

Abstract
Pioglitazone is used globally for the treatment of type 2 diabetes mellitus (T2DM) and is one of the most effective therapies for improving glucose homeostasis and insulin resistance in T2DM patients. However, its mechanism of action in the tissues and pathways that regulate glucose metabolism are incompletely defined. Here we investigated the direct effects of pioglitazone on hepatocellular pyruvate metabolism and the dependency of these observations on the purported regulators of mitochondrial pyruvate transport, MPC1 and MPC2. In cultured H4IIE hepatocytes, pioglitazone inhibited [2-14C]-pyruvate oxidation and pyruvate-driven oxygen consumption and, in mitochondria isolated from both hepatocytes and human skeletal muscle, pioglitazone selectively and dose-dependently inhibited pyruvate-driven ATP synthesis. Pioglitazone also suppressed hepatocellular glucose production (HGP), without influencing the mRNA expression of key HGP regulatory genes. Targeted siRNA silencing of MPC1 and 2 caused a modest inhibition of pyruvate oxidation and pyruvate-driven ATP synthesis, but did not alter pyruvate-driven HGP and, importantly, it did not influence the actions of pioglitazone on either pathway. In summary, these findings outline a novel mode of action of pioglitazone relevant to the pathogenesis of T2DM and suggest that targeting pyruvate metabolism may lead to the development of effective new T2DM therapies.
Keywords: gluconeogenesis, mitochondria, MPCs, pioglitazone, pyruvate
Introduction
The thiazolidinedione (TZD) class of drugs is widely used for the treatment of type 2 diabetes mellitus (T2DM) [1] and has been shown to be effective in preventing the conversion of prediabetes to diabetes [2]. Pioglitazone (PIO) is an archetypal member of the TZD family that promotes hypoglycemia through enhanced peripheral (muscle and fat) insulin-stimulated glucose uptake and suppressing hepatic glucose output [3,4]. The metabolic activity of TZDs was initially believed to be secondary to a molecular interaction with peroxisome proliferator-activated receptor gamma (PPAR-γ), a nuclear receptor that regulates gene expression in response to ligand binding [5,6]. Improvements in insulin sensitivity are thought to occur as a result of PPAR-γ-mediated adipose tissue expansion [7] and an increased release of insulin-sensitizing adipokines [8]. However, growing evidence supports direct PPAR-γ-independent actions of TZDs in insulin-sensitive tissues that are relevant to the pathogenesis and treatment of T2DM [9].
The molecular and biochemical mechanisms surrounding the PPAR-γ sparing activities of TZDs remain largely unknown. Some studies have demonstrated a direct effect of high doses of TZDs on complex I and III function in mitochondria isolated from muscle [10] and liver [11–13], while others have shown that TZDs can bind directly to outer mitochondrial membrane proteins [14,15]. A potentially unifying hypothesis to explain the direct mitochondrial effects of TZDs and their clinical efficacy is that they modify pyruvate metabolism. Pyruvate is a key molecule in cellular energy metabolism and lies at the intersection of multiple metabolic pathways linking glycolysis to glucose oxidation. In liver, pyruvate metabolism and transport is a critical determinant of gluconeogenic flux and plays an important role in regulating fasting and postprandial hyperglycemia in patients with T2DM.
Interest in mitochondrial pyruvate transport has increased following the recent discovery of the mitochondrial pyruvate carrier proteins (MPCs), thought to be responsible for import of pyruvate into the mitochondria [16,17]. Two groups have described a role for the MPCs in the regulation of hepatic glucose metabolism in rodents [18,19], supporting data that mitochondrial pyruvate transport is an important orchestrator of gluconeogenesis [20]. Divakaruni and colleagues recently provided evidence that TZDs interact with MPCs in skeletal muscle cells and inhibit pyruvate-driven oxygen consumption [21]. However, whether TZDs play a role in liver pyruvate metabolism, or whether the MPCs are required for their activity in hepatocytes, is unknown.
