

Abstract
Hybrid analytical instrumentation constructed around mass spectrometry (MS) are becoming preferred techniques for addressing many grand challenges in science and medicine. From the omics sciences to drug discovery and synthetic biology, multidimensional separations based on MS provide the high peak capacity and high measurement throughput necessary to obtain large-scale measurements which are used to infer systems-level information. In this review, we describe multidimensional MS configurations as technologies which are big data drivers and discuss some new and emerging strategies for mining information from large-scale datasets. A discussion is included on the information content which can be obtained from individual dimensions, as well as the unique information which can be derived by comparing different levels of data. Finally, we discuss some emerging data visualization strategies which seek to make highly dimensional datasets both accessible and comprehensible.
Keywords: systems biology, integrated omics sciences, structural elucidation, peak capacity, mass defect analysis, metabolites and metabolomics
INTRODUCTION
All grand challenges where mass spectrometry (MS) plays a role are characterized by the big data paradigm (Table 1). Proteomics seeks to detect and measure all proteins found in an organism (1), which based on several recent drafts, numbers between 16,000 and 19,000 for basic human proteins (2,3,4), but could be as high as several million once protein variants and modifications are taken into account (5). The inclusion of spatially-resolved protein information from imaging studies will increase this number even further (6). The human metabolome is represented by many diverse classes of small molecule metabolites, of which a little over 40,000 have been annotated so far with support from MS techniques (7,8,9), but estimates place the possible number of human metabolites as high as 180,000 for lipids alone (10). Another grand challenge, systems biology, seeks to form connections between all of the various classes of biomolecules in both space and time towards the comprehensive diagnosis of disease states (11), and MS is at the forefront of integrated omics approaches that will help realize this vision (12,13,14). Drug discovery initiatives are aimed at finding the proverbial needles in haystacks in a molecular landscape of over 1060 possible chemical structures (15,16), which is a haystack containing a novemdecillion straws of hay, or about 40 orders-of-magnitude greater than the number of grains of sand on Earth (17). To address this formidable challenge, researchers are turning to high-throughput screening using MS-based assays which are capable of screening up to 100,000 compounds a day from combinatorial small molecule libraries (18,19). Genomics, which is driven by massively-paralleled DNA sequencers (20), is currently feeling the burden of big data, with a projected 2–40 million terabytes of genomic data being generated in the next ten years, representing anywhere from 100 million to 2 billion complete human genomes sequenced. According to a recent report, this volume of genomics data will surpass that of YouTube, Twitter, and the future Square Kilometre Array by 2025 (21).
Table 1.
Big data drivers in mass spectrometry
| Grand Challenge | Description | Scope of Data Volume |
|---|---|---|
| Systems Biology | Map the interconnectivity of all biomolecules in space and time. | >100,000 discrete biomolecules, over 109 possible binary connections |
| Omics Sciences | Genomics Proteomics Metabolomics Lipidomics Glycomics |
>20,000 human genes ca. 20,000 base human proteins; 106 possible protein variants >40,000 annotated human metabolites; >200,000 possible metabolites |
| Drug Discovery | Find chemicals with desirable pharmacological properties | 1060 possible drug targets; 1011 virtual drug-like chemical structures mapped |
| Synthetic Biology | Engineer and chemically characterize surrogate biosystems for translational research. | Elements of all of the above plus xenometabolites and temporal sampling on the order of seconds. |
The above examples only underscore one aspect of big data: big numbers, but big data challenges are much more complex than dealing with large scale datasets. The three Vs are often invoked to identify a big data challenge: 1) a large volume of data, 2) being generated at high velocity, 3) and characterized by a variety of different subsets of data (22). Lusher et al. define a big data challenge more broadly in terms of “whether the [researchers are] able to extract the relevant information from their rapidly growing data resources” (23). Thus, the definition of big data is relative to the field in which the data is being generated. A relevant example is the massive data generated in the field of astronomy, datasets on the order of petabytes (1,000 terabytes), which are efficiently handled due to decoupling data storage with data analysis, bridged through cloud computing (24). Another example is CERN’s Large Hadron Collider data analysis network which is built upon a massively distributed system architecture (25). Mass spectrometry as a field is beginning to embrace the concept of a decentralized computing infrastructure, but it is not there yet.
In this review, we trace the challenges of big data in analytical chemistry from its origins in the enumeration of chemical isomers to the (arguably) current locus in the field of multidimensional analysis based on mass spectrometry. The need for multidimensional analysis is rationalized based on the small molecule enumeration problem (26), and beyond high dimensionality and enhanced peak capacities, the correlation between discrete dimensions of data is presented to illustrate the rich information content that is afforded by scaling to higher dimensions. Finally, we provide an overview of some recent and creative visualization strategies that provide comprehensible access to higher order dimensional information in a low-dimensional format.
THE FOUNDATION OF BIG DATA IN CHEMICAL RESEARCH
GENERATING CHEMICAL KNOWLEDGE
In many of his works, the futurist Buckminster Fuller argued that human knowledge was increasing at an unprecedented rate in human history, and this provided unique opportunities in the areas of science, engineering, and design. Fuller supported his argument by plotting a timeline of the discovery of the chemical elements, reproduced in Figure 1A, and noted that knowledge was essentially increasing exponentially over time (27). In light of new data, the discovery of chemical elements has occurred at a relatively fixed rate for the past 50 years, reflecting the difficulty in creating stable nuclei of the super-heavy elements. Knowledge stems from a myriad of sources, and is more aptly illustrated from discoveries made in the absence of limitations. An updated observation of knowledge doubling in the chemical sciences can be seen in Figure 1B which plots the indexing of new chemical substances in the CAS Registry over time. As chemical space is vast, the number of unique chemical registry numbers is increasing near exponentially since the CAS Registry system was first introduced in 1965, reaching 100 million substances at the year of this publication (28), and it is expected that 200 million substances will be indexed within the next 5 years. The open access repository, PubChem (29), is growing even faster, with over 150 million chemical substances indexed since its introduction in 2004 (30). Over 60 million of these have been validated as unique chemical compounds (31). These and other efforts (32,33,34,35) to catalog all chemical compounds discovered represents our known chemical universe (36), which based on one estimate represents less than 1 in 1050 possible molecular structures for small organic compounds alone (37). To put this number in context, 1 in 1050 is a greater disparity of scale than the height of a person compared to the diameter of the observable universe, or 1 in 1026 m (38). While enumeration of all possible molecular structures is impossible, some recent progress has been made towards enumerating compounds of 17 atoms or less containing C, H, N, O, S, and halogens, which has resulted in 166.4 billion virtual drug-like compounds of ca. 350 Da or less (39,40). Such new compound discovery and annotation efforts are big data challenges which represent the chemical sciences in the purest sense, and form the basis for the nascent fields of chemography (41) and cheminformatics (42,43). Figure 1C contains a histogram of the number of chemical abstracts indexed by the CAPlus system which is accessed through SciFinder. As of the time of writing this review, over 45 million chemical abstracts have been indexed (44), which based on current trends is expected to double within the next decade. This number of publications is on scale with the estimated 50 million total number of peer-reviewed articles across all disciplines as of 2009 (45), of which a little over half were represented in CAPlus at that time (ca. 30M). Extrapolating this observation to the present suggests that currently there are at least 75M journal articles in existence at the time of writing this review. In a broader sense, this trend illustrates dissemination of greater quantities of chemical data into chemical information, which follows an exponential growth rate. If the canonical goal of the analytical sciences is to separate, identify, and quantify chemical substances from a variety of sources, then there is an incredibly vast amount of chemical space left to explore.
Figure 1.

Histograms which illustrate the increasing rate of knowledge and innovation relevant to the analytical sciences. These histograms describe growth numbers for (A) chemical element discovered, (B) chemical substances registered in the CAS Registry System, (C) chemical abstracts indexed in CAPlus, (D) the number of transistors in each generation of microprocessor, also known as Moore’s Law, (E) data storage capacity for each generation of consumer disk drive, and (F) the accessible internet bandwidth for residential customers.
TRANSLATING CHEMICAL INFORMATION
The ability to acquire and analyze large amounts of data is driven by advances in the computer sciences. The lower panels of Figure 1 illustrate the so-called “digital laws”, which describe 1) exponential scaling of computer processor speeds (Moore’s Law, Figure 1A) (46), 2) increasing capacity of consumer-grade data storage (Figure 1B) (47), and 3) available bandwidth of residential broadband internet (Figure 1C) (48). Collectively, these digital laws represent innovation and, on a broader level, humanity’s current capacity to process, store, and disseminate information. From the doubling brackets annotated on each graph, it is apparent that computer processor speed (related to the number of transistors) now doubles about every 4 to 6 years, whereas data storage capacity is doubling every 2 to 3 years. These observations infer two key points for large-scale analytical data mining: 1) data volume will increase faster than the informatics tools necessary to interpret the data, and 2) broader data accessibility will provide new opportunities to handle large analytical datasets. The first is already being realized in many areas of MS-based research, while the second point is beginning to emerge from two notable concepts: crowdsourcing, and cloud computing.
