

Abstract
The schweinfurthins are plant-derived stilbenes with an intriguing profile of anti-cancer activity. To obtain analogues of the schweinfurthins that might preserve the biological activity but have greater water solubility, a formal replacement of the central olefin with an amide has been explored. Two pairs of amides have been prepared, each containing the same hexahydroxanthene “left half” joined through an amide linkage to two different “right halves.” In each series, the amide has been inserted in both possible orientations, placing the carbonyl group on the tricyclic ABC ring system and the amine on the D-ring, or placing the amine on the hexahydroxanthene and the carbonyl group on the D-ring. The four new schweinfurthin analogues have been tested in the NCI 60 cell line screen, and in both cases the more active isomer carried the carbonyl group on the C-ring.
Keywords: Schweinfurthin, stilbene, amide isostere, cancer
TOC image
Introduction
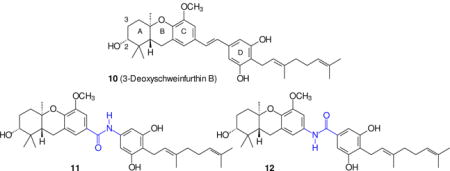
The natural products commonly grouped as schweinfurthins include six compounds isolated from the plant Macaranga schweinfurthii1–3 (A–D, 1–4, I and J), four isolated from the plant Macaranga alnifolia4 (E–H, 5–8), and vedelianin (9) which was isolated much earlier from Macaranga vedeliana.5 When tested in the National Cancer Institute screen of 60 human cancer cell lines, those containing a hexahydroxanthene substructure as the ABC ring system showed an intriguing pattern of biological activity. Their unique fingerprint of activity does not parallel that of any chemotherapeutic agent in current clinical use, and approximates those of only a few, far more complex natural products (notably cephalostatins,6 OSW-1,7 ritterazines,8 and stellettins9). Mechanistic investigations from several groups have suggested that the schweinfurthins may engage several different targets, including oxysterol binding proteins,10 trans-Golgi-network trafficking,11 and cholesterol biosynthesis and its cellular export.12 The combination of a unique profile of activity with the limited success of efforts to obtain more of the schweinfurthins by isolation from the plant source, has led us to an extended effort to prepare these natural products by chemical synthesis. Our efforts to date have afforded seven of the natural products (schweinfurthins A,13 B,14 C,15 E,14 F,16 G16 and vedelianin17), as well as 3-deoxyschweinfurthin B18 (10) which we have used as a lead for a number of structure-activity studies.19,20 All of the optically active synthetic materials were prepared in high ee through reagent level control of absolute stereochemistry via an enantioselective epoxidation followed by a cascade cyclization to the tricyclic core. In fact, both enantiomers of schweinfurthin F were prepared,16 and the significantly different selectivity of the two enantiomers in the 60 cell line screen was used to assign the natural absolute stereochemistry as R, R, R and not S, S, S.
A possible drawback to the use of schweinfurthin-based drugs in chemotherapy is their limited solubility in water. Based on the activity of various analogues we have prepared, it appears that features essential for activity include a trans-fused A-B ring system, an oxygen substituent at C-5 of the C-ring, at least one free phenol on the D-ring, and conjugation from the C-ring to the D-ring as found in the trans (but not the cis19) olefin. On the other hand, the C-3 oxygen of schweinfurthin B is expendable without significant loss of activity and the 6′-position in the D-ring can accommodate a wide variety of substituents. Thus we have come to view the hexahydroxanthene found in the ABC ring system as the primary pharmacophore, although the D ring substituents contribute to activity in a lesser way. These observations led us to examine whether the central stilbene olefin could be replaced with a more polar group such as an amide to maintain activity while improving water solubility. The restricted rotation around the amide bond should favor a transoid structure, and may allow some communication of electronic effects from the C-ring to the D-ring. As a test of that hypothesis, in this paper we report the preparation of four new “schweinfurthin amides” as well as our initial studies on their biological activity.
Results and Discussion
From the outset of this effort, it was viewed as essential to prepare the new schweinfurthin analogues as a matched pair (Scheme 1), that is, one analogue with the carbonyl group on the tricyclic ABC ring system and the amide nitrogen on the D-ring (11), and a second isomeric analogue where the substituents were reversed (12). The obvious disconnection at the amide bond leads to the acids 13 and 16 and the amines 14 and 15. If some variation on the Curtius or Hoffman reaction21 could be employed despite the presence of a variety of other functional groups, it would be possible to derive the amine 14 from the acid 16 and the amine 15 from the acid 13. This would enhance the efficiency of the overall effort by the dual use of these carboxylic acids. While the ether and acetal groups in compound 13 were viewed as relatively stable to both acidic and basic conditions, the stability of the isoprenoid olefins in compounds of the general structure 16 was of more concern. Nevertheless, given the possibility for use of compound 16 in both target amides, the initial goal became preparation of a protected acid 16.
Scheme 1.
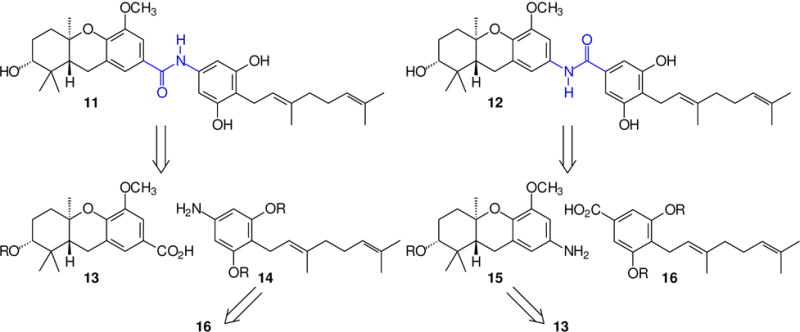
Retrosynthesis of a representative schweinfurthin amide.
The synthetic sequence used to prepare the target “right half” acid began with commercial 3,5-dihydroxybenzoic acid (17). Treatment of this resorcinol with bromine under acidic conditions gave the para bromo derivative 18 in nearly quantitative yield.22 Treatment of compound 18 with MOMCl and diisopropylethylamine resulted in smooth formation of the triMOM derivative 19. While acyloxy esters commonly have been employed in prodrugs,23 application of the MOM group as a carboxylic acid protecting group in place of a methyl ester or protected primary alcohol shortened the synthetic sequence significantly relative to our previous efforts. The MOM ester 19 proved to be sufficiently stable to allow halogen-metal exchange and alkylation with geranyl bromide to give the ester 20 in moderate yield.24
Selective hydrolysis of the MOM ester by treatment with LiOH gave the free carboxylic acid 21, and set the stage for preparation of an aryl amine via C-C bond cleavage and a formal insertion. One might consider either a Hoffmann or Curtius rearrangement to accomplish this transformation, but the presence of the isoprenoid olefin led us to favor the Curtius strategy. While there is precedent for conversion of an olefin to an aziridine via reaction with a nitrene,25,26 the prevailing view is that the Curtius rearrangement is a concerted process rather than one which involves discrete nitrene formation.21 In the event, reaction of the acid 21 with diphenyl phosphoryl azide (DPPA) followed by treatment with LiOH, gave the desired amine 22 in moderate yield. The complementary carboxylic acid 24 could be prepared by simple oxidation of the aldehyde 23, a hexahydroxanthene we have used in past syntheses of numerous schweinfurthin analogues.14 An EDC-mediated condensation of the acid 24 and the amine 22 gave the expected amide 25, and a final acid-catalyzed hydrolysis of the MOM protecting groups gave the target compound 11.