The aim of the present study was to assess: (a) whether PIO negatively regulates pyruvate oxidation and pyruvate-driven mitochondrial ATP production in hepatocytes, (b) the impact of PIO on pyruvate-driven hepatic glucose production (HGP), and (c) the role of the MPC proteins in mediating the action of PIO on pyruvate metabolism in hepatocytes. Our data demonstrate that PIO is a specific inhibitor of pyruvate oxidation in hepatocytes and significantly lowers pyruvate-driven glucose production. However, neither MPC1 nor MPC2 were necessary to mediate the effect of PIO on pyruvate metabolism, suggesting that alternative targets may be involved in the action of TZDs in hepatocytes.
Results
Pioglitazone inhibits pyruvate oxidation in hepatocytes
Although TZDs and related compounds have been reported to inhibit pyruvate-driven uncoupled (maximal) respiration in a number of different cell types [21,22], the direct effects of PIO have not been specifically tested in liver cells. We chose to focus on PIO because it is the only FDA-approved TZD currently in widespread clinical use and, to our knowledge, no data exist regarding the direct effect of this compound on hepatic pyruvate metabolism or glucose production. We initially investigated basal substrate oxidation, in the absence of chemical uncouplers, in control H4IIE hepatocytes using [2-14C]-pyruvate and [U14C]-palmitate. Four-hour PIO treatment inhibited pyruvate oxidation at 10 μM, while pharmacological inhibition of pyruvate transport with UK5099 (UK) similarly suppressed pyruvate oxidation, albeit at much lower drug concentrations (50 nM; Fig. 1A). In agreement with one previous publication [21], this finding was reproducible in the murine skeletal muscle L6 cell line (Fig. 1B). Neither PIO nor UK treatment influenced palmitate oxidation in H4IIE cells, which was robustly suppressed by Etomoxir (10 μM; Fig. 1C), confirming that the impairment of pyruvate oxidation in hepatocytes was substrate-specific.
Fig. 1.

Pioglitazone inhibits pyruvate oxidation in hepatocytes. (A) Rates of 14C-pyruvate oxidation in H4IIE cells incubated for 4 h in serum- and glucose-free media supplemented with sodium pyruvate (100 μM) and glutamine (2 mM), treated with various concentrations of PIO and UK. *P < 0.05, **P < 0.01, **P < 0.001 vs VEH. (B) 14C-pyruvate oxidation in L6 murine skeletal muscle cells treated with VEH, PIO (10 μM), or UK (5 μM). *P < 0.05, **P < 0.01 vs VEH; †P < 0.05 vs PIO. (C) 14C-palmitate oxidation (100 μM + 2 mM L-carnitine) in H4IIE cells treated with VEH, PIO, UK, or Etomoxir (10 μM). ***P < 0.001 vs VEH. (D) Oxygen consumption rates of intact H4IIE cells following 4-h treatment with either VEH, PIO, or UK during a basal period (500 μM pyruvate) and after the sequential addition of oligomycin (4 μM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP; 4 μM), and rotenone (1 μM). **P < 0.01 PIO vs VEH; †P < 0.05, †††P < 0.001 UK vs VEH; ^P < 0.05 PIO vs UK. (E) Cell viability, expressed as relative light units, following 24-h treatment with various concentrations of PIO or UK. For all panels, values are mean ± SEM of ≥3 independent experiments.
To further validate our radiolabeled substrate oxidation assays, we performed complimentary pyruvate-driven respirometry experiments in intact cells using the XF Seahorse. Following 15-min incubation with treatments, PIO (10 μM) and UK (5 μM) blunted both the basal and maximal (FCCP-stimulated) oxygen consumption rates (Fig. 1D), while the average respiration associated with ATP turnover also tended to be lower in PIO- than VEH-treated cells (218 ± 18 vs 305 ± 24 pmol·min−1; P = 0.09). To exclude the possibility of toxicity-related actions of treatments, cell viability was assessed using a luciferase-reporter assay based on the cellular reduction potential. We found no detrimental effects on cell viability of PIO or UK across the range of concentrations used in this study (Fig. 1E).