CROUDSOURCING MS DATA
Crowdsourcing describes a division of labor concept where the combined efforts of a large group of individuals are applied towards solving a complex problem. Typically, the contributing individuals are not experts in the problem they are tasked to solve, but rather “citizen scientists” who utilizes their natural human capabilities to address scientific challenges. Crowdsourcing in science as a concept is not new. In 1714, the British government issued the Longitude prize to anyone that could determine, by relatively simple means, the longitude of a ship at sea, which fostered advances in cartography and celestial navigation, while creating the new science of marine chronometry (49). Recently, the problem of 3D protein folding has been addressed by means of a novel online video game, Foldit, whereby players attempt to fold proteins, many of which have unsolved crystal structures. Foldit is an interactive puzzle game that is built upon Rosetta folding algorithms (50,51), allowing online players to move subdomains, as well as “shake” and “wiggle” a protein structure in order to minimize its energy (52). Foldit players have so far been able to solve the 3D structure of a retroviral protease (53,54), as well as improve the biological activity of a computationally-designed enzyme (55). In mass spectrometry, although sparse, there have been a few notable efforts to crowdsource large-scale efforts. There are several MS database initiatives that source from user-submitted data, including MassBank, HMDB, and mzCloud. Brady et al. described a gameplay approach similar to Foldit whereby players of the web-based “Spectral Game” are asked to match mass spectra with their corresponding molecular structures (56). Utilizing citizen scientists, Du et al. described a study whereby property owners collected soil samples which were screened by LC-MS for potential natural products. These efforts yielded a novel fungal metabolite, maximiscin, which exhibited antitumor activity in a mouse model (57). An open call for more crowdsourced data analysis resources in MS-based proteomics has recently been made (58), which, based on other similar efforts in structural and network biology (59,60) will foster more innovation and standardization across the field. Pragmatically speaking, it makes sense to query large groups of individuals for data analysis, as the capacity of the human brain surpasses that of conceptualized mathematical algorithms for discerning complex patterns across datasets, e.g., images (61). We envision the above strategies developed for structural biology and the nascent efforts now underway to crowdsource data analysis in mass spectrometry will be critical to inferring important patterns or information from massive datasets, including those from discrete fields (e.g., transcriptomics, proteomics, etc.), but ultimately linking datasets spanning the breadth of systems biology. Whereas the promise of data-driven discovery has classically been framed in the context of autonomous algorithms combing through large amounts of data, the human element in such efforts should not be understated (62).
CLOUD COMPUTING IN MS RESEARCH
Cloud computing, or specifically the concept of conducting data-intensive computational work across a distributed network of computers (63), is rapidly being adopted in many MS-based workflows (64). One example is XCMS online, which offers cloud-based processing of LC-MS metabolomics datasets from feature extraction to normalization of data dimensions and statistical analysis of the results, and is capable of handling terabytes of data (65,66). Another example is OpenMSI, which outsources the US Department of Energy’s NERSC supercomputing facility towards processing highly-dimensional imaging MS datasets, which in a single experiment can exceed 50 GB in size (67,68). One of the earliest big data initiatives in MS, proteomics, has seen several recent offerings of open access software tools for processing tandem MS data (24,69,70,71), and notable among these is the Trans-Proteomic Pipeline (72) which supports outsourcing of the software to the Amazon Web Services cloud computing infrastructure (73). These and other efforts to decentralize the computational resources required to handle MS-based data will help facilitate the development of educational and research programs where large investments in computer infrastructure is no longer necessary, and will help offset the need for institutions to invest heavily in instrumentation altogether. Open-source repositories of MS data are helping to make this possible (74,75). In the near future, it is conceivable that entire research programs will conduct MS research on digitally streamed data without direct access to a mass spectrometer.
MULTIDIMENSIONAL METHODS BASED ON MASS SPECTROMETRY
THE GENESIS OF BIG DATA IN MS
Mass spectrometry is well-suited to address big data challenges, as the throughput and information density of the technique are both extraordinarily high. Figure 2 highlights the peak capacity (right panel) and the peak production rate (left panel) of some MS-based analytical techniques which are used, and have the potential to be used, in big data initiatives. Peak capacities are commonly reported for condensed phase separations such as liquid and gas chromatography (76,77), but less common in ion mobility (IM) and MS research, and so specific considerations are taken to generate these metrics in this present work. For example, MS calculations for peak capacity are based on methods developed by Frahm et al., which account for instrument resolving powers and isotope redundancy (78) and utilize typical, rather than optimal parameters for each method (79). For IM, peak capacities are obtained from measured values of different techniques where available (80,81,82), or otherwise calculated from reported resolving powers (83,84). For IM coupled to MS (IM-MS), there is a correlation between size and mass, and so IM-MS data in Figure 2 is scaled by a factor of 0.25 (25% unique space occupancy) to reflect this reduced orthogonality, which is based on experiments conducted in the authors’ laboratory. The power and potential of multidimensional analytical separations becomes evident when comparing both the peak capacities and peak production rates of individual analytical separation dimensions with those of multidimensional techniques. A cursory look at IM separations at the top of the scale reveals that the peak capacities are quite low, less than 100 (85), but IM can produce data at rates exceeding 100 peaks a second (86). The next “data dense” separation technique is liquid chromatography, where peak capacities exceed 100 for single-dimensional LC, and several thousand if 2D LC is utilized (87,88). The latter is on scale with 2D electrophoresis (89,90), as well as traditional MS techniques such as triple quadrupoles and ion trap instrumentation. High resolution MS techniques exhibit peak capacities approaching 100,000 or greater, and are capable of very high peak production rates ranging from 100,000 peaks/second for Orbitrap MS (FTMS), to over 100 million peaks/second for time-of-flight (TOF) MS. Ion cyclotron resonance (FTICR) is capable of peak capacities approaching 1 million. Peak capacities beyond ca. 1 million peaks are only available through tandem MS experiments, and/or by coupling multiple dimensions of separation to MS. For example, ion mobility coupled to TOF (IM-MS) offers a similar peak capacity as FTICR, but can generate about an order-of-magnitude more peaks at 10 million peaks/second (86). The addition of LC to MS and IM-MS combined with tandem MS/MS achieves between 1 million and 100 million peaks depending on the specific configuration, with production rates of 1,000 spectra or more per second (80,91). Mass spectrometry imaging (MSI) adds spatial information to the analysis, increasing peak capacity to over 1 billion peaks, with a ca. 100,000 peaks per second production rate (92). A combination of MSI experiments to IM-MS and tandem MS/MS analysis, generates over 10 billion peaks at a rate of ca. 1 million peaks per second (93). Finally, 3-dimensional MSI which obtains layered spatial information is capable of very high data density, here illustrated by a recent example of over half a million MSI pixels coupled to TOF, which generates over 100 billion peaks at a rate of more than 100,000 peaks per second (94). If ion mobility experiments and/or tandem MS/MS were included, then a theoretical peak capacity in excess of 1 trillion is possible with 3D MSI. This small sampling of possible analytical configurations does not take into consideration the inclusion of experimental time-points or cohorts, which are a component of comprehensive biological studies. Conservatively, multidimensional MS data generation is on the order of thousands of peaks per second, with capabilities for resolving tens of thousands of molecular signals in a single experimental sequence.
Figure 2.

Peak capacities (right panel) and peak capacity production rates (left panel) for hybrid multidimensional mass spectrometry and related techniques. Specific techniques and combinations are selected based on the availability of information in the literature.
THE IMPORTANCE OF MULTIDIMENSIONAL MS
The fundamental analytical approach to system complexity is to reduce a complex problem into manageable subsets of data. In analytical chemistry, this involves elucidating the chemical structure of an analyte from an initially unknown sample, or conversely, by characterizing a complex sample by descriptors of its molecular constituents. For MS, a conventional approach to molecular identification is to use the mass measurement to reduce several million possible molecular formulas to a few thousand or less towards initial characterization (95). Other orthogonal pieces of information are then used in a complementary fashion to assign a structural identity to the unknown analyte. This challenge is often understated, and so it is instructive to revisit the challenge of structural elucidation by MS using a relevant example.
Figure 3 illustrates an example of molecular identification using an MS-centric approach. For molecules less than 2000 Da, there are over 8 billion possible molecular formulas which can be assigned to an unknown analyte (96). Typically, an initial sample fractionation step such as chromatography is utilized in order to reduce complexity, but also to avoid ion suppression effects which are inherent in all MS ion sources (97). The latter motivation is important because it is often stated that post-ionization separations such as ion mobility can offset the need for condensed phase separations (electrophoresis or chromatography), but such strategies cannot address limitations of the ion source itself. Once a mass measurement is obtained, the number of possible structures can be reduced significantly. In this example, a mass of 354 Da at unit resolution represents over 200,000 possible chemical structures indexed in PubChem, which contains over 60M verified compounds as of 2015. At 100 ppm (354.16±0.03 Da), the number of structures reduces to ca. 44k, and at 10 ppm (354.158±0.003 Da) there are just over 11k indexed structures. With a combination of high mass accuracy (ca. 1 ppm) and invoking filtering rules based on isotope pattern matching and probable structures (96), a molecular formula can be assigned to the analyte with high confidence. Marshall and coworkers have demonstrated that a mass accuracy on the order of 0.1 mDa (0.2 ppm at 500 Da) is necessary to unambiguously assign a molecular formula based on mass measurement alone (98), and this level of high mass accuracy has been demonstrated for FT-ICR (99) and Orbitrap MS (100). Whereas these examples demonstrate that it is tractable to assign a unique molecular formula based on the mass measurement alone, the molecular formula is not a specific descriptor of the analyte. For example, Figure 3 demonstrates that there are still over 10,000 possible isomeric structures in PubChem for this particular chemical formula (C20H22N2O4). In order to translate molecular formula information to a unique chemical structure, other orthogonal pieces of information must be utilized, such as tandem MS data.
