organelle functional modules in Arabidopsis. Current Genomics. 2016;17(5):427–438. doi: 10.2174/1389202917666160726151048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.J. Peng, Bai, K., Shang, X., Wang, G., Xue, H., Jin, S., Cheng, L., Wang, Y., & Chen, J., “Predicting Disease-related Genes using Integrated Biomedical Networks,” BMC Genomics, vol. 18, no. 1, pp. 1043, 2017. [DOI] [PMC free article] [PubMed]
- 7.J. Peng, H. Li, Y. Liu, L. Juan, Q. Jiang, Y. Wang, and J. Chen, “InteGO2: a web tool for measuring and visualizing gene semantic similarities using Gene Ontology,” BMC Genomics, vol. 17 Suppl 5, pp. 530, 2016. [DOI] [PMC free article] [PubMed]
- 8.J. Peng, T. Wang, J. Wang, Y. Wang, and J. Chen, “Extending gene ontology with gene association networks,” Bioinformatics, vol. 32, no. 8, pp. 1185–94, Apr 15, 2016. [DOI] [PubMed]
- 9.Sun J, Shi H, Wang Z, Zhang C, Liu L, Wang L, He W, Hao D, Liu S, Zhou M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol BioSyst. 2014;10(8):2074–2081. doi: 10.1039/C3MB70608G. [DOI] [PubMed] [Google Scholar]
- 10.Resnik P. Using information content to evaluate semantic similarity in a taxonomy. 1995. [Google Scholar]
- 11.Lin D. An information-theoretic definition of similarity. 1998. pp. 296–304.
- 12.J. Z. Wang, Z. Du, R. Payattakool, P. S. Yu, and C. F. Chen, “A new method to measure the semantic similarity of GO terms,” Bioinformatics, vol. 23, no. 10, pp. 1274–81, May 15, 2007. [DOI] [PubMed]
- 13.Mathur S, Dinakarpandian D. Finding disease similarity based on implicit semantic similarity. J Biomed Inform. 2012;45(2):363–371. doi: 10.1016/j.jbi.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 14.L. Cheng, J. Li, P. Ju, J. Peng, and Y. Wang, “SemFunSim: a new method for measuring disease similarity by integrating semantic and gene functional association,” PLoS One, vol. 9, no. 6, pp. e99415, 2014. [DOI] [PMC free article] [PubMed]
- 15.Huang Y, Chen X, You Z, Huang D, Chan K. ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget. 2016;7(18):25902–25914. doi: 10.18632/oncotarget.8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, Huang Y, Wang X, You Z, Chan K. FMLNCSIM: fuzzy measure-based lncRNA functional similarity calculation model. Oncotarget. 2016;7(29):45948–45958. doi: 10.18632/oncotarget.10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Gong B, Chen X, Liu T, Wu C, Zhang F, Li C, Li X, Rao S, Li X. DOSim: an R package for similarity between diseases based on disease ontology. BMC Bioinformatics. 2011;12:266. doi: 10.1186/1471-2105-12-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.L. Cheng, Y. Jiang, Z. Wang, H. Shi, J. Sun, H. Yang, S. Zhang, Y. Hu, and M. Zhou, “DisSim: an online system for exploring significant similar diseases and exhibiting potential therapeutic drugs,” Sci Rep, vol. 6, pp. 30024, Jul 26, 2016. [DOI] [PMC free article] [PubMed]
- 20.Schriml LM, Arze C, Nadendla S, Chang YW, Mazaitis M, Felix V, Feng G, Kibbe WA. Disease ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40(Database issue):D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortutay C, Vihinen M. Identification of candidate disease genes by integrating gene Ontologies and protein-interaction networks: case study of primary immunodeficiencies. Nucleic Acids Res. 2009;37(2):622–628. doi: 10.1093/nar/gkn982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.J. M. Stuart, E. Segal, D. Koller, and S. K. Kim, “A gene-coexpression network for global discovery of conserved genetic modules,” Science, vol. 302, no. 5643, pp. 249–55, Oct 10, 2003. [DOI] [PubMed]
- 23.Amberger J, Bocchini C, Hamosh A. A new face and new challenges for online Mendelian inheritance in man (OMIM®) Hum Mutat. 2011;32(5):564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 24.Dhammi IK, Kumar S. Medical subject headings (MeSH) terms. Indian J Orthop. 2014;48(5):443–444. doi: 10.4103/0019-5413.139827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell JA, Aronson AR, Mork JG, Folk LC, Humphrey SM, Ward JM. Gene indexing: characterization and analysis of NLM's GeneRIFs. AMIA Annu Symp Proc. 2003:460–4. [PMC free article] [PubMed]
- 26.Becker KG, Barnes KC, Bright TJ, Wang SA. The genetic association database. Nat Genet. 2004;36(5):431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 27.Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL, Rosenstein MC, Wiegers TC, Mattingly CJ. The comparative Toxicogenomics database: update 2013. Nucleic Acids Res. 2013;41(Database issue):D1104–D1114. doi: 10.1093/nar/gks994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.C. Pesquita, D. Faria, A. O. Falcao, P. Lord, and F. M. Couto, “Semantic similarity in biomedical ontologies,” PLoS Comput Biol, vol. 5, no. 7, pp. e1000443, 2009. [DOI] [PMC free article] [PubMed]
- 29.L. Cheng, G. Wang, J. Li, T. Zhang, P. Xu, and Y. Wang, “SIDD: A Semantically Integrated Database towards a Global View of Human Disease,” PLOS ONE, vol. 8, no. 10, pp. e75504, 2013. [DOI] [PMC free article] [PubMed]
- 30.Camon E, Magrane M, Barrell D, Lee V, Dimmer E, Maslen J, Binns D, Harte N, Lopez R, Apweiler R. The gene ontology annotation (goa) database: sharing knowledge in uniprot with gene ontology. Nucleic Acids Res. 2004;32(suppl 1):D262–D266. doi: 10.1093/nar/gkh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42(Database issue):D1070–D1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng L, Sun J, Xu W, Dong L, Hu Y, Zhou M. OAHG: an integrated resource for annotating human genes with multi-level ontologies. Sci Rep. 2016;10:34820. doi: 10.1038/srep34820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21(7):1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sqalli MH, Al-Saeedi M, Binbeshr F, Siddiqui M. UCloud: A simulated Hybrid Cloud for a university environment. 2012. pp. 170–172.
- 35.K. I. Goh, M. E. Cusick, D. Valle, B. Childs, M. Vidal, and A. L. Barabasi, “The human disease network,” Proc Natl Acad Sci U S A, vol. 104, no. 21, pp. 8685–90, May 22, 2007. [DOI] [PMC free article] [PubMed]
- 36.Zhang F, Ren C, Lau KK, Zheng Z, Lu G, Yi Z, Zhao Y, Su F, Zhang S, Zhang B, Sobie EA, Zhang W, Walsh MJ. A network medicine approach to build a comprehensive atlas for the prognosis of human cancer. Brief Bioinform, Aug. 2016;24 [DOI] [PMC free article] [PubMed]
- 37.F. Zhang, B. Gao, L. Xu, C. Li, D. Hao, S. Zhang, M. Zhou, F. Su, X. Chen, H. Zhi, and X. Li, “Allele-specific behavior of molecular networks: understanding small-molecule drug response in yeast,” PLoS One, vol. 8, no. 1, pp. e53581, 2013. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The system can be accessed from http://www.bio-annotation.cn:8080/DisSetSim/.