After amide 11 was prepared, synthesis of amide 12 was relatively straightforward because the second sequence could take advantage of some intermediates already in hand. As shown in Scheme 3, the carboxylic acid 24 gave the desired amine 26 upon treatment with DPPA. An EDC mediated condensation of acid 21 and amine 26 gave the desired amide framework (27), and MOM hydrolysis gave the final target, amide 12, in reasonable yield.
Scheme 3.
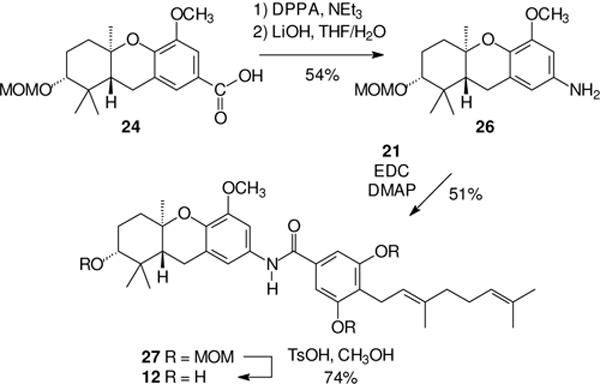
Synthesis of an isomeric schweinfurthin amide.
For a more robust comparison of the impact of formal replacement of the stilbene olefin with an amide unit, a second pair of schweinfurthin amides was prepared. We recently reported the synthesis and biological activity of some indole schweinfurthin analogues27,28 and used that effort as an inspiration for this second pair. As shown in Scheme 4, the parent indole carboxylic acid ester 28 was first dimethylated (29) to protect potentially sensitive functionality and to allow direct comparison with the activity of an indole schweinfurthin previously prepared. After hydrolysis of the methyl ester 29, the resulting carboxylic acid 30 was converted to the corresponding amine 31 by reaction with DPPA. Formation of the amide 32 was accomplished under standard conditions, as was final hydrolysis of the MOM protecting groups. The “reversed” amide 34 was obtained upon condensation of the amine 26 with the carboxylic acid 30, and a final hydrolysis of the MOM groups gave the target amide 35.
Scheme 4.
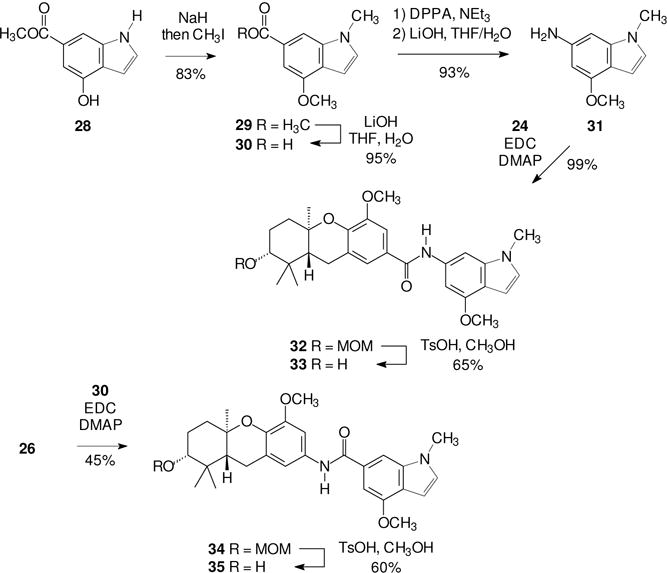
Synthesis of schweinfurthin-indole amides.
Once the four new schweinfurthin amides were in hand, each was tested in the NCI’s 60 cell line assay.29 These compounds showed schweinfurthin-like activity as measured by similar patterns of selectivity, with Pearson correlation coefficients of 0.65 to 0.78 (Table 1), and with the potency of compounds 11 and 33 comparable to the parent stilbenes. Both of the isomeric amides 12 and 35 were less potent and demonstrated a lower differential activity across the 60 cell lines assayed. In terms of calculated logP, there is no difference in water solubility between the two amide isomers in each pair, so the observed differences in biological activity must track to other molecular properties.
Table 1.
NCI 60 Data Summary
| Compound | Mean GI50 (μM) | Differential at GI50 (log10) | Pearson Correlation Coefficient to 1 at GI50 | LogP (calculated)a |
| 1 | 0.36 | 3.11 | 1.00 | 8.29 |
| 10 | 0.87 | 3.25 | 0.77 | 8.94 |
| 11 | 0.21 | 2.73 | 0.78 | 7.89 |
| 12 | 1.1 | 1.50 | 0.65 | 7.89 |
| 33 | 0.66 | 2.41 | 0.78 | 4.61 |
| 35 | 6.6 | 2.12 | 0.70 | 4.61 |
Values were determined using the calculator at http://www.molinspiration.com/services/properties.html
In conclusion, we have developed synthetic sequences that allow formal replacement of the central stilbene olefin of the schweinfurthins with an amide linkage. This substitution preserves the selectivity and potency of the parent schweinfurthins, and should increase the water solubility of these analogues relative to the parent compounds as measured by calculated logP values. In both series tested, the more active isomers 11 and 33 bore the carbonyl group on the C-ring, and the amine on the D-ring, suggesting that electron donation to the D-ring is more beneficial to activity than electron withdrawal. Further studies will be needed to determine if this is a consistent trend, but these findings provide some guidance to the design of still more pharmaceutically useful schweinfurthin analogues.
Experimental Section
General Experimental Procedures
Both tetrahydrofuran (THF) and diethyl ether (Et2O) were freshly distilled from sodium and benzophenone, whereas methylene chloride (CH2Cl2) was distilled from calcium hydride prior to use. The solutions of n–BuLi were purchased from a commercial source and titrated with diphenylacetic acid prior to use. All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions in nonaqueous solvents were conducted in flame-dried glassware under a positive pressure of argon and with a magnetic stir bar. NMR spectra were obtained at 400 or 500 MHz for 1H, 101 or 126 MHz for 13C NMR, in CDCl3 with (CH3)4Si (1H, 0.00 ppm) or CDCl3 (1H, 7.26 ppm; 13C NMR; 77.16 ppm) or CD3OD (1H, 3.31 ppm; 13C NMR; 49.00 ppm), as the internal standards. High-resolution mass spectra were obtained by GC-TOF. Silica gel (60 Å, 0.040 – 0.063 mm) was used for flash column chromatography.