Pioglitazone inhibits pyruvate-driven ATP synthesis in isolated mitochondria
Prior findings on the inhibitory effects of TZDs on pyruvate metabolism indicated that PIO required intact cells to exert these effects [21]. Therefore, we next investigated whether the inhibitory actions of PIO treatment toward pyruvate metabolism were preserved in isolated mitochondria harvested from H4IIE cells. Consistent with our OCR findings, mitochondrial ATP synthesis from pyruvate + malate was dose-dependently (1–10 μM) lowered following acute PIO treatment, but was not altered when ATP synthesis was supported by glutamate, succinate, or palmitoylcarnitine substrates (Fig. 2A). As a crucial control, UK robustly suppressed pyruvate-driven ATP synthesis without effect on other substrates over the same concentration range. Interestingly, the PIO-induced inhibition of ATP synthesis was overcome when pyruvate concentrations exceeded 5 mM, although higher concentrations of pyruvate appeared inhibitory to ATP synthesis (Fig. 2B). Indeed, the maximal rate of pyruvate-driven ATP synthesis observed across a wide range of substrate concentrations was ~ 30% and 53% lower with PIO and UK treatment, respectively, compared to vehicle. To highlight the translational potential of these findings, we confirmed these results using mitochondria isolated from human skeletal muscle (Fig. 2F).
Fig. 2.

Pioglitazone inhibits pyruvate-driven ATP synthesis in isolated mitochondria. (A–D) Rate of ATP synthesis in mitochondria freshly isolated from H4IIE cells and acutely treated with the indicated concentrations of PIO or UK. Treatments were added 15 min prior to the measurement of ATP synthesis, which was supported by ADP (0.6 mM) and 500 μM pyruvate + 2 mM malate (A), 500 μM glutamate + 2 mM malate (B), 500 μM succinate (C), or 0.7 μM palmitoylcarnitine + 2 mM malate (D). *P < 0.05, ***P < 0.001 vs VEH. (E) Rate of ATP synthesis in isolated H4IIE mitochondria stimulated by various concentrations of pyruvate and treated with VEH, PIO, (10 μM) or UK (5 μM). **P < 0.01 PIO/UK vs VEH, †P < 0.05, ††P < 0.01 PIO vs UK. (F) Rate of ATP synthesis in mitochondria isolated from human skeletal muscle supported by either pyruvate/malate (PM) or glutamate/malate (GM) and treated acutely with VEH or PIO. *P < 0.05 vs VEH. For all panels, values are mean ± SEM of ≥3 independent experiments.
Pioglitazone inhibition of pyruvate oxidation does not require the MPC proteins
We next investigated whether expression of the mitochondrial pyruvate carrier proteins MPC1 and MPC2 was paramount to the observed effects of PIO in H4IIE hepatocytes, as has been suggested by others [21]. In agreement with previous observations following stable protein repression with lentiviral shRNA [21] or tissue-specific deletion in mice [18,19], silencing by siRNA of MPC1 resulted in a concomitant reduction in MPC2 protein levels and vice versa (Fig. 3A, B). Silencing was most effective with siMPC2, which reduced MPC1 and MPC2 protein levels to approximately 20% and 1% of control cells, respectively (Fig. 3A, B), a degree of silencing that is at least comparable to that achieved in several recent studies using similar approaches [21,23]. In agreement with prior data in hepatocytes [19], basal pyruvate oxidation was approximately 20% lower in siMPC2 cells (Fig. 3C). However, the relative inhibition of pyruvate oxidation by PIO and UK was identical between siSCR and siMPC2 cells (Fig. 3C), strongly arguing against a role for the MPCs in the effects of PIO on pyruvate metabolism.
Fig. 3.

Pioglitazone inhibition of pyruvate oxidation does not require the MPC proteins. (A, B) Representative western blot (A) and densitometry quantification (B) of MPC1 and MPC2 protein expression, normalized to GAPDH, in H4IIE cells following siRNA knockdown of each protein. (C) Rate of 14C-pyruvate oxidation in siSCR or siMPC2 H4IIE cells treated with either VEH, PIO (10 μM), or UK (5 μM). **P < 0.01, ***P < 0.001 vs VEH; ^P < 0.05, ^^P < 0.01 vs siSCR. (D) Rate of pyruvate- or glutamate-driven ATP synthesis in mitochondria isolated from siSCR or siMPC2 H4IIE cells. **P < 0.01 vs siSCR. (E, F) Percentage inhibition of pyruvate-driven ATP synthesis by indicated concentrations of PIO or UK in mitochondria isolated from siSCR or siMPC2 H4IIE cells. *P < 0.05, **P < 0.01, ***P < 0.001 vs previous PIO concentration; †P < 0.05 vs siSCR. Values are mean ± SEM of ≥3 independent experiments.