Figure 3.

An example of the molecular characterization workflow using multidimensional mass spectrometry. From top to bottom: over 8 billion possible molecular formulas exist between 0 and 2000 Da. Obtaining the mass measurement allows database searching, which here is illustrated by the over 61 million compounds indexed in PubChem at the time of this review. Subsequent levels of mass accuracy reduce the number of possible molecular formulas from over 200,000 (unit resolution), to ca. 10,000 at 1 ppm mass accuracy for the example mass of 354 Da. Using higher mass accuracy and/or a heuristic filtering approach obtains a unique molecular formula, which is still represents several thousand isomeric compounds. Obtaining a unique molecular identity requires additional measurement dimensions, such as tandem MS/MS, LC, IM, and perhaps measurements from other analytical techniques (spectroscopy, NMR, etc.).
To place this example in the context of big data, Figure 4 illustrates the support and limitations that large-scale databases can provide for MS-based characterizations. The current scope of the PubChem compound database between 0 and1000 Da is projected in the histogram contained in Figure 4A. Two previous surveys of the database are also shown (96,101), which provide a sense of the volume of data that is currently being generated. Of note is that smaller molecules are being added at a faster rate, which has shifted the distribution of compounds represented since the 2007 survey. The survey conducted in this study reveals a bimodal distribution at low mass, which we interpret as reflecting the greater effort being made in comprehensive annotation of the smaller molecules. Over time, we anticipate that the distribution will normalize and shift back to higher mass, where more possible isomeric structures reside in chemical space. The dotted line in Figure 4A depicts the theoretically possible unique molecular formulas based on valence rules (96). At low mass, the number of possible isomeric structures for each molecular formula decreases significantly, but regardless, there are still a very large number of isomeric structures represented in the PubChem database below ca. 400 Da. For example, valence and chemical stability rules suggest that the number of possible molecular formulas at ca. 250 Da for an organic molecule (C, H, N, and O) is on the order of 5,000, but empirically there are over 200,000 validated chemical structures in this mass range. Even at this tractable mass range, the challenge of assigning a unique structure to an analyte is still quite formidable. The inset in Figure 4A shows the distribution of molecules within a 10 Da mass window, and at this level of zoom the so-called “forbidden zones” resulting from quantized mass spacing are clearly visible (102). Thus, while MS has very high peak capacity, much of the possible signal clusters within narrow regions of mass space to an extent that is specific to the atomic composition originating from the mass defect [properly, the mass excess (78)]. In this 10 Da window, there are over half a million verified chemical structures. Figure 4B illustrates three panels of increasing levels of zoom in which the histogram resolutions are scaled to different mass accuracies. A 1 Da mass window at 100 ppm focuses on a single cluster of molecular structures within the database, containing over 200,000 structures. Higher mass bin resolution (10 ppm) brings this number of structures down to ca. 48k, and at 1 ppm mass accuracy, the number of structures can be reduced to about 10,000, here represented by 5 possible molecular formulas or less for each 1 ppm resolution bin. For most molecular formulas in this range, there are only 10s to 100s of structures represented, but highlighted is the dramatic case described previously where one molecular formula (C20H22N2O4) represents over 10,000 isomeric structures indexed in the PubChem Compound Database.
Figure 4.

An illustration of the amount of information density present at different levels of mass measurement accuracy, using the validated entries in the PubChem compound database. (A) The distribution of molecules in the PubChem compound database between 0 and 1000 Da, as surveyed in this current work and in two previous surveys from 2007 and 2011. As new compounds are discovered and archived, the distribution has shifted to lower mass, with most entries currently centered between 100 and 600 Da. Theoretical molecular formulas determined from chemical stability rules are illustrated by the dotted line, indicating that most of these entries are isomers. The inset zooms in on a 10 Da window where over half a million compounds are represented. (B) At increasing levels of mass accuracy, the number of possible molecular formulas can be reduced to a few thousand, but in one extreme case shown at 1 ppm, one formula is represented by over 10,000 isomers in the database. Mass spectrometry can significantly reduce complexity, but it cannot fully address molecular characterization without other dimensions of information.
Integrating additional separation dimensions with MS is thus necessary to address sample complexity. While many combinations of condensed and gas-phase separations have been demonstrated to work well with MS, there are inherent technological limitations imposed for specific combinations. For example, seamless coupling of LC to MS requires a continuous liquid stream ion source operated at ambient pressure, such as electrospray or atmospheric pressure chemical ionization (103). On the other hand, MSI is conventionally coupled to MS by means of a pulsed ion beam or laser source to provide high speed, discrete ionization of spatial locations on the sample (104,105). Developments in MSI-MS using liquid sampling probes are now beginning to emerge (106,107), which reduces sample pretreatment at a cost of throughput and spatial resolution. Coupling ion mobility to mass spectrometry (IM-MS) requires transferring ions across disparate pressure regions and analyses in an efficient manner, and this challenge has been addressed through electrodynamic ion optics and the nesting of analytical timescales (108,109,84). Despite some limitations, there are numerous combinations of separations which have been adapted to MS, each of which provide a unique level of information when combined.
COMPARISON OF ORTHOGONAL SEPARATION DIMENSIONS
MASS DEFECT ANALYSIS
The accurate mass measurement provides a highly specific measurement of an intrinsic property of the analyte, the exact mass, but as noted previously, the mass measurement alone does not provide specific information beyond the chemical composition. Several MS studies have exploited the additional information which can be gained from derivative comparisons (i.e., change in mass) using the single dimension of MS analysis, namely through the correlation of the small mass shift from nominal mass owing to the mass defect. Mass defect refers to the change in the nominal mass due to the binding energy of chemical bonds, which manifests as the decimal mass measurement in high resolution MS data (110). Since this mass shift reflects the chemical composition of the analyte, an orthogonal comparison between the nominal mass and the mass defect will group the measurements into chemical class families, which provides a convenient means of locating related chemical species in a complex MS spectrum. MS measurements are based on the IUPAC mass scale which normalizes the measurement to carbon-12 (12C = 12.00000 Da), but mass defect analysis commonly utilizes a rescaled mass axis to help identify small mass differences relative to a reference mass that represents a specific molecule of interest. Two mass defect scales which have been utilized are the Kendrick scale and the averagine scale. The Kendrick scale normalizes the mass axis to CH2 which is 14.00000 Da (111), and has found widespread utility in analyzing the chemical constituents in petroleum (112,113), as well as in MS analysis of lipids (114). An example of a Kendrick mass defect plot is contained in Figure 5, which aligns families of compounds into easily-discernible groups, as is shown for the separation of constituents contained in crude oil (112). The averagine scale is normalized to the “averagine” subunit, which is a theoretical amino acid based on the statistically-weighted occurrence of amino acids in the PIR protein database as of 1995 (C4.9384H7.7583N1.3577O1.4773S0.0417) (115,116). The averagine mass scale has been used in MS proteomics to aid in peptide identification (117), although axis rescaling is not necessary, and peptide mass defect analysis has been carried out using the conventional IUPAC scale (118). Of note is a recent effort to theoretically enumerate all possible tryptic peptides of 3.5 kDa or less to support among other initiatives, mass defect-based identifications (119,120). Mass defect analysis is commonly used in drug metabolism studies where it is desirable to search out exogenous metabolites which possess chemical compositions that are related to the drug species of interest (121,122,123). The mass defect is also particularly effective at identifying surfactant and halogenated compound contaminants in complex samples (124,125).
Figure 5.

A Kendrick mass defect plot of crude oil. The plot projects the nominal (integer) Kendrick mass on the x-axis, and the difference between nominal and exact Kendrick mass on the y-axis. In this way, chemically-similar compounds align horizontally, while chemical class families group into distinct regions on the plot. Here, odd mass species (e.g., carbon-13) are projected to validate the assignments made for the primary even mass species in the original work. The numbers below each class header at right correspond to the number of degrees of unsaturation within each chemical family. Reprinted with permission from Hughey CA, Hendrickson CL, Rodgers RP, Marshall AG, Qian K. 2001. Analytical Chemistry 73: 4676–81. Copyright 2001 American Chemical Society.