(2R,4aR,9aR)-N-(4-((E)-3,7-Dimethylocta-2,6-dienyl)-3,5-dihydroxyphenyl)-2-hydroxy-5-methoxy-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-7-carboxamide (11). NSC#791211
To a stirred solution of the acetal 25 (39 mg, 56 μmol) in MeOH (5.6 mL) was added p-TsOH·H2O (61 mg, 0.32 mmol). The flask was sealed and stirred for 28 hours at rt. Analysis by tlc suggested that the reaction was not complete so additional p-TsOH·H2O (15 mg, 79 μmol) was added and the solution was stirred for an additional 17 hours. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded amide 11 (16.3 mg, 52%) as an off-white solid: 1H NMR (400 MHz, CD3OD) δ 7.33 (d, J = 1.9 Hz, 1H), 7.30 (d, J = 1.8 Hz, 1H), 6.71 (s, 2H), 5.29 – 5.21 (m, 1H), 5.12 – 5.05 (m, 1H), 3.85 (s, 3H), 3.38 – 3.32 (m, 1H), 3.28 (d, J = 7.4 Hz, 2H), 2.79 – 2.73 (m, 2H), 2.10 – 2.01 (m, 3H), 1.99 – 1.91 (m, 2H), 1.84 – 1.74 (m, 5H), 1.67 – 1.62 (m, 5H), 1.57 (s, 3H), 1.22 (s, 3H), 1.09 (s, 3H), 0.87 (s, 3H); 13C NMR (101 MHz, CD 13 3OD) δ C NMR (101 MHz, CD3OD) δ 168.6, 157.2 (2C), 149.9, 147.2, 138.0, 134.8, 132.0, 127.3, 125.6, 124.8, 123.8, 122.8, 113.1, 110.0, 101.4 (2C), 78.9, 78.6, 56.5, 48.2, 41.0, 39.5, 38.8, 28.9, 27.9, 27.8, 25.9, 24.1, 23.0, 20.3, 17.7, 16.3, 14.9; HRMS (TOF MS ES+) m/z calculated for C34H46NO6 (M + H)+ 564.3325, found 564.3326.
4-((E)-3,7-Dimethylocta-2,6-dienyl)-3,5-dihydroxy-N-((2R,4aR,9aR)-2-hydroxy-5-methoxy-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthen-7-yl)benzamide (12), NSC#791210
To a stirred solution of the acetal 27 (32 mg, 46 μmol) in MeOH (4.6 mL) was added p-TsOH·H2O (50 mg, 0.26 mmol). The flask was sealed and stirred for 27 hours at rt. After tlc analysis suggested that the reaction was not complete, additional p-TsOH·H2O (15 mg, 79 μmol) was added and the solution was stirred for an additional 18 hours. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded amide 12 (19.2 mg, 74%) as an off-white solid: 1H NMR (400 MHz, CD3OD) δ 7.13 (s, 1H), 6.95 (s, 1H), 6.80 (s, 2H), 5.25 (t, J = 7.1 Hz, 1H), 5.08 (t, J = 7.0 Hz, 1H), 3.80 (s, 3H), 3.35 (d, J = 7.2 Hz, 3H), 2.72 (s, 1H), 2.70 (s, 1H), 2.11 – 2.00 (m, 3H), 2.00 – 1.91 (m, 2H), 1.84 – 1.74 (m, 5H), 1.69 – 1.64 (m, 2H), 1.63 (s, 3H), 1.57 (s, 3H), 1.21 (s, 3H), 1.08 (s, 3H), 0.87 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 169.4, 157.4 (2C), 149.7, 140.9, 135.5, 134.8, 132.0, 131.9, 125.5, 124.0, 123.8, 120.6, 115.6, 106.8 (2C), 105.6, 78.7, 77.9, 56.4, 48.5, 41.0, 39.5, 38.9, 29.0, 27.9, 27.8, 25.9, 24.3, 23.4, 20.1, 17.7, 16.3, 14.8. HRMS (TOF MS ES+) m/z calculated for C34H46NO6 (M + H)+ 564.3325, found 564.3336.
Methoxymethyl 4-bromo-3,5-bis(methoxymethoxy)benzoate (19)
To a stirred solution of 4-bromo-3,5-dihydroxybenzoic acid (1.64 g, 7.0 mmol) and N,N-diisopropylethylamine (4.0 mL 23.2 mmol) in CH2Cl2 (24 mL) at 0 °C was added chloromethyl methyl ether (2.14 mL, 28.2 mmol) dropwise via syringe. The solution was allowed to equilibrate to rt overnight and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The organic layers were combined and washed with brine, and then dried over Na2SO4. The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 30% EtOAc in hexanes) afforded 19 (1.16 g, 89%) as an off-white solid: 1H NMR (500 MHz, CDCl3) δ 7.52 (s, 2H), 5.48 (s, 2H), 5.31 (s, 4H), 3.54 (s, 3H), 3.53 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 165.4, 155.0 (2C), 130.2, 110.3 (2C), 110.1, 95.3 (2C), 91.4, 58.1, 56.7 (2C). HRMS (TOF MS ES+) m/z calculated for C13H1779BrO7Na (M + Na)+ 387.0055, found 387.0060.
(E)-Methoxymethyl 4-(3,7-dimethylocta-2,6-dienyl)-3,5-bis(methoxymethoxy)benzoate (20).
An oven-dried flask was charged with ester 19 (0.5 g, 1.37 mmol) in anhydrous THF (10 mL) at –78 °C. To this solution was added n-BuLi (2.5M in hexanes, 0.58 mL, 1.4 mmol) dropwise via syringe. The resulting solution was stirred at –78 °C for 20 minutes and then CuBr·DMS (0.31 g, 1.5 mmol) was added. The solution was stirred an additional 20 minutes at –78 °C and then geranyl bromide (0.33 g, 1.5 mmol) was added dropwise via syringe. The solution was allowed to equilibrate to rt overnight and then the reaction was quenched by the addition of saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The organic extracts were combined, washed with brine, and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 30% EtOAc in hexanes) afforded compound 20 (0.34 g, 60%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.47 (s, 2H), 5.46 (s, 2H), 5.24 (s, 4H), 5.20 – 5.15 (m, 1H), 5.05 (t, J = 6.9 Hz, 1H), 3.53 (s, 3H), 3.48 (s, 6H), 3.45 (d, J = 7.1 Hz, 2H), 2.03 (dd, J = 14.7, 6.9 Hz, 2H), 1.99 – 1.90 (m, 2H), 1.79 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 166.0, 155.7 (2C), 135.6, 131.5, 128.7, 126.5, 124.4, 121.8, 109.4 (2C), 94.7 (2C), 91.0, 57.9, 56.3 (2C), 39.9, 26.9, 25.8, 23.2, 17.8, 16.3. HRMS (TOF MS ES+) m/z calculated for C23H34O7Na (M + Na)+ 445.2202, found 445.2197.