Because it has been suggested that pyruvate transport in MPC1/2-silenced cells is bypassed by its cytosolic transamination to, and subsequent mitochondrial uptake of, alanine or malate, we investigated whether pyruvate metabolism was more severely compromised in isolated mitochondria from MPC2-silenced cells. Pyruvate-driven ATP synthesis was 36% lower in siMPC2 cells, while ATP synthesis from glutamate was unaffected (Fig. 3D). Consistent with our findings from intact cells, the dose-dependent inhibition of pyruvate-driven ATP synthesis by PIO was identical between siSCR and siMPC2 cells (Fig. 3E). These novel data further highlight that (a) MPC proteins are not required for the effect of PIO on pyruvate-driven oxidation and ATP production in hepatocytes, and (b) the lack of effect of MPC silencing on PIO action cannot be explained by alternative routes of pyruvate-derived carbon entry into isolated mitochondria. Interestingly, inhibition of pyruvate-driven ATP synthesis by UK was somewhat less potent in siMPC2 cells compared to siSCR (Fig. 3F), which may be indicative of distinct mechanisms of action of UK compared to PIO.
Pioglitazone does not influence PDH activation status
As MPC silencing did not influence the ability of PIO to inhibit oxidative pyruvate metabolism, we reasoned that PIO could be acting downstream of pyruvate transport. The principle pathway for oxidative pyruvate disposal involves its decarboxylation to acetyl-CoA via the pyruvate dehydrogenase complex (PDH), although whether TZDs directly influence PDH activity has not previously been addressed. To evaluate whether PDH is a target for PIO, we adapted the methodology of Constantin-Teodosiu et al.[24], which provided a sensitive assay of the active fraction of PDH in our cell culture system. Neither the active fraction nor total activity of PDH was altered by 2 h of PIO (10 μM) treatment, while the pyruvate dehydrogenase kinase inhibitor dichloroacetate (PDH activator) increased the active fraction without altering total activity (Fig. 4). These data are the first direct assessment of the effects of PIO on PDH activity in hepatocytes and demonstrate that the inhibition of pyruvate oxidation with PIO cannot be explained simply by a direct downregulation of PDH activation status.
Fig. 4.
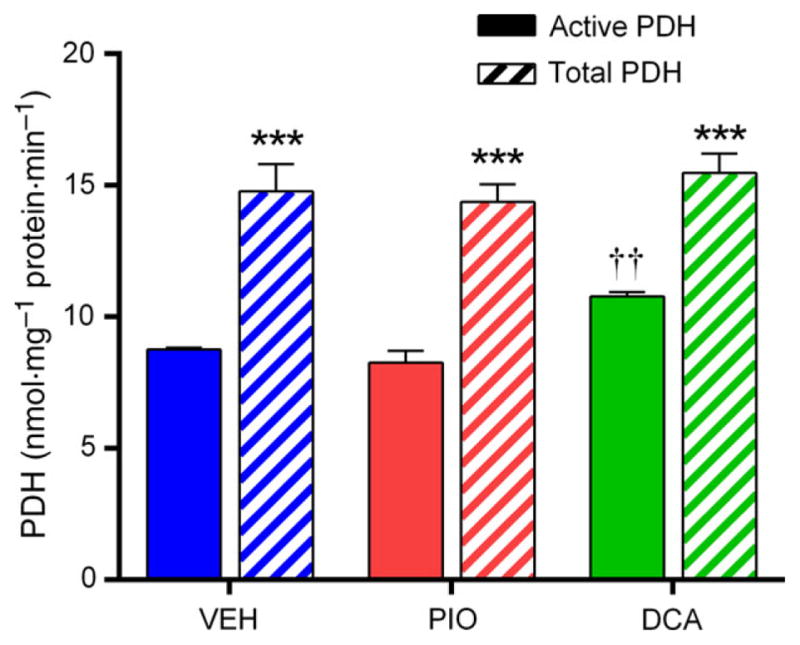
Pioglitazone does not influence PDH activation status. Pyruvate dehydrogenase activation status was assayed in whole cell lysates following 2 h of incubation of H4IIE cells in serum- and glucose-free media (1 mM sodium pyruvate) with VEH, PIO (10 μM), or dichloroacetate (5 mM). Complete PDH activation was achieved through preincubation of cell lysate with CaCl2 (100 μM), MgCl2 (10 mM), DCA (5 mM), spermine (500 μM), glucose (10 mM), and hexokinase (2 U·mL−1). ***P < 0.001 total vs active; ††P < 0.05 vs VEH and PIO. Values are mean ± SEM of ≥3 independent experiments.