ION MOBILTY AND MOBILITY-MASS CORRELATIONS
Ion mobility has emerged in recent years as a mature analytical technique capable of rapid separations which seamlessly integrate into a variety of MS instrument platforms (84). The benefit of the separation is through increasing the peak capacity of the analysis as well as providing an additional level of discrimination by filtering out desirable signals from complex matrices. Ion mobility also provides complimentary information to MS in the form of structural information by means of the gas-phase collision cross section (CCS), which is now a relatively routine parameter to determine from a variety of IM experiments. In contrast to MS, however, the structural information from ion mobility is a less-specific descriptor of the analyte than mass, as the CCS is an orientationally-averaged shape parameter that reduces the 3D structure to a 2D area (126). CCS is also an extrinsic property which depends on the specific parameters of the experiment, and so CCS values are reported with statistics (standard deviation and number of observations) in order to gauge the relative specificity of the measurement. Despite these limitations, CCS databases have recently been demonstrated to have utility in assisting in molecular identification by providing an additional parameter for characterization (127,128). Aided by computational methods (129), the CCS can also be used to infer more detailed structural information regarding the analyte (130,131,132).
COMBINED IM-MS AND MASS DEFECT ANALYSIS
While IM alone provides some level of specificity in the measurement, additional information is gained when combined with MS. At a fundamental level, IM-MS separates analytes by size and mass, which collectively describes the relative gas-phase densities of different structural populations (133,134). These mobility-mass correlations are useful for discerning related structural families towards chemical-class specific filtering (135,136) and characterization schemes (137). As noted previously, class information can also be inferred from analysis using the mass defect originating from the MS measurement. A recent study conducted in the authors’ laboratory for lipids is contained in Figure 6, which compares mass defect analysis to IM-MS correlations for five classes of lipids (138). Raw IM-MS data is visualized in a heat map projection (panel A), which projects the mass-to-charge (x-axis) versus the ion mobility drift time (y-axis), while retaining the third dimension of signal intensity as a color map. In this particular example, a tricolor gradient is used to differentiate the islands of high signal abundance for polar lipids and a series of alkyl ammonium salts spiked in as internal calibrants which form distinct class-specific trends in the raw data (137). Spectral features are then extracted, in this case for singly-charged analytes, and drift times are converted to CCS (panel B). Using the accurate mass measurement, the data can also be projected as mass versus mass defect (panel C), which reveals chemical relationships in the extracted ion signals. Here, the inorganic ammonium salts are easily differentiated from the lipids, and two lipid subclasses, sphingolipids and phospholipids, can be differentiated from each other using either the IM-MS projection (Figure 6B) or the MS-mass defect projection (Figure 6C), illustrating that similar chemical class information can be obtained from both IM-MS and high accuracy MS. Thus IM-MS provides an additional level of information that goes beyond the sum of its parts.
Figure 6.

A multidimensional analysis of five lipid classes (two sphingolipids and three phospholipids) using data obtained from an IM-MS experiment. (A) The raw IM-MS spectrum is projected as a heat map, with m/z on the x-axis, IM drift time on the y-axis, and signal intensity on the color mapping scale. (B) Feature extraction of singly-charged ions is performed, resulting in a “conformational space” plot of mass versus collision cross-section. (C) The accurate mass measurement (ca. 5 ppm in this work) can also be subjected to a mass defect scaling, resulting in a mass vs. mass defect plot. (D) Both the IM-MS conformational space map and the mass defect plot reveal groupings the data based on their respective lipid class, with sphingolipids separated from phospholipids.
NOVEL STRATEGIES FOR VISUALIZING BIG DATA
As the rate and volume of data increases, visualizing important information derived from the data becomes increasingly challenging. Because of the fundamental limitations of human perception, large-scale data cannot be adequately comprehended unless they are reduced to lower-dimensional projections. Successful data visualization strategies thus simplify the level of data complexity and also incorporate familiar and intuitive visual cues which help infer connections between or otherwise provide access to the unseen higher dimensions. A few recent and noteworthy means of visualizing large datasets are reviewed briefly here.
CLOUD PLOTS
A contemporary metabolomics experiment incorporating MS analysis will typically detect on the order of several thousand metabolites representing a diverse array of chemical classes (139,140,141). Because of limited time and information, the majority of these detected metabolites will only be characterized by a few nonspecific descriptors originating from the multidimensional experiment, such as retention time, signal intensity, and molecular mass (142). As such, it is highly desirable to find underlying relationships between discrete metabolites which can be directly correlated to the experimental measurements. Recently, Patti et al. describe a novel visualization format known as a cloud plot, where detected metabolites are projected as “bubbles” onto a plot of retention time vs. mass-to-charge (143). A cloud plot of metabolite data from a sepsis study is contained in Figure 7 where the vertical m/z scaling represents a positive or negative fold change for each metabolite, and the size of each bubble represents the fold change of the corresponding metabolite. In this way, the cloud plot projects simultaneously five dimensions of information: retention time (x-axis), mass-to-charge (y-axis), directional fold-change of signal intensity (positive or negative y-axis projection), magnitude of fold-change (bubble size), and the corresponding p-value from a statistical t-test (bubble color). The LC chromatogram and gradient method are also superimposed on the plot, providing additional information regarding the separation method and by association, the relative chemical hydrophobicity of each detected metabolite. In this particular example, 29,920 extracted spectral features (unique retention time and m/z) are distilled down to the 487 most significant metabolites (p-value ≤ 1.0 x 10−4, fold-change ≥ 3), which are then projected on the cloud plot, providing a means of directly accessing only the most relevant signals originating from the experiment. The cloud plot is an interactive graphic implemented in XCMS online such that clicking on each individual metabolite bubble reveals detailed information such as the putative metabolite assignment from a METLIN database query (144). While developed specifically for metabolomics data, the underlying concept of cloud plots as an intuitive and interactive visualization tool has broad relevance in all multidimensional MS initiatives.
Figure 7.

A cloud plot of LC-MS metabolomics data obtained from a mouse sepsis model. In a cloud plot, the LC retention time is projected on the x-axis, positive/negative fold-change m/z on the y-axis and individual spectral features are plotted as bubbles with size indicating the magnitude of the fold change, and color indicating the statistical significance. Outlined bubbles indicate positive database matches for that particular feature. Also shown is the superimposed LC chromatogram and LC gradient, which provides hydrophobic/hydrophilic information regarding the analytes. In this way, the cloud plot projects a large number of measurement dimensions onto an intuitive graphic which provides ready access to each level of information. Reprinted with permission from Patti GJ, Tautenhahn R, Rinehart D, Cho K, Shriver LP, et al. 2013. Analytical Chemistry 85: 798–804. Copyright 2013 American Chemical Society.
SELF-ORGANIZING MAPS
The majority of MS-based metabolite data has been interpreted through the use of comparative, multivariate statistics which performs binary comparisons of dimensionally-transformed data in order to find underlying relationships between detected signals. Of these, principal component analysis (PCA) is the most widely utilized in metabolomics. PCA enables the visualization of clusters of related signals in the data, but does not provide direct, quantitative information regarding analyte similarity, as the data has been mathematically transformed prior to conducting comparisons (145). One complimentary approach to identifying relationships between detected signals in large datasets is to cluster similar signals using a self-organizing map (SOM). SOMs have found use in untargeted MS-based metabolomics research, where biological relationships are inferred from the clustered metabolites (146,147). While an SOM originating from a single metabolomics experiment can provide insight on the metabolites expressed in one experimental context, the information content is greatly increased when differential analysis is utilized. SOM differential analysis allows numerous comparisons between SOMs originating from discrete samples and/or time points of the experiment. Another benefit of the SOM approach is the ability to “decrypt” specific nodes on the map to extract primary feature information, such as retention time and m/z (148,149). A recent example of using SOMs to systems-level mapping of molecules is contained in Figure 8 for an “organ-on-chip” human liver bioreactor exposed to acetaminophen (APAP) (150). This particular synthetic organ consists of a network of hollow fibers around which are seeded with cultured cells harvested from a human cadaver liver. Cell culture media is perfused through the hollow fibers and time points from the waste stream are analyzed by IM-MS which provides a comprehensive analysis of the metabolites secreted from the cells. In this example, two sets of SOMs are created, representing positive (panel A) and negative (panel B) fold-change of detected features. Following APAP exposure, several regions of the map change in intensity, representing a fold-change for a group of metabolite features. Groups of nodes can be decrypted at any time to obtain primary information which can be utilized for molecular identification (panel C). In this case, the upregulation of APAP bound glucuronic acid is observed in addition to the dysregulation of bile acid, the latter of which is a classic marker for liver stress.
Figure 8.