(E)-4-(3,7-Dimethylocta-2,6-dienyl)-3,5-bis(methoxymethoxy)benzoic acid (21)
To a stirred solution of ester 20 (0.470 g, 1.11 mmol) in THF (5 mL) and H2O (5 mL) was added LiOH·H2O (93 mg, 2.23 mmol). The flask was stirred at rt overnight and then the solution was acidified with HCl. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried over Na2SO4 and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded compound 21 (0.421 g, 81%) as a white powder: 1H NMR (500 MHz, CDCl3) δ 7.51 (s, 2H), 5.25 (s, 4H), 5.21 – 5.16 (m, 1H), 5.05 (t, J = 6.8 Hz, 1H), 3.49 (s, 6H), 3.46 (d, J = 7.0 Hz, 2H), 2.07 – 2.00 (m, 2H), 1.99 – 1.93 (m, 2H), 1.79 (s, 3H), 1.64 (s, 3H), 1.56 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 171.7, 155.7 (2C), 135.7, 131.5, 128.1, 126.9, 124.4, 121.7, 109.8 (2C), 94.7 (2C), 56.3 (2C), 39.9, 26.8, 25.8, 23.2, 17.8, 16.3. HRMS (TOF MS ES+) m/z calculated for C21H30O6Na (M + Na)+ 401.1940, found 401.1946.
(E)-4-(3,7-Dimethylocta-2,6-dienyl)-3,5-bis(methoxymethoxy)aniline (22)
An oven-dried flask was charged with carboxylic acid 21 (100 mg, 0.26 mmol) in anhydrous benzene (26 mL). To this stirred solution was added triethylamine (0.368 mL, 2.60 mmol) followed by the dropwise addition of DPPA (0.569 mL, 2.60 mmol) via syringe. The solution was stirred at rt for 15 minutes, and then the flask was fitted with a reflux condenser and heated at reflux overnight. The benzene was removed under reduced pressure and the residue was dissolved in THF (2.6 mL). To this solution was added aqueous 4N LiOH (1.3 mL, 5.30 mmol) and the resulting mixture was stirred vigorously at rt for 1 hour. The mixture was diluted with H2O and the layers were separated. The aqueous layer was extracted with EtOAc (3x) and the combined organic extracts were washed with brine, and dried (Na2SO4). The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded amine 22 (45 mg, 49%) as a brown oil: 1H NMR (400 MHz, CDCl3) δ 6.19 (s, 2H), 5.22 – 5.18 (m, 1H), 5.15 (s, 4H), 5.13 – 5.06 (m, 1H), 3.48 (s, 6H), 3.30 (d, J = 7.0 Hz, 2H), 2.10 – 2.01 (m, 2H), 2.00 – 1.92 (m, 2H), 1.78 (s, 3H), 1.67 (s, 3H), 1.59 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 156.6 (2C), 145.8, 134.0, 131.3, 124.6, 123.8, 110.7, 96.0 (2C), 94.6 (2C), 56.0 (2C), 40.0, 26.9, 25.8, 22.2, 17.8, 16.1. HRMS (TOF MS ES+) m/z calculated for C20H32NO4 (M + H)+ 350.2331, found 350.2324.
(2R,4aR,9aR)-5-Methoxy-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-7-carboxylic acid (24)
To a stirred solution of aldehyde 23 (250 mg, 0.72 mmol) and 2-methyl-2-butene (1.50 mL, 14.4 mmol) in t-BuOH (7 mL) was added NaH2PO4 (301 mg, 2.50 mmol) and NaClO2 (390 mg, 4.3 mmol) in H2O (2.4 mL) via syringe. The solution was stirred vigorously at rt for 3.5 hours and then washed with saturated aqueous NH4Cl. The mixture was extracted with EtOAc (3x) and the combined organic extracts were washed with brine and dried (Na2SO4). The mixture was filtered and filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded carboxylic acid 24 (138 mg, 53%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.57 (s, 1H), 7.42 (s, 1H), 4.78 (d, J = 6.9 Hz, 1H), 4.65 (d, J = 6.9 Hz, 1H), 3.90 (s, 3H), 3.41 (s, 3H), 3.28 (dd, J = 11.6, 3.9 Hz, 1H), 2.80 – 2.69 (m, 2H), 2.16 (d, J = 12.8 Hz, 1H), 2.05 – 1.97 (m, 1H), 1.86 – 1.77 (m, 1H), 1.71 (dd, J = 11.5, 6.5 Hz, 1H), 1.65 – 1.54 (m, 1H), 1.27 (s, 3H), 1.10 (s, 3H), 0.91 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 172.0, 148.8, 148.0, 125.3, 122.5, 120.3, 110.5, 96.3, 84.1, 78.2, 56.2, 55.8, 47.0, 38.4, 37.6, 27.5, 25.4, 23.2, 20.1, 15.3. HRMS (TOF MS ES-) m/z calculated for C20H27O6 (M - H)− 363.1808, found 363.1798.