Pioglitazone inhibits hepatocellular glucose production independent of MPC expression
Although a number of studies have demonstrated that high (≥100 μM) TZD concentrations may inhibit hepatocellular glucose production in vitro [25,26], no data exist regarding the direct effects of pharmacologically relevant concentrations of pioglitazone on hepatocellular glucose production. In control H4IIE hepatocytes, we observed a dose-dependent suppression of the rate of pyruvate-driven glucose output during overnight incubation with PIO or UK (Fig. 5A, B). In our system, glucose output was detectable within 4 h, was linear over at least 24 h (Fig. 5D), and was robustly inhibited by insulin (Fig. 5C). Low levels of pyruvate/lactate (0.2/2 mM), alone or in combination with glutamine/alanine, were insufficient to support detectable rates of glucose production, suggesting that alternative pathways of glucose production were not significant net contributors to glucose production in these cells.
Fig. 5.

Pioglitazone inhibits hepatocellular glucose production. (A–C) Rate of glucose production in H4IIE cells incubated overnight in glucose- and serum-free media supplemented with pyruvate (2 mM), lactate (20 mM), and glutamine (2 mM), and treated with various concentrations of PIO (A), UK (B), or insulin (C). *P < 0.05, **P < 0.01, ***P < 0.001 vs VEH; †P < 0.05, ††P < 0.01 vs prior concentration. (D) Time-course of glucose production in VEH and PIO treated H4IIE cells. **P < 0.01, ***P < 0.001 vs VEH. (E) Effect of PIO and insulin on the mRNA expression of key gluconeogenic genes in H4IIE cells following overnight treatment in serum- and glucose-free media. FBP, fructose 1,6-biphosphatase; G6P, glucose-6-phosphatase; PEPCK, phosphoenolpyruvate carboxykinase; PCX, pyruvate carboxylase. *P < 0.05, **P < 0.01, ***P < 0.001 vs VEH. NS, not significant. Values are mean ± SEM of ≥2 independent experiments.
Insulin is thought to regulate glucose production at the transcriptional level by the suppression of rate-limiting gluconeogenic enzymes, but unlike insulin, the inhibition of pyruvate-driven glucose production by PIO occurred in the absence of any alteration of FBP, G6PC, PCK1, or PCX (Fig. 5E). Unlike pyruvate oxidation, and in contrast to one previous report [19], rates of pyruvate-driven glucose production were unaltered in siMPC2 cells (Fig. 6A). Moreover, the inhibition of glucose production by PIO was similar in siSCR and siMPC2 cells, regardless of whether rates were supported solely by pyruvate/lactate, or additionally supplemented with 20 mM glutamine or alanine (Fig. 6C–E). Interestingly, 0.5 μM UK was more potent at suppressing HGP in siMPC2 cells (Fig. 6F–H). Taken together, these data again highlight that the mechanism of action of PIO does not depend on the MPC proteins and furthermore that it is distinct from the effects of UK5099 and perhaps unrelated to pyruvate transport.
Fig. 6.

Mitochondrial pyruvate carrier silencing does not influence hepatocellular glucose production. (A) Rate of glucose production in siSCR and siMPC2 H4IIE cells supported by pyruvate (2 mM) and lactate (20 mM). (B) Representative western blot of MPC1 and MPC2 protein expression H4IIE cells following siRNA knockdown of MPC2. (C–H) Rates of glucose production in siSCR and siMPC2 H4IIE cells supported by pyruvate (2 mM) and lactate (20 mM) alone (A and F) or supplemented with 20 mM glutamine (D and G) or alanine (E and H) and treated with the indicated concentrations of PIO (C–E) or UK (F–H). *P < 0.05, **P < 0.01, ***P < 0.001 vs VEH, ^P < 0.05, ^^P < 0.01, ^^^P < 0.001 vs both other treatments. †P < 0.05, ††P < 0.01 vs siSCR. Values are mean ± SEM of ≥2 independent experiments.