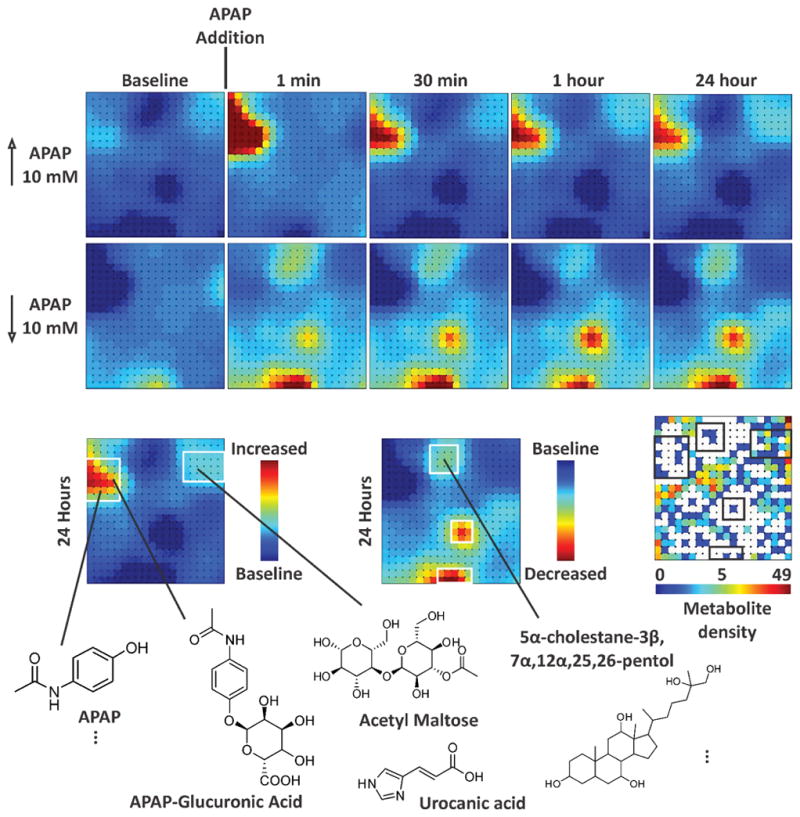
These few examples underscore the importance of effective data visualization strategies for mass spectrometry-based data intensive experiments. Whereas big data holds the promise of new discoveries, the reality of achieving these discoveries will be contingent upon our ability to effectively navigate and find connections between multiple dimensions of information.
CONCLUSION
Mass spectrometry is a rapidly changing field which now encompasses a myriad of allied analytical techniques and disciplines. The current state-of-the-art in hybrid MS instrumentation generates data that is dense with information, and can be obtained must faster than it can be interpreted. This is the big data challenge, and can be considered the breadth and scope of analytical space, which includes the information from each separation dimension, the number of total separation dimensions, and the derived information from comparing dimensions to one another, all placed within the context of a spatial and temporal location. In this context, one may argue that, similar to chemical space, analytical space is both immense and vastly unexplored, providing unique opportunities for future innovation and discovery. We can speculate many things about the future of mass spectrometry based analytical sciences, but one prediction we are certain about is given the continuing integration of high resolution instrumentation into hybrid architectures, the field of mass spectrometry will retain a place at the forefront of big data.
Acknowledgments
The authors are grateful to the following sources of financial support for this work: the National Science Foundation under Award number CHE-1229341, the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers 5UH3TR000491-04 and 3UH3TR000491-04S1, and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM92218. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also acknowledge intramural support for this work provided by the Vanderbilt University Center for Innovative Technology, the Vanderbilt Institute for Chemical Biology, the Vanderbilt Institute for Integrative Biosystems Research and Education, and the Vanderbilt University College of Arts and Sciences.
Footnotes
DISCLOSURE STATEMENT
The authors are unaware of any potential bias which may affect the objectivity of the review, but do acknowledge collaborative arrangements with Agilent Technologies (Santa Clara, CA) and Waters Corporation (Milford, MA). The Vanderbilt University Center for Innovative Technology is designated as an Agilent Thought Leader Laboratory and a Waters Centre of Innovation.
LITERATURE CITED
- 1.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 2.Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–87. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 3.Kim M-S, Pinto SM, Getnet D, Nirujogi RS, Manda SS, et al. A draft map of the human proteome. Nature. 2014;509:575–81. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, et al. Tissue-based map of the human proteome. Science. 2015:347. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 5.Uhlén M, Ponten F. Antibody-based Proteomics for Human Tissue Profiling. Molecular & Cellular Proteomics. 2005;4:384–93. doi: 10.1074/mcp.R500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Marx V. Mapping proteins with spatial proteomics. Nat Meth. 2015;12:815–19. doi: 10.1038/nmeth.3555. [DOI] [PubMed] [Google Scholar]
- 7.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, et al. HMDB: the Human Metabolome Database. Nucleic Acids Research. 2007;35:D521–D26. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Research. 2013;41:D801–D07. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Junot C, Fenaille F, Colsch B, Bécher F. High resolution mass spectrometry based techniques at the crossroads of metabolic pathways. Mass Spectrometry Reviews. 2014;33:471–500. doi: 10.1002/mas.21401. [DOI] [PubMed] [Google Scholar]
- 10.Yetukuri L, Ekroos K, Vidal-Puig A, Oresic M. Informatics and computational strategies for the study of lipids. Molecular BioSystems. 2008;4:121–27. doi: 10.1039/b715468b. [DOI] [PubMed] [Google Scholar]
- 11.Hood L, Heath JR, Phelps ME, Lin B. Systems Biology and New Technologies Enable Predictive and Preventative Medicine. Science. 2004;306:640–43. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson JK, Wilson ID. Understanding ‘Global’ Systems Biology: Metabonomics and the Continuum of Metabolism. Nature Reviews Drug Discovery. 2003;2:668–76. doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- 13.Feng X, Liu X, Luo Q, Liu B-F. Mass spectrometry in systems biology: An overview. Mass Spectrometry Reviews. 2008;27:635–60. doi: 10.1002/mas.20182. [DOI] [PubMed] [Google Scholar]
- 14.Graessel A, Hauck SM, von Toerne C, Kloppmann E, Goldberg T, et al. A Combined Omics Approach to Generate the Surface Atlas of Human Naive CD4+ T Cells during Early T-Cell Receptor Activation. Molecular & Cellular Proteomics. 2015;14:2085–102. doi: 10.1074/mcp.M114.045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bohacek RS, McMartin C, Guida WC. The art and practice of structure-based drug design: A molecular modeling perspective. Medicinal Research Reviews. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 16.Peironcely JE, Reijmers T, Coulier L, Bender A, Hankemeier T. Understanding and Classifying Metabolite Space and Metabolite-Likeness. PLoS ONE. 2011;6:e28966. doi: 10.1371/journal.pone.0028966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfram|Alpha. Queried “number of grains of sand on Earth”. 2015 Sep 28; ( www.wolframalpha.com)
- 18.Gurard-Levin ZA, Scholle MD, Eisenberg AH, Mrksich M. High-Throughput Screening of Small Molecule Libraries using SAMDI Mass Spectrometry. ACS Combinatorial Science. 2011;13:347–50. doi: 10.1021/co2000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Rond T, Danielewicz M, Northen T. High throughput screening of enzyme activity with mass spectrometry imaging. Current Opinion in Biotechnology. 2015;31:1–9. doi: 10.1016/j.copbio.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends in Genetics. 2008;24:133–41. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 21.Stephens ZD, Lee SY, Faghri F, Campbell RH, Zhai C, et al. Big Data: Astronomical or Genomical? PLoS Biology. 2015;13:e1002195. doi: 10.1371/journal.pbio.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laney D. 3D Data Management: Controlling Data Volume, Velocity, and Variety. META Group “Application Delivery Strategies” Whitepaper, File 949 2001 [Google Scholar]
- 23.Lusher SJ, McGuire R, van Schaik RC, Nicholson CD, de Vlieg J. Data-driven medicinal chemistry in the era of big data. Drug Discovery Today. 2014;19:859–68. doi: 10.1016/j.drudis.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Askenazi M, Webber JT, Marto JA. mzServer: Web-based Programmatic Access for Mass Spectrometry Data Analysis. Molecular & Cellular Proteomics. 2011:10. doi: 10.1074/mcp.M110.003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bird I. Computing for the Large Hadron Collider. Annual Review of Nuclear and Particle Science. 2011;61:99–118. [Google Scholar]
- 26.Reymond J-L, Ruddigkeit L, Blum L, van Deursen R. The enumeration of chemical space. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012;2:717–33. [Google Scholar]
- 27.Marks RW, Fuller RB. The Dymaxion World of Buckminster Fuller. Garden City, NY: Anchor Press/Doubleday; 1973. [Google Scholar]
- 28.Wang L. Chemical and Engineering News. American Chemical Society; 2015. Chemical Abstract Service Marks Multiple Milestones. [Google Scholar]
- 29.Bolton EE, Wang Y, Thiessen PA, Bryant SH. Chapter 12 - PubChem: Integrated Platform of Small Molecules and Biological Activities. In: Ralph AW, David CS, editors. Annual Reports in Computational Chemistry. Elsevier; 2008. pp. 217–41. [Google Scholar]
- 30. [Accessed September 28, 2015];PubChem Substance Database. ( https://pubchem.ncbi.nlm.nih.gov)
- 31. [Accessed September 28, 2015];PubChem Compound Database. ( https://pubchem.ncbi.nlm.nih.gov)
- 32.Pence HE, Williams A. ChemSpider: An Online Chemical Information Resource. [Accessed September 28, 2015];Journal of Chemical Education. 2010 87:1123–24. ( www.chemspider.com) [Google Scholar]
- 33.NIST Chemistry WebBook. Linstrom PJ, Mallard WG, editors. [Accessed September 28, 2015];NIST Standard Reference Database Number 69. webbook.nist.gov.