(2R,4aR,9aR)-N-(4-((E)-3,7-Dimethylocta-2,6-dienyl)-3,5-bis(methoxymethoxy)phenyl)-5-methoxy-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-7-carboxamide (25)
To a stirred solution of carboxylic acid 24 (49 mg, 0.13 mmol) and amine 22 (39 mg, 0.11 mmol) in anhydrous CH2Cl2 (1 mL) was added EDC (64 mg, 0.34 mmol) and 4-(dimethylamino)pyridine (2.7 mg, 22 μmol). The solution was stirred at rt for 19 hours and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 50% EtOAc in hexanes) afforded amide 25 (42 mg, 54%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H), 7.31 (d, J = 1.9 Hz, 1H), 7.21 (d, J = 1.8 Hz, 1H), 7.18 (s, 2H), 5.23 – 5.16 (m, 5H), 5.11 – 5.04 (m, 1H), 4.76 (d, J = 6.9 Hz, 1H), 4.64 (d, J = 6.9 Hz, 1H), 3.87 (s, 3H), 3.50 – 3.46 (m, 6H), 3.40 (s, 3H), 3.37 (d, J = 7.1 Hz, 2H), 3.26 (dd, J = 11.5, 4.0 Hz, 1H), 2.76 – 2.68 (m, 2H), 2.15 – 2.10 (m, 1H), 2.08 – 1.99 (m, 3H), 1.98 – 1.93 (m, 3H), 1.85 – 1.79 (m, 1H), 1.78 (s, 3H), 1.69 – 1.66 (m, 1H), 1.65 (s, 3H), 1.61 – 1.52 (m, 4H), 1.24 (s, 3H), 1.08 (s, 3H), 0.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.4, 155.9 (2C), 149.1, 146.1, 137.3, 134.6, 131.3, 126.1, 124.5, 122.9, 122.4, 120.4, 116.4, 108.7, 100.4 (2C), 96.3, 94.7 (2C), 84.0, 77.8, 56.2, 56.2 (2C), 55.7, 47.0, 39.9, 38.4, 37.5, 27.5, 26.9, 25.8, 25.4, 23.3, 22.5, 20.0, 17.7, 16.2, 15.2. HRMS (TOF MS ES+) m/z calculated for C40H58NO9 (M + H)+ 696.4112, found 696.4116.
2R,4aR,9aR)-5-Methoxy-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthen-7-amine (26)
An oven-dried flask was charged with carboxylic acid 24 (75 mg, 0.21 mmol) in anhydrous benzene (21 mL). To this stirred solution was added triethylamine (0.287 mL, 2.10 mmol) followed by the dropwise addition of DPPA (0.443 mL, 2.10 mmol) via syringe. The resulting solution was stirred at rt for 15 minutes, and then the flask was fitted with a reflux condenser and heated at reflux overnight. The benzene was removed under reduced pressure and the residue was dissolved in THF (2.0 mL). To this solution was added aqueous 4N LiOH (1.0 mL, 4.12 mmol) and the resulting mixture was stirred vigorously at rt for 1 hour. The mixture was diluted with H2O and the layers were separated. The aqueous layer was extracted with EtOAc (3x) and the combined organic extracts were washed with brine, and dried over Na2SO4. The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (20 to 60% EtOAc in hexanes) afforded amine 26 (37 mg, 54%) as a brown oil: 1H NMR (400 MHz, CDCl3) δ 6.15 (d, J = 2.5 Hz, 1H), 6.06 (d, J = 2.4 Hz, 1H), 4.76 (d, J = 6.8 Hz, 1H), 4.64 (d, J = 6.8 Hz, 1H), 3.78 (s, 3H), 3.40 (s, 3H), 3.25 (dd, J = 11.6, 4.2 Hz, 1H), 2.61 (s, 1H), 2.59 (s, 1H), 2.08 (dt, J = 12.8, 3.4 Hz, 1H), 1.96 (ddd, J = 13.2, 7.6, 3.7 Hz, 1H), 1.77 (td, J = 13.4, 3.2 Hz, 1H), 1.69 (dd, J = 10.0, 8.1 Hz, 1H), 1.63 – 1.49 (m, 1H), 1.21 (s, 3H), 1.06 (s, 3H), 0.88 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 149.4, 139.0, 135.7, 123.3, 107.4, 99.0, 96.3, 84.3, 76.1, 56.0, 55.7, 47.3, 38.3, 37.8, 27.5, 25.4, 23.4, 19.7, 15.2; HRMS (TOF MS ES+) m/z calculated for C19H30NO4 (M + H)+ 336.2175, found 336.2182.
4-((E)-3,7-Dimethylocta-2,6-dienyl)-N-((2R,4aR,9aR)-5-methoxy-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthen-7-yl)-3,5-bis(methoxymethoxy)benzamide (27)
To a stirred solution of carboxylic acid 21 (42 mg, 0.11 mmol) and amine 26 (31 mg, 92 μmol) in anhydrous CH2Cl2 (1 mL) was added EDC (53 mg, 0.28 mmol) and 4-(dimethylamino)pyridine (2.3 mg, 18 μmol). The solution was stirred at rt for 20 hours and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 60% EtOAc in hexanes) afforded amide 27 (32 mg, 51%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.75 (s, 1H), 7.27 (s, 2H), 7.19 (d, J = 2.1 Hz, 1H), 6.91 (d, J = 2.2 Hz, 1H), 5.26 (s, 4H), 5.24 – 5.18 (m, 1H), 5.12 – 5.04 (m, 1H), 4.79 (d, J = 6.8 Hz, 1H), 4.66 (d, J = 6.8 Hz, 1H), 3.86 (s, 3H), 3.50 (s, 6H), 3.46 (d, J = 7.2 Hz, 2H), 3.43 (s, 3H), 3.28 (dd, J = 11.6, 4.1 Hz, 1H), 2.72 (s, 1H), 2.70 (s, 1H), 2.13 (dt, J = 12.7, 3.3 Hz, 1H), 2.09 – 2.03 (m, 2H), 2.01 – 1.95 (m, 3H), 1.85 – 1.77 (m, 4H), 1.73 (t, J = 9.1 Hz, 1H), 1.66 (s, 3H), 1.63 – 1.55 (m, 4H), 1.26 (s, 3H), 1.09 (s, 3H), 0.91 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 165.5, 155.9 (2C), 148.9, 139.8, 135.5, 134.2, 131.5, 130.2, 127.2, 124.4, 122.8, 121.9, 113.4, 106.7 (2C), 103.4, 96.3, 94.7 (2C), 84.2, 76.8, 56.3 (2C), 56.2, 55.8, 47.2, 39.9, 38.4, 37.7, 27.5, 26.8, 25.8, 25.4, 23.4, 23.0, 19.8, 17.8, 16.3, 15.2. HRMS (TOF MS ES+) m/z calculated for C40H58NO9 (M + H)+ 696.4112, found 696.4122.