Discussion
The major novel finding of this study is that the TZD pioglitazone inhibits pyruvate metabolism in hepatocytes and in isolated mitochondria, and that this effect is not modulated by the mitochondrial pyruvate carrier proteins MPC1 or MPC2. We show that PIO impairs both oxidative and gluconeogenic pathways of pyruvate metabolism in a dose-dependent manner in vitro and ex vivo. These data are clinically relevant because TZDs are widely used in the treatment of T2DM, but their direct mechanism(s) of action in their target tissues (e.g., skeletal muscle and liver), is largely unknown. These findings may facilitate the development of novel TZD-like compounds that are clinically efficacious but lack the undesirable side effects of traditional TZDs.
In human subjects with T2DM, pioglitazone improves insulin sensitivity in skeletal muscle and liver and reduces hepatic glucose production [4,27]. Previously, these effects were proposed to be related to transcriptional changes in gene networks regulating adipose tissue differentiation and metabolism [28]. However, in isolated tissues and cells, a consistent finding has been that TZDs can directly inhibit aerobic fuel consumption via PPAR-γ-independent mechanisms [10,29] or transcriptional changes [9]. Our data build upon this work by demonstrating that in hepatocytes, pharmacological concentrations of PIO acutely inhibit pyruvate-driven oxygen consumption, coupled mitochondrial ATP production and whole cell pyruvate oxidation by 30–35%. It has been suggested that PIO may impair substrate oxidation through direct inhibition of the mitochondrial complexes [11]. However, our finding that metabolism of nonpyruvate substrates in either whole hepatocytes or isolated mitochondria was not influenced by PIO argues against any global PIO-mediated inhibition of mitochondrial metabolism, at least in the pharmacological range of PIO concentrations tested here. Therefore, the reduction in 14CO2 production in cells offered 14C-pyruvate is more likely attributable to a decreased rate of pyruvate-derived acetyl-group provision to the TCA cycle.
In this respect, one prospective target of PIO could be the pyruvate dehydrogenase complex (PDH), the enzyme responsible for regulating the conversion of pyruvate to acetyl-CoA. Previous efforts to evaluate the effects of TZDs on the PDH have employed the use of methyl-pyruvate to distinguish the activity of MPCs and PDH [21]. This approach is hampered by the apparent difference in the metabolism of methyl-pyruvate to that of pyruvate [30]. For the first time, we have determined the direct effect of PIO on the activation status of the PDH, but could find no inhibitory actions of PIO on either the total or active forms of the enzyme complex. It is important to note that while this indicates a lack of any direct effect of PIO on the PDH catalytic activation status, it does not strictly dictate that flux through the enzyme was unaffected. However, should PDH flux indeed be preserved upon PIO treatment, the inhibition of pyruvate oxidation could be attributable to a suppression of non-PDH-mediated acetyl-group provision. A recent NMR study of pyruvate metabolism in rat livers suggested that a significant source of pyruvate entry into the TCA cycle was through pyruvate carboxylase, particularly in the fasted state [31]. In this respect, it is conceivable that the 35% reduction in pyruvate oxidation we observe is attributable to a suppression of pyruvate carboxylase flux. Indeed, our observation that PIO also suppressed pyruvate-driven glucose production supports the hypothesis that flux through pyruvate carboxylase is reduced by PIO. However, the requirement for anaplerotic pyruvate carboxylase flux in isolated mitochondria was likely obviated in our experiments by the provision of 2 mM exogenous malate, arguing against the direct involvement of pyruvate carboxylase in the PIO-mediated inhibition of pyruvate metabolism.