- 34.Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, et al. The ChEMBL bioactivity database: an update. [Accessed September 28, 2015];Nucleic Acids Research. 2014 42:D1083–D90. doi: 10.1093/nar/gkt1031. ( https://www.ebi.ac.uk/chembl/) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, et al. DrugBank 4.0: shedding new light on drug metabolism. [Accessed September 28, 2015];Nucleic Acids Research. 2014 42:D1091–D97. doi: 10.1093/nar/gkt1068. ( http://www.drugbank.ca) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–61. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 37.Virshup AM, Contreras-García J, Wipf P, Yang W, Beratan DN. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. Journal of the American Chemical Society. 2013;135:7296–303. doi: 10.1021/ja401184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bars I, Terning J. Extra Dimensions in Space and Time. New York, NY: Springer-Verlag; 2010. p. 218. [Google Scholar]
- 39.Ruddigkeit L, Blum LC, Reymond J-L. Visualization and Virtual Screening of the Chemical Universe Database GDB-17. Journal of Chemical Information and Modeling. 2013;53:56–65. doi: 10.1021/ci300535x. [DOI] [PubMed] [Google Scholar]
- 40.Reymond J-L. The Chemical Space Project. Accounts of Chemical Research. 2015;48:722–30. doi: 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- 41.Oprea TI, Gottfries J. Chemography: The Art of Navigating in Chemical Space. Journal of Combinatorial Chemistry. 2001;3:157–66. doi: 10.1021/cc0000388. [DOI] [PubMed] [Google Scholar]
- 42.Engel T. Basic Overview of Chemoinformatics†. Journal of Chemical Information and Modeling. 2006;46:2267–77. doi: 10.1021/ci600234z. [DOI] [PubMed] [Google Scholar]
- 43.Varnek A, Baskin II. Chemoinformatics as a Theoretical Chemistry Discipline. Molecular Informatics. 2011;30:20–32. doi: 10.1002/minf.201000100. [DOI] [PubMed] [Google Scholar]
- 44.Scifinder. Chemical Abstracts Service; Columbus, OH: Sep, 2015. [Accessed September 28, 2015]. [Google Scholar]
- 45.Jinha AE. Article 50 million: an estimate of the number of scholarly articles in existence. Learned Publishing. 2010;23:258–63. [Google Scholar]
- 46.Powell JR. The Quantum Limit to Moore’s Law. Proceedings of the IEEE. 2008;96:1247–48. [Google Scholar]
- 47.Walter C. Kryder’s Law. Scientific American. 2005;293:32–22. doi: 10.1038/scientificamerican0805-32. [DOI] [PubMed] [Google Scholar]
- 48.Eldering CA, Sylla ML, Eisenach JA. Is there a Moore’s law for bandwidth? Communications Magazine, IEEE. 1999;37:117–21. [Google Scholar]
- 49.Sobel D. Longitude: The True Story of a Lone Genius Who Solved the Greatest Scientific Problem of His Time. Bloomsbury Publishing; 2010. [Google Scholar]
- 50.Misura KMS, Chivian D, Rohl CA, Kim DE, Baker D. Physically realistic homology models built with rosetta can be more accurate than their templates. Proceedings of the National Academy of Sciences. 2006;103:5361–66. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaufmann KW, Lemmon GH, DeLuca SL, Sheehan JH, Meiler J. Practically Useful: What the Rosetta Protein Modeling Suite Can Do for You. Biochemistry. 2010;49:2987–98. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khatib F, Cooper S, Tyka MD, Xu K, Makedon I, et al. Algorithm discovery by protein folding game players. Proceedings of the National Academy of Sciences. 2011;108:18949–53. doi: 10.1073/pnas.1115898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khatib F, DiMaio F, Cooper S, Kazmierczyk M, Gilski M, et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players. Nat Struct Mol Biol. 2011;18:1175–77. doi: 10.1038/nsmb.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilski M, Kazmierczyk M, Krzywda S, Zabranska H, Cooper S, et al. High-resolution structure of a retroviral protease folded as a monomer. Acta Crystallographica Section D. 2011;67:907–14. doi: 10.1107/S0907444911035943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eiben CB, Siegel JB, Bale JB, Cooper S, Khatib F, et al. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players. Nat Biotech. 2012;30:190–92. doi: 10.1038/nbt.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bradley J-C, Lancashire R, Lang A, Williams A. The Spectral Game: leveraging Open Data and crowdsourcing for education. Journal of Cheminformatics. 2009;1:9. doi: 10.1186/1758-2946-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Du L, Robles AJ, King JB, Powell DR, Miller AN, et al. Crowdsourcing Natural Products Discovery to Access Uncharted Dimensions of Fungal Metabolite Diversity. Angewandte Chemie International Edition. 2014;53:804–09. doi: 10.1002/anie.201306549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin SF, Falkenberg H, Dyrlund TF, Khoudoli GA, Mageean CJ, Linding R. PROTEINCHALLENGE: Crowd sourcing in proteomics analysis and software development. Journal of Proteomics. 2013;88:41–46. doi: 10.1016/j.jprot.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 59.Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP) — round x. Proteins: Structure, Function, and Bioinformatics. 2014;82:1–6. doi: 10.1002/prot.24452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, et al. Wisdom of crowds for robust gene network inference. Nat Meth. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bishop CM. Pattern Recognition and Machine Learning. Springer; 2006. [Google Scholar]
- 62.Smalheiser NR. Informatics and hypothesis-driven research. EMBO reports. 2002;3:702–02. doi: 10.1093/embo-reports/kvf164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashem IAT, Yaqoob I, Anuar NB, Mokhtar S, Gani A, Ullah Khan S. The rise of “big data” on cloud computing: Review and open research issues. Information Systems. 2015;47:98–115. [Google Scholar]
- 64.Chen T, Zhao J, Ma J, Zhu Y. Web Resources for Mass Spectrometry-based Proteomics. Genomics, Proteomics & Bioinformatics. 2015;13:36–39. doi: 10.1016/j.gpb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Analytical Chemistry. 2012;84:5035–39. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rinehart D, Johnson CH, Nguyen T, Ivanisevic J, Benton HP, et al. Metabolomic data streaming for biology-dependent data acquisition. Nature Biotechnology. 2014;32:524–27. doi: 10.1038/nbt.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rübel O, Greiner A, Cholia S, Louie K, Bethel EW, et al. OpenMSI: A High-Performance Web-Based Platform for Mass Spectrometry Imaging. Analytical Chemistry. 2013;85:10354–61. doi: 10.1021/ac402540a. [DOI] [PubMed] [Google Scholar]
- 68.Fischer CR, Ruebel O, Bowen BP. An accessible, scalable ecosystem for enabling and sharing diverse mass spectrometry imaging analyses. Archives of Biochemistry and Biophysics. 2015 doi: 10.1016/j.abb.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 69.Malm E, Srivastava V, Sundqvist G, Bulone V. APP: an Automated Proteomics Pipeline for the analysis of mass spectrometry data based on multiple open access tools. BMC Bioinformatics. 2014;15:441. doi: 10.1186/s12859-014-0441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mohammed Y, Mostovenko E, Henneman AA, Marissen RJ, Deelder AM, Palmblad M. Cloud Parallel Processing of Tandem Mass Spectrometry Based Proteomics Data. Journal of Proteome Research. 2012;11:5101–08. doi: 10.1021/pr300561q. [DOI] [PubMed] [Google Scholar]
- 71.Muth T, Peters J, Blackburn J, Rapp E, Martens L. ProteoCloud: A full-featured open source proteomics cloud computing pipeline. Journal of Proteomics. 2013;88:104–08. doi: 10.1016/j.jprot.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 72.Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics – Clinical Applications. 2015;9:745–54. doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Slagel J, Mendoza L, Shteynberg D, Deutsch EW, Moritz RL. Processing Shotgun Proteomics Data on the Amazon Cloud with the Trans-Proteomic Pipeline. Molecular & Cellular Proteomics. 2015;14:399–404. doi: 10.1074/mcp.O114.043380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Riffle M, Eng JK. Proteomics data repositories. Proteomics. 2009;9:4653–63. doi: 10.1002/pmic.200900216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez-Riverol Y, Alpi E, Wang R, Hermjakob H, Vizcaíno JA. Making proteomics data accessible and reusable: Current state of proteomics databases and repositories. PROTEOMICS. 2015;15:930–50. doi: 10.1002/pmic.201400302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karger BL, Snyder LR, Horvath C. Introduction to Separation Science. John Wiley & Sons; 1973. p. 608. [Google Scholar]
- 77.Giddings JC. Two-dimensional separations: concept and promise. Analytical Chemistry. 1984;56:1258A–70A. doi: 10.1021/ac00276a003. [DOI] [PubMed] [Google Scholar]