Methyl 4-methoxy-1-methyl-1H-indole-6-carboxylate (29)
An oven-dried flask was charged with commercial methyl 4-hydroxyindole-6-carboxylate (50 mg, 0.26 mmol) in anhydrous DMF (1 mL) at 0 °C. To this solution was added NaH (60% in mineral oil, 31 mg, 0.79 mmol) and the solution was stirred for 30 minutes at 0 °C. After CH3I (40 μL, 0.63 mmol) was added dropwise to the solution via syringe, the solution was allowed to equilibrate to rt overnight. The reaction was quenched with the addition of H2O (3 mL) and the mixture was extracted with EtOAc (5x). The organic extracts were combined, dried over Na2SO4, and filtered, and the filtrate was concentrated under reduced pressure. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 20% EtOAc in hexanes) afforded the dimethylated compound 29 (47.8 mg, 83%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.78 (s, 1H), 7.20 (s, 1H), 7.11 (d, J = 3.0 Hz, 1H), 6.61 (d, J = 2.7 Hz, 1H), 4.00 (s, 3H), 3.95 (s, 3H), 3.84 (s, 3H).30
4-Methoxy-1-methyl-1H-indole-6-carboxylic acid (30)
To a stirred solution of indole 29 (47.8 mg, 0.22 mmol) in THF (1 mL) and H2O (1 mL) was added LiOH·H2O (18 mg, 0.44 mmol). The flask was stirred at rt overnight and then the solution was washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried over Na2SO4 and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 40% EtOAc in hexanes) afforded the carboxylic acid 30 (42.4 mg, 95%) as a white powder: 1H NMR (500 MHz, CDCl3) δ 7.88 (s, 1H), 7.27 (s, 1H), 7.15 (d, J = 2.9 Hz, 1H), 6.64 (d, J = 2.9 Hz, 1H), 4.03 (s, 3H), 3.86 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 173.1, 153.1, 137.4, 131.0, 123.7, 123.3, 107.0, 100.2, 99.2, 55.7, 33.4. HRMS (TOF MS ES-) m/z calculated for C11H10NO3 (M - H)− 204.0661, found 204.0669.
4-Methoxy-1-methyl-1H-indol-6-amine (31)
An oven-dried flask was charged with carboxylic acid 30 (31 mg, 0.15 mmol) in anhydrous benzene (15 mL). To this stirred solution was added triethylamine (210 μL, 1.50 mmol) followed by the dropwise addition of DPPA (326 μL, 1.50 mmol) via syringe. The solution was stirred at rt for 15 minutes, and then the flask was fitted with a reflux condenser and heated at reflux overnight. The benzene was removed under reduced pressure and the residue was dissolved in THF (1.5 mL). To this solution was added aqueous 4N LiOH (755 μL, 3.00 mmol) and the resulting mixture was stirred vigorously at rt for 2 hours. After the mixture was diluted with H2O, the layers were separated. The aqueous layer was extracted with EtOAc (3x) and the combined organic extracts were washed with brine, and dried over Na2SO4. The mixture was filtered and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded amine 31 (24.7 mg, 93%) as a brown oil: 1H NMR (500 MHz, CDCl3) δ 6.75 (d, J = 3.1 Hz, 1H), 6.43 (d, J = 3.1 Hz, 1H), 6.24 (s, 1H), 6.01 (s, 1H), 3.90 (s, 3H), 3.63 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 154.0, 143.1, 139.3, 125.4, 112.7, 98.3, 91.8, 88.2, 55.4, 33.0. HRMS (TOF MS ES+) m/z calculated for C10H13N2O (M + H)+ 177.1028, found 177.1033.
(2R,4aR,9aR)-5-Methoxy-N-(4-methoxy-1-methyl-1H-indol-6-yl)-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-7-carboxamide (32)
To a stirred solution of carboxylic acid 24 (20 mg, 57 μmol) and amine 31 (12 mg, 68 μmol) in anhydrous CH2Cl2 (1 mL) was added EDC (33 mg, 0.17 mmol) and 4-(dimethylamino)pyridine (1.4 mg, 11 μmol). The solution was stirred at rt for 24 hours and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried over Na2SO4 and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded 32 (20.4 mg, 99%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.96 (s, 1H), 7.61 (s, 1H), 7.36 (d, J = 1.4 Hz, 1H), 7.25 (s, 1H), 6.93 (d, J = 3.1 Hz, 1H), 6.63 (s, 1H), 6.53 (d, J = 3.0 Hz, 1H), 4.77 (d, J = 6.9 Hz, 1H), 4.65 (d, J = 6.9 Hz, 1H), 3.94 (s, 3H), 3.90 (s, 3H), 3.74 (s, 3H), 3.41 (s, 3H), 3.27 (dd, J = 11.6, 4.1 Hz, 1H), 2.78 – 2.69 (m, 2H), 2.15 (dt, J = 12.7, 3.2 Hz, 1H), 2.03 – 1.97 (m, 1H), 1.81 (td, J = 13.8, 3.2 Hz, 1H), 1.71 (dd, J = 11.1, 6.9 Hz, 1H), 1.59 (d, J = 14.0 Hz, 1H), 1.26 (s, 3H), 1.09 (s, 3H), 0.90 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 165.6, 153.4, 149.3, 146.1, 138.2, 133.8, 127.6, 126.6, 122.5, 120.4, 116.3, 108.9, 98.5, 96.4, 95.0, 94.3, 84.1, 77.8, 56.4, 55.8, 55.6, 47.1, 38.4, 37.7, 33.2, 27.6, 25.4, 23.4, 20.0, 15.3. HRMS (TOF MS ES+) m/z calculated for C30H39N2O6 (M + H)+ 523.2808, found 523.2812.
(2R,4aR,9aR)-2-Hydroxy-5-methoxy-N-(4-methoxy-1-methyl-1H-indol-6-yl)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthene-7-carboxamide (33), NSC#795315
To a stirred solution of amide 32 (15 mg, 29 μmol) in MeOH (3 mL) was added p-TsOH·H2O (11 mg, 29 μmol). The flask was sealed and stirred for 2 days at rt. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were dried over Na2SO4 and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 80% EtOAc in hexanes) afforded 33 (9 mg, 65%) as a clear oil: 1H NMR (400 MHz, CDCl3) δ 7.91 (s, 1H), 7.60 (s, 1H), 7.35 (s, 1H), 7.26 (s, 1H), 6.93 (d, J = 1.1 Hz, 1H), 6.62 (s, 1H), 6.53 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H), 3.74 (s, 3H), 3.43 (d, J = 8.8 Hz, 1H), 2.80 – 2.73 (m, 2H), 2.15 (d, J = 14.3 Hz, 1H), 1.93 – 1.82 (m, 2H), 1.73 – 1.66 (m, 1H), 1.64 – 1.56 (m, 2H), 1.26 (s, 3H), 1.11 (s, 3H), 0.89 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 164.6, 152.4, 148.3, 145.1, 137.2, 132.8, 126.6, 125.6, 121.5, 119.4, 115.3, 107.9, 97.5, 94.0, 93.3, 77.1, 76.9, 55.4, 54.6, 45.8, 37.6, 36.8, 32.3, 27.5, 26.5, 22.5, 19.1, 13.5. HRMS (TOF MS ES+) m/z calculated for C28H35N2O5 (M + H)+ 479.2546, found 479.2541.