Thiazolidinedione treatment in humans negatively regulates HGP [4,27], while in nondiabetic rats, troglitazone reduces HGP within 1 h in vivo [32]. In isolated hepatocytes from starved rats, troglitazone and pioglitazone were shown to inhibit HGP from lactate and pyruvate [25,33]. In addition to confirming these data in cell culture, our data also indicate that, unlike insulin, the effect of PIO on HGP is independent of changes in the mRNA expression of rate-limiting HGP enzymes, further supporting the hypothesis that PIO lowers HGP in vitro through direct modulation of pyruvate-specific metabolism in the mitochondria. Importantly, we observed a significant effect of PIO at much lower concentrations (2.5 μM) than those used in the studies highlighted above, which are well below the 35 μM Cmax of PIO observed in rats given a single oral dose of 10 mg·kg−1 [34]. As previously discussed, the reduction in HGP with PIO treatment may be related to changes in pyruvate carboxylase flux. A possibility that could explain the reduction in both oxidative and gluconeogenic routes of pyruvate disposal is that PIO inhibits mitochondrial pyruvate transport. Indeed, PIO was less effective at inhibiting pyruvate-driven ATP production at higher pyruvate concentrations, whereupon the passive component of mitochondrial pyruvate entry becomes more predominant [35]. This finding in itself points toward pyruvate transport, rather than the direct inhibition of pyruvate carboxylase or pyruvate dehydrogenase (Km toward pyruvate for both enzymes is < 500 μM [36,37]) as the more likely target for PIO. Recently two new proteins, MPC1 and MPC2, were identified as critical mediators of pyruvate transport in yeast, drosophila, and cultured mammalian fibroblasts [16,18]. It has previously been demonstrated that PIO binds to multiple proteins on the mitochondrial membrane [14], and one study has described a mechanistic link between TZDs, the MPC proteins, and pyruvate metabolism [21] which the authors postulated indicated a requirement of the MPCs for the inhibition by TZDs of pyruvate-driven oxygen consumption. Should the effects of PIO on pyruvate metabolism indeed be moderated through an interaction with the MPCs, a reduction in MPC protein levels with knockdown should result in an increased potency of PIO toward pyruvate metabolism [21]. On the contrary, we found that the inhibition of both pyruvate oxidation and pyruvate-driven glucose production by PIO was unaffected by MPC silencing, suggesting that these proteins are unlikely to be involved in the PIO-induced alteration of pyruvate metabolism.
In the current study, we assayed various components of mitochondrial pyruvate metabolism, in the same cellular system and under comparable media conditions. This allows for comment on the extent to which these processes may be regulated by pyruvate availability and/or transport. Assuming a 2:1 stoichiometry for pyruvate conversion to glucose, our data suggest that the calculated rates of both pyruvate oxidation and pyruvate-driven glucose production would require similar rates of mitochondrial pyruvate entry (~ 275 vs ~ 300 pmol·mg protein−1·min−1). In this respect, it is noteworthy that MPC silencing appeared to influence only pyruvate oxidation and pyruvate-supported ATP synthesis, while pyruvate-driven glucose production was unaltered. This may be indicative of an effect of MPC knockdown on oxidative pyruvate disposal that is distinct from pyruvate transport. Indeed, it has previously been speculated that the MPC proteins, although likely integral to pyruvate metabolism, may not directly or exclusively regulate mitochondrial pyruvate entry [38]. Further support for this comes from the robust finding, in this study and others [19,21], that both UK5099 and TZDs are able to exert additional inhibitory effects on pyruvate metabolism in MPC-silenced cells, beyond that observed with MPC knockdown alone. Moreover, liver-specific knockout mice for both MPC1 and MPC2 have normal glucose tolerance, fasting glucose, and rates of hepatic glucose appearance Ra in vivo [18,19]. Overall, the preserved inhibition of pyruvate metabolism by UK5099 in MPC-silenced cells cannot be reconciled with a paradigm in which the MPCs are the only proteins responsible for mitochondrial pyruvate import.