- 78.Frahm JL, Howard BE, Heber S, Muddiman DC. Accessible proteomics space and its implications for peak capacity for zero-, one- and two-dimensional separations coupled with FT-ICR and TOF mass spectrometry. Journal of Mass Spectrometry. 2006;41:281–88. doi: 10.1002/jms.1024. [DOI] [PubMed] [Google Scholar]
- 79.Barner-Kowollik C, Gruendling T, Falkenhagen J, Weidner S. Mass Spectrometry in Polymer Chemistry. Wiley; 2012. [Google Scholar]
- 80.Canterbury JD, Yi X, Hoopmann MR, MacCoss MJ. Assessing the Dynamic Range and Peak Capacity of Nanoflow LC FAIMS MS on an Ion Trap Mass Spectrometer for Proteomics. Analytical Chemistry. 2008;80:6888–97. doi: 10.1021/ac8004988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider B, Nazarov E, Covey T. Peak capacity in differential mobility spectrometry: effects of transport gas and gas modifiers. International Journal for Ion Mobility Spectrometry. 2012;15:141–50. [Google Scholar]
- 82.Merenbloom SI, Bohrer BC, Koeniger SL, Clemmer DE. Assessing the Peak Capacity of IMS IMS Separations of Tryptic Peptide Ions in He at 300 K. Analytical Chemistry. 2007;79:515–22. doi: 10.1021/ac061567m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.May JC, McLean JA. The influence of drift gas composition on the separation mechanism in traveling wave ion mobility spectrometry: insight from electrodynamic simulations. International Journal for Ion Mobility Spectrometry. 2013;16:85–94. doi: 10.1007/s12127-013-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.May JC, McLean JA. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Analytical Chemistry. 2015;87:1422–36. doi: 10.1021/ac504720m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Causon TJ, Hann S. Theoretical evaluation of peak capacity improvements by use of liquid chromatography combined with drift tube ion mobility-mass spectrometry. Journal of Chromatography A. 2015;1416:47–56. doi: 10.1016/j.chroma.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 86.McLean JA, Ruotolo BT, Gillig KJ, Russell DH. Ion Mobility-Mass Spectrometry: a New Paradigm for Proteomics. International Journal of Mass Spectrometry. 2005;240:301–15. [Google Scholar]
- 87.Neue UD. Theory of peak capacity in gradient elution. Journal of Chromatography A. 2005;1079:153–61. doi: 10.1016/j.chroma.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 88.Neue UD. Peak capacity in unidimensional chromatography. Journal of Chromatography A. 2008;1184:107–30. doi: 10.1016/j.chroma.2007.11.113. [DOI] [PubMed] [Google Scholar]
- 89.Moore AW, Jorgenson JW. Comprehensive three-dimensional separation of peptides using size exclusion chromatography/reversed phase liquid chromatography/optically gated capillary zone electrophoresis. Analytical Chemistry. 1995;67:3456–63. doi: 10.1021/ac00115a014. [DOI] [PubMed] [Google Scholar]
- 90.Tia S, Herr AE. On-chip technologies for multidimensional separations. Lab on a Chip. 2009;9:2524–36. doi: 10.1039/b900683b. [DOI] [PubMed] [Google Scholar]
- 91.Bruce JE, Anderson GA, Wen J, Harkewicz R, Smith RD. High-Mass-Measurement Accuracy and 100 Sequence Coverage of Enzymatically Digested Bovine Serum Albumin from an ESI-FTICR Mass Spectrum. Analytical Chemistry. 1999;71:2595–99. doi: 10.1021/ac990231s. [DOI] [PubMed] [Google Scholar]
- 92.Prentice BM, Chumbley CW, Caprioli RM. High-speed MALDI MS/MS imaging mass spectrometry using continuous raster sampling. Journal of Mass Spectrometry. 2015;50:703–10. doi: 10.1002/jms.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stauber J, MacAleese L, Franck J, Claude E, Snel M, et al. On-tissue protein identification and imaging by MALDI-Ion mobility mass spectrometry. Journal of the American Society for Mass Spectrometry. 2010;21:338–47. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 94.Trede D, Schiffler S, Becker M, Wirtz S, Steinhorst K, et al. Exploring Three-Dimensional Matrix-Assisted Laser Desorption/Ionization Imaging Mass Spectrometry Data: Three-Dimensional Spatial Segmentation of Mouse Kidney. Analytical Chemistry. 2012;84:6079–87. doi: 10.1021/ac300673y. [DOI] [PubMed] [Google Scholar]
- 95.Kind T, Fiehn O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanalytical Reviews. 2010;2:23–60. doi: 10.1007/s12566-010-0015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007;8:1–20. doi: 10.1186/1471-2105-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Annesley TM. Ion Suppression in Mass Spectrometry. Clinical Chemistry. 2003;49:1041–44. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 98.Kim S, Rodgers RP, Marshall AG. Truly “exact” mass: Elemental composition can be determined uniquely from molecular mass measurement at 0.1 mDa accuracy for molecules up to 500 Da. International Journal of Mass Spectrometry. 2006;251:260–65. [Google Scholar]
- 99.Savory JJ, Kaiser NK, McKenna AM, Xian F, Blakney GT, et al. Parts-Per-Billion Fourier Transform Ion Cyclotron Resonance Mass Measurement Accuracy with a “Walking” Calibration Equation. Analytical Chemistry. 2011;83:1732–36. doi: 10.1021/ac102943z. [DOI] [PubMed] [Google Scholar]
- 100.Scheltema RA, Kamleh A, Wildridge D, Ebikeme C, Watson DG, et al. Increasing the mass accuracy of high-resolution LC-MS data using background ions – a case study on the LTQ-Orbitrap. PROTEOMICS. 2008;8:4647–56. doi: 10.1002/pmic.200800314. [DOI] [PubMed] [Google Scholar]
- 101.Green FM, Gilmore IS, Seah MP. Mass Spectrometry and Informatics: Distribution of Molecules in the PubChem Database and General Requirements for Mass Accuracy in Surface Analysis. Analytical Chemistry. 2011;83:3239–43. doi: 10.1021/ac200067s. [DOI] [PubMed] [Google Scholar]
- 102.Nefedov AV, Mitra I, Brasier AR, Sadygov RG. Examining Troughs in the Mass Distribution of All Theoretically Possible Tryptic Peptides. Journal of Proteome Research. 2011;10:4150–57. doi: 10.1021/pr2003177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yergey AL, Edmonds CG, Lewis IAS, Vestal ML. Liquid Chromatography/Mass Spectrometry: Techniques and Applications. Springer US; 2013. [Google Scholar]
- 104.Norris JL, Caprioli RM. Analysis of Tissue Specimens by Matrix-Assisted Laser Desorption/Ionization Imaging Mass Spectrometry in Biological and Clinical Research. Chemical Reviews. 2013;113:2309–42. doi: 10.1021/cr3004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watrous JD, Dorrestein PC. Imaging mass spectrometry in microbiology. Nat Rev Micro. 2011;9:683–94. doi: 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lanekoff I, Burnum-Johnson K, Thomas M, Cha J, Dey S, et al. Three-dimensional imaging of lipids and metabolites in tissues by nanospray desorption electrospray ionization mass spectrometry. Analytical and Bioanalytical Chemistry. 2015;407:2063–71. doi: 10.1007/s00216-014-8174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Calligaris D, Caragacianu D, Liu X, Norton I, Thompson CJ, et al. Application of desorption electrospray ionization mass spectrometry imaging in breast cancer margin analysis. Proceedings of the National Academy of Sciences. 2014;111:15184–89. doi: 10.1073/pnas.1408129111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giles K, Pringle SD, Worthington KR, Little DL, WJ, Bateman RH. Applications of a Traveling Wave-Based Radio-Frequency-Only Stacked Ring Ion Guide. Rapid Communications in Mass Spectrometry. 2004;18:2401–14. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 109.Baker ES, Clowers BH, Li F, Tang K, Tolmachev AV, et al. Ion mobility spectrometry—mass spectrometry performance using electrodynamic ion funnels and elevated drift gas pressures. Journal of the American Society for Mass Spectrometry. 2007;18:1176–87. doi: 10.1016/j.jasms.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sleno L. The use of mass defect in modern mass spectrometry. Journal of Mass Spectrometry. 2012;47(2):226–236. doi: 10.1002/jms.2953. [DOI] [PubMed] [Google Scholar]
- 111.Kendrick E. A Mass Scale Based on CH2 = 14.0000 for High Resolution Mass Spectrometry of Organic Compounds. Analytical Chemistry. 1963;35:2146–54. [Google Scholar]
- 112.Hughey CA, Hendrickson CL, Rodgers RP, Marshall AG, Qian K. Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra. Analytical Chemistry. 2001;73:4676–81. doi: 10.1021/ac010560w. [DOI] [PubMed] [Google Scholar]
- 113.Marshall AG, Rodgers RP. Petroleomics: The Next Grand Challenge for Chemical Analysis. Accounts of Chemical Research. 2004;37:53–59. doi: 10.1021/ar020177t. [DOI] [PubMed] [Google Scholar]