4-Methoxy-N-((2R,4aR,9aR)-5-methoxy-2-(methoxymethoxy)-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthen-7-yl)-1-methyl-1H-indole-6-carboxamide (34)
To a stirred solution of amine 26 (11.5 mg, 34 μmol) and carboxylic acid 30 (16.5 mg, 80 μmol) in anhydrous CH2Cl2 (1 mL) was added EDC (26 mg, 0.14 mmol) and 4-(dimethylamino)pyridine (1 mg, 7 μmol). The solution was stirred at rt for 24 hours and then washed with saturated aqueous NH4Cl. The layers were separated and the aqueous layer was extracted with CH2Cl2 (3x). The combined organic extracts were washed with brine, dried over Na2SO4, and filtered, and the filtrate was concentrated in vacuo. Final purification using an ISCO Combiflash Rf chromatography gradient (0 to 100% EtOAc in hexanes) afforded amide 34 (8 mg, 45%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.51 (s, 1H), 7.23 (d, J = 2.3 Hz, 1H), 7.10 (d, J = 3.1 Hz, 1H), 6.99 (d, J = 1.0 Hz, 1H), 6.93 (d, J = 2.3 Hz, 1H), 6.62 (d, J = 2.6 Hz, 1H), 4.77 (d, J = 6.8 Hz, 1H), 4.65 (d, J = 6.8 Hz, 1H), 4.02 (s, 3H), 3.88 (s, 3H), 3.84 (s, 3H), 3.41 (s, 3H), 3.28 (dd, J = 11.5, 4.1 Hz, 1H), 2.71 (s, 1H), 2.68 (s, 1H), 2.13 (dt, J = 12.7, 3.3 Hz, 1H), 1.99 (ddd, J = 11.4, 7.5, 3.5 Hz, 1H), 1.80 (td, J = 13.8, 3.3 Hz, 1H), 1.64 – 1.57 (m, 1H), 1.26 (s, 3H), 1.08 (s, 3H), 0.90 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.9, 153.5, 149.0, 139.7, 137.6, 130.6, 130.1, 129.7, 122.9, 121.9, 113.3, 103.3, 102.7, 99.0, 98.1, 96.3, 84.2, 67.2, 56.2, 55.8, 55.7, 47.2, 38.4, 37.7, 33.4, 27.5, 25.5, 23.4, 19.9, 15.2. HRMS (TOF MS ES+) m/z calculated for C30H39N2O6 (M + H)+ 523.2808, found 523.2818.
N-((2R,4aR,9aR)-2-Hydroxy-5-methoxy-1,1,4a-trimethyl-2,3,4,4a,9,9a-hexahydro-1H-xanthen-7-yl)-4-methoxy-1-methyl-1H-indole-6-carboxamide (35), NSC#795316
To a stirred solution of amide 34 (8.6 mg, 16 μmol) in MeOH (1.6 mL) was added p-TsOH·H2O (6 mg, 31 μmol). The flask was sealed and stirred for 2 days at rt. The solution was diluted with EtOAc and then washed with saturated aqueous NaHCO3. The layers were separated and the aqueous layer was extracted with EtOAc (3x). The combined organic extracts were dried over Na2SO4 and filtered, and the filtrate was concentrated in vacuo. Final purification using an an ISCO Combiflash Rf chromatography gradient (0 to 100% EtOAc in hexanes) afforded amide 35 (4.7 mg, 60%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.52 (s, 1H), 7.23 (d, J = 2.1 Hz, 1H), 7.09 (d, J = 3.0 Hz, 1H), 7.00 (s, 1H), 6.94 (d, J = 2.0 Hz, 1H), 6.62 (d, J = 3.1 Hz, 1H), 4.02 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.42 (dd, J = 11.1, 2.8 Hz, 1H), 2.71 (s, 1H), 2.68 (s, 1H), 2.16 – 2.09 (m, 1H), 1.91 – 1.79 (m, 2H), 1.71 (t, J = 9.0 Hz, 1H), 1.65 – 1.56 (m, 2H), 1.25 (s, 3H), 1.09 (s, 3H), 0.88 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.9, 153.5, 149.0, 139.7, 137.6, 130.7, 130.1, 129.8, 122.8, 122.0, 113.3, 103.5, 102.7, 99.0, 98.2, 78.2, 56.2, 55.7, 47.0, 38.5, 37.9, 33.4, 28.5, 27.5, 23.5, 19.9, 14.4. One carbon signal was obscured by the chloroform resonance; when the spectrum was recorded in C6D6 this was observed at 76.4 ppm. HRMS (ESI) m/z calcd for C28H35N2O5 (M + H)+ 479.2546, found 479.2540.
Figure 1.
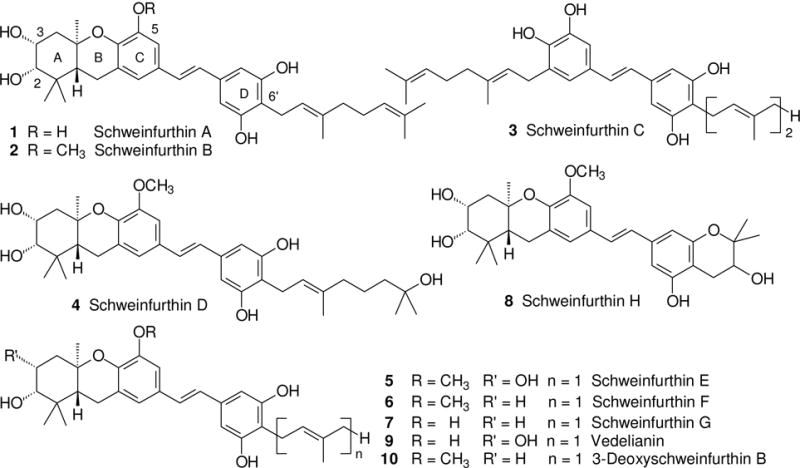
Some natural schweinfurthins (1–9) and a lead analogue (10).
Scheme 2.
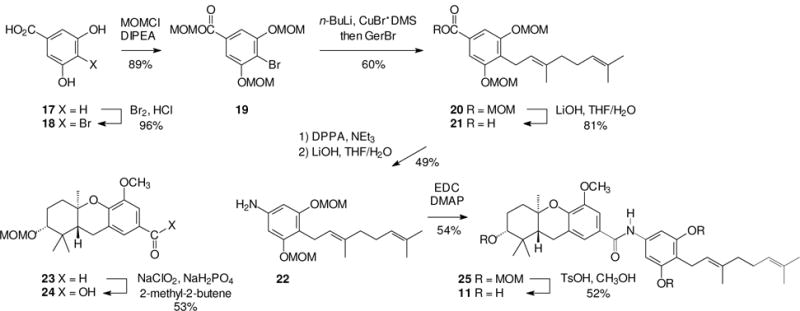
Synthesis of the first amide analogue of a schweinfurthin.
Acknowledgments
We thank the Center for Biocatalysis and Bioprocessing for a fellowship (to D.P.S.) through the predoctoral Training Program in Biotechnology (T32 GM008365). Financial support from the Roy J. Carver Charitable Trust (#01-224) through its Research Program of Excellence (to D.F.W.) is greatly appreciated. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (1ZIA-BC01147005). We thank the Developmental Therapeutics Program of NCI for NCI 60 testing, and Jason Evans for COMPARE analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available: The 1H and 13C NMR spectra of all new compounds, and the full 60 cell assay data (including the GI50, TGI, and LC50 values for each cell line) for compounds 11, 12, 33, and 35, can be found here. This material is available free of charge via the Internet at
Competing financial interests. JAB and DFW are named inventors on several patents that describe schweinfurthin analogues, held by the University of Iowa and/or NIH.
References
- 1.Beutler JA, Shoemaker RH, Johnson T, Boyd MR. J Nat Prod. 1998;61:1509–1512. doi: 10.1021/np980208m. [DOI] [PubMed] [Google Scholar]
- 2.Beutler JA, Jato J, Cragg GM, Boyd MR. Natural Product Letters. 2000;14:399–404. [Google Scholar]
- 3.Klausmeyer P, Van QN, Jato J, McCloud TG, Beutler JA. J Nat Prod. 2010;73:479–481. doi: 10.1021/np9006348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoder BJ, Cao S, Norris A, Miller JS, Ratovoson F, Razafitsalama J, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:342–346. doi: 10.1021/np060484y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thoison O, Hnawia E, Guerittevoegelein F, Sevenet T. Phytochemistry. 1992;31:1439–1442. [Google Scholar]
- 6.Pettit GR, Inoue M, Kamano Y, Herald DL, Arm C, Dufresne C, Christie ND, Schmidt JM, Doubek DL, Krupa TS. J Am Chem Soc. 1988;110:2006–2007. [Google Scholar]
- 7.Kubo S, Mimaki Y, Terao M, Sashida Y, Nikaido T, Ohmoto T. Phytochemistry. 1992;31:3969–3973. doi: 10.1016/0031-9422(92)83295-a. [DOI] [PubMed] [Google Scholar]
- 8.Komiya T, Fusetani N, Matsunaga S, Kubo A, Kaye FJ, Kelley MJ, Tamura K, Yoshida M, Fukuoka M, Nakagawa K. Cancer Chemother Pharmacol. 2003;51:202–208. doi: 10.1007/s00280-002-0558-8. [DOI] [PubMed] [Google Scholar]
- 9.Tasdemir D, Mangalindan GC, Concepcion GP, Verbitski SM, Rabindran S, Miranda M, Greenstein M, Hooper JNA, Harper MK, Ireland CM. J Nat Prod. 2002;65:210–214. doi: 10.1021/np0104020. [DOI] [PubMed] [Google Scholar]
- 10.Burgett AWG, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Fortner KC, Mimaki Y, Kuroda M, Murphy JP, Schwalb DJ, Petrella EC, Cornella-Taracido I, Schirle M, Tallarico JA, Shair MD. Nature Chemical Biology. 2011;7:639–647. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao XF, Zheng WJ, Sugi NH, Agarwala KL, Xu QL, Wang ZC, Tendyke K, Lee WN, Parent L, Li W, Cheng HS, Shen YC, Taylor N, Dezso Z, Du H, Kotake Y, Zhao ND, Wang J, Postema M, Woodall-Jappe M, Takase Y, Uenaka T, Kingston DGI, Nomoto K. Cancer Biology & Therapy. 2015;16:589–601. doi: 10.1080/15384047.2015.1019184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuder CH, Weivoda MM, Zhang Y, Zhu J, Neighbors JD, Wiemer DF, Hohl RJ. Lipids. 2015;50:1195–1207. doi: 10.1007/s11745-015-4083-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Topczewski JJ, Kodet JG, Wiemer DF. J Org Chem. 2011;76:909–919. doi: 10.1021/jo1022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topczewski JJ, Neighbors JD, Wiemer DF. J Org Chem. 2009;74:6965–6972. doi: 10.1021/jo901161m. [DOI] [PubMed] [Google Scholar]
- 15.Treadwell EM, Cermak SC, Wiemer DF. J Org Chem. 1999;64:8718–8723. [Google Scholar]
- 16.Mente NR, Neighbors JD, Wiemer DF. J Org Chem. 2008;73:7963–7970. doi: 10.1021/jo800951q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Topczewski JJ, Wiemer DF. Tetrahedron Lett. 2011;52:1628–1630. doi: 10.1016/j.tetlet.2011.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neighbors JD, Beutler JA, Wiemer DF. J Org Chem. 2005;70:925–931. doi: 10.1021/jo048444r. [DOI] [PubMed] [Google Scholar]
- 19.Ulrich NC, Kodet JG, Mente NR, Kuder CH, Beutler JA, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2010;18:1676–1683. doi: 10.1016/j.bmc.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topczewski JJ, Callahan MP, Kodet JG, Inbarasu JD, Mente NR, Beutler JA, Wiemer DF. Bioorg Med Chem. 2011;19:7570–7581. doi: 10.1016/j.bmc.2011.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aubé J, Fehl C, Liu R, McLeod MC, Motiwala HF. Comprehensive Organic Synthesis: Second Edition. 2014;6:598–635. [Google Scholar]
- 22.Hume PA, Furkert DP, Brimble MA. Organic Letters. 2013;15:4588–4591. doi: 10.1021/ol402191t. [DOI] [PubMed] [Google Scholar]
- 23.Wiemer AJ, Wiemer DF. Top Curr Chem. 2015;360:115–160. doi: 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gardner KD, Wiemer DF. J Org Chem. 2016;81:1585–1592. doi: 10.1021/acs.joc.5b02756. [DOI] [PubMed] [Google Scholar]
- 25.Jones JE, Ruppel JV, Gao G-Y, Moore TM, Zhang XP. J Org Chem. 2008;73:7260–7265. doi: 10.1021/jo801151x. [DOI] [PubMed] [Google Scholar]
- 26.Gao G-Y, Jones JE, Vyas R, Harden JD, Zhang XP. J Org Chem. 2006;71:6655–6658. doi: 10.1021/jo0609226. [DOI] [PubMed] [Google Scholar]
- 27.Kodet JG, Beutler JA, Wiemer DF. Bioorg Med Chem. 2014;22:2542–2552. doi: 10.1016/j.bmc.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodet JG, Wiemer DF. J Org Chem. 2013;78:9291–9302. doi: 10.1021/jo4014244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigrowolff A, Graygoodrich M, Campbell H, Mayo J, Boyd M. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 30.Kim M, Vedejs E. J Org Chem. 2004;69:6945–6948. doi: 10.1021/jo040191e. [DOI] [PubMed] [Google Scholar]