- 114.Lerno LA, German JB, Lebrilla CB. Method for the Identification of Lipid Classes Based on Referenced Kendrick Mass Analysis. Analytical Chemistry. 2010;82:4236–45. doi: 10.1021/ac100556g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Senko MW, Beu SC, McLafferty FW. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. Journal of the American Society for Mass Spectrometry. 1995;6:229–33. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 116.Wu CH, Yeh L-SL, Huang H, Arminski L, Castro-Alvear J, et al. The Protein Information Resource. Nucleic Acids Research. 2003;31:345–47. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yao X, Diego P, Ramos AA, Shi Y. Averagine-Scaling Analysis and Fragment Ion Mass Defect Labeling in Peptide Mass Spectrometry. Analytical Chemistry. 2008;80:7383–91. doi: 10.1021/ac801096e. [DOI] [PubMed] [Google Scholar]
- 118.Toumi ML, Desaire H. Improving Mass Defect Filters for Human Proteins. Journal of Proteome Research. 2010;9:5492–95. doi: 10.1021/pr100291q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nefedov AV, Mitra I, Brasier AR, Sadygov RG. Examining Troughs in the Mass Distribution of All Theoretically Possible Tryptic Peptides. Journal of Proteome Research. 2011;10:4150–57. doi: 10.1021/pr2003177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mitra I, Nefedov AV, Brasier AR, Sadygov RG. Improved Mass Defect Model for Theoretical Tryptic Peptides. Analytical Chemistry. 2012;84:3026–32. doi: 10.1021/ac203255e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cuyckens F, Hurkmans R, Castro-Perez JM, Leclercq L, Mortishire-Smith RJ. Extracting metabolite ions out of a matrix background by combined mass defect, neutral loss and isotope filtration. Rapid Communications in Mass Spectrometry. 2009;23:327–32. doi: 10.1002/rcm.3881. [DOI] [PubMed] [Google Scholar]
- 122.Zhang H, Zhang D, Ray K, Zhu M. Mass defect filter technique and its applications to drug metabolite identification by high-resolution mass spectrometry. Journal of Mass Spectrometry. 2009;44:999–1016. doi: 10.1002/jms.1610. [DOI] [PubMed] [Google Scholar]
- 123.Zhu M, Ma L, Zhang D, Ray K, Zhao W, et al. Detection and Characterization of Metabolites in Biological Matrices Using Mass Defect Filtering of Liquid Chromatography/High Resolution Mass Spectrometry Data. Drug Metabolism and Disposition. 2006;34:1722–33. doi: 10.1124/dmd.106.009241. [DOI] [PubMed] [Google Scholar]
- 124.Li X, Brownawell BJ. Analysis of Quaternary Ammonium Compounds in Estuarine Sediments by LC ToF-MS: Very High Positive Mass Defects of Alkylamine Ions as Powerful Diagnostic Tools for Identification and Structural Elucidation. Analytical Chemistry. 2009;81:7926–35. doi: 10.1021/ac900900y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nagy K, Sandoz L, Craft BD, Destaillats F. Mass-defect filtering of isotope signatures to reveal the source of chlorinated palm oil contaminants. Food Additives & Contaminants: Part A. 2011;28:1492–500. doi: 10.1080/19440049.2011.618467. [DOI] [PubMed] [Google Scholar]
- 126.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. New York: John Wiley & Sons; 1988. p. 560. [Google Scholar]
- 127.Lietz CB, Yu Q, Li L. Large-Scale Collision Cross-Section Profiling on a Traveling Wave Ion Mobility Mass Spectrometer. Journal of The American Society for Mass Spectrometry. 2014;25:2009–19. doi: 10.1007/s13361-014-0920-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Paglia G, Williams JP, Menikarachchi L, Thompson JW, Tyldesley-Worster R, et al. Ion Mobility Derived Collision Cross Sections to Support Metabolomics Applications. Analytical Chemistry. 2014;86:3985–93. doi: 10.1021/ac500405x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wyttenbach T, Pierson NA, Clemmer DE, Bowers MT. Ion mobility analysis of molecular dynamics. Annual Revew of Physical Chemistry. 2014;65:175–96. doi: 10.1146/annurev-physchem-040513-103644. [DOI] [PubMed] [Google Scholar]
- 130.Zhong Y, Hyung S-J, Ruotolo BT. Ion mobility–mass spectrometry for structural proteomics. Expert Review of Proteomics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lapthorn C, Pullen F, Chowdhry BZ. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: Separating and assigning structures to ions. Mass Spectrometry Reviews. 2012;32:43–71. doi: 10.1002/mas.21349. [DOI] [PubMed] [Google Scholar]
- 132.Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–94. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 133.Berant Z, Karpas Z. Mass-mobility correlation of ions in view of new mobility data. Journal of the American Chemical Society. 1989;111:3819–24. [Google Scholar]
- 134.Fenn LS, Kliman M, Mahsut A, Zhao SR, McLean JA. Characterizing ion mobility-mass spectrometry conformation space for the analysis of complex biological samples. Analytical and Bioanalytical Chemistry. 2009;394:235–44. doi: 10.1007/s00216-009-2666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Harvey D, Crispin M, Bonomelli C, Scrivens J. Ion Mobility Mass Spectrometry for Ion Recovery and Clean-Up of MS and MS/MS Spectra Obtained from Low Abundance Viral Samples. Journal of The American Society for Mass Spectrometry. 2015;26:1754–67. doi: 10.1007/s13361-015-1163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li H, Bendiak B, Siems W, Gang D, Hill H., Jr Ion mobility-mass correlation trend line separation of glycoprotein digests without deglycosylation. International Journal for Ion Mobility Spectrometry. 2013;16:105–15. doi: 10.1007/s12127-013-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.May JC, Goodwin CR, Lareau NM, Leaptrot KL, Morris CB, et al. Conformational Ordering of Biomolecules in the Gas Phase: Nitrogen Collision Cross Sections Measured on a Prototype High Resolution Drift Tube Ion Mobility-Mass Spectrometer. Analytical Chemistry. 2014;86:2107–16. doi: 10.1021/ac4038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.May JC, McLean JA. Lipid Map. Analytical Scientist. 2014:0814. [Google Scholar]
- 139.Ceglarek U, Leichtle A, Brügel M, Kortz L, Brauer R, et al. Challenges and developments in tandem mass spectrometry based clinical metabolomics. Molecular and Cellular Endocrinology. 2009;301:266–71. doi: 10.1016/j.mce.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 140.Benton HP, Ivanisevic J, Mahieu NG, Kurczy ME, Johnson CH, et al. Autonomous Metabolomics for Rapid Metabolite Identification in Global Profiling. Analytical Chemistry. 2015;87:884–91. doi: 10.1021/ac5025649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Patti GJ, Yanes O, Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13:263–69. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Johnson CH, Ivanisevic J, Benton HP, Siuzdak G. Bioinformatics: The Next Frontier of Metabolomics. Analytical Chemistry. 2015;87:147–56. doi: 10.1021/ac5040693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Patti GJ, Tautenhahn R, Rinehart D, Cho K, Shriver LP, et al. A View from Above: Cloud Plots to Visualize Global Metabolomic Data. Analytical Chemistry. 2013;85:798–804. doi: 10.1021/ac3029745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gowda H, Ivanisevic J, Johnson CH, Kurczy ME, Benton HP, et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Analytical Chemistry. 2014;86:6931–39. doi: 10.1021/ac500734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Goodacre R, Neal MJ, Kell DB. Quantitative Analysis of Multivariate Data Using Artificial Neural Networks: A Tutorial Review and Applications to the Deconvolution of Pyrolysis Mass Spectra. Zentralblatt für Bakteriologie. 1996;284:516–39. doi: 10.1016/s0934-8840(96)80004-1. [DOI] [PubMed] [Google Scholar]
- 146.Franceschi P, Wehrens R. Self-organizing maps: A versatile tool for the automatic analysis of untargeted imaging datasets. PROTEOMICS. 2014;14:853–61. doi: 10.1002/pmic.201300308. [DOI] [PubMed] [Google Scholar]
- 147.Patterson AD, Li H, Eichler GS, Krausz KW, Weinstein JN, et al. UPLC-ESI-TOFMS-Based Metabolomics and Gene Expression Dynamics Inspector Self-Organizing Metabolomic Maps as Tools for Understanding the Cellular Response to Ionizing Radiation. Analytical Chemistry. 2008;80:665–74. doi: 10.1021/ac701807v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goodwin CR, Sherrod SD, Marasco CC, Bachmann BO, Schramm-Sapyta N, et al. Phenotypic Mapping of Metabolic Profiles Using Self-Organizing Maps of High-Dimensional Mass Spectrometry Data. Analytical Chemistry. 2014;86:6563–71. doi: 10.1021/ac5010794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Goodwin Cody R, Covington Brett C, Derewacz Dagmara K, McNees CR, Wikswo John P, et al. Structuring Microbial Metabolic Responses to Multiplexed Stimuli via Self-Organizing Metabolomics Maps. Chemistry & Biology. 2015;22:661–70. doi: 10.1016/j.chembiol.2015.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sherrod SD, McLean JA. Systems-Wide High Dimensional Data Acquisition and Informatics Using Structural Mass Spectrometry Strategies. Clinical Chemistry. 2015 doi: 10.1373/clinchem.2015.238261. article in press. [DOI] [PMC free article] [PubMed] [Google Scholar]