A key novel finding in the current study is that MPC silencing had no effect of hepatocellular glucose production. This is in contrast to McCommis and colleagues [19], who reported a ~ 70% reduction of glucose production upon MPC2 knockdown in isolated hepatocytes. One potential explanation for the discrepancy in this finding is the influence of glycogenolysis on glucose production. In the aforementioned study [19], glucose production was augmented with ~ 30 nM glucagon, which has been shown to maximally activate glycogenolysis in murine hepatocytes [39]. Therefore, it is possible that the observed reduction in glucose production in hepatocytes from MPC2-null animals reflects, in part, a reduction in glycogenolysis, rather than compromised pyruvate-driven gluconeogenesis. In support of this, liver glycogen content and rates of glucagon-stimulated glucose production in the absence of pyruvate were significantly lower in MPC2 knockout animals [19]. We chose to evaluate the gluconeogenic component of HGP only, i.e., in the absence of glucagon, as glycogenolysis does not contribute to the inappropriate rise in hepatic glucose output observed in T2DM [40]. The lack of effect of MPC silencing on HGP in our study is in agreement with the finding that mice expressing a truncated MPC2 protein demonstrated impaired pyruvate oxidation but normal gluconeogenic capacity [41]. Moreover, the pyruvate transport inhibitor UK5099 (Ki = 50 nM; [42]) blocked only approximately 50% of pyruvate-driven HGP at a concentration of 500 nM in our study. It has been proposed that this may reflect compensation by alternative gluconeogenic pathways when pyruvate transport is impaired. For example, cycling of pyruvate through cytosolic alanine transaminase may facilitate pyruvate-derived acetyl-CoA production independent of mitochondrial pyruvate transport [18,19]. However, we found that provision of supplementary alanine did not recover PIO- or UK5099-inhibited rates of glucose production, regardless of the presence/absence of the MPC proteins. The latter is consistent with the observation that pyruvate transamination likely requires mitochondrial pyruvate transport [43]. Alternatively, glutaminolysis may support TCA cycle function and glucose production in response to UK5099 treatment [18,43], although again, we found no difference in the effects of PIO, UK, or MPC silencing between HGP with glutamine-deplete and glutamine-rich media. Finally, it could be argued that our results may be influenced by the potentially transient or incomplete nature of MPC protein silencing. However, although some studies have documented that small quantities of MPCs are sufficient to maintain pyruvate transport activity [44], we routinely achieved 95% silencing of MPC2 protein in this study, which is comparable to numerous studies [21,23]. Indeed, similarly efficient removal of MPC1 (90%) in vivo was sufficient to almost completely abolish the incorporation of 2-14C-pyruvate into lipids [44].
The acute metabolic effects of PIO we report here are comparable in magnitude to the improvements observed in whole body insulin sensitivity in T2DM patients after several weeks of PIO treatment [4] and thus, may be clinically relevant. Indeed, our data in isolated human skeletal muscle mitochondria indicate that the effect of PIO on pyruvate-driven ATP production is not restricted to the liver. In man, PIO increases whole body (primarily skeletal muscle) insulin sensitivity secondary to increased nonoxidative glucose disposal (i.e., glycogen synthesis) without increasing glucose oxidation [4]. Therefore, a blunting of mitochondrial pyruvate uptake and/or oxidation in skeletal muscle could explain the shift in glucose oxidation toward glycogen storage in T2DM patients treated with PIO. In future experiments, it will be important to study in detail the effects of TZDs in multiple metabolic tissues, including the liver, skeletal muscle, and adipose tissue. In summary, we have shown that low concentrations of PIO inhibit pyruvate oxidation and glucose production in hepatocytes, with this effect being independent of the newly identified MPC proteins and unrelated to transcriptional changes in metabolic or mitochondrial genes. Our data suggest the possibility that PIO interacts with a mitochondrial protein that regulates both oxidative and gluconeogenic pyruvate metabolism, but more research will be needed to identify the molecular target of PIO.
Experimental procedures
Cell culture and human muscle biopsies
Rat H4IIE hepatocytes were used in this study. These cells have been shown by many groups to metabolize glucose and lipid in a physiologically relevant manner [45–47] and are sensitive to physiological concentrations of insulin and metformin [46], making them a highly applicable and convenient system for the study of hepatic metabolism in the context of T2DM. Low passage cells were purchased from ATCC and routinely cultured in low glucose (5 mM) Dulbecco’s modified Eagle’s medium (DMEM) media (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) in the absence of antibiotics, up to passage 30. To confirm the validity of our experiments in human tissue, vastus lateralis skeletal muscle biopsies were obtained from human subjects after an overnight fast in accordance with the UTHSCSA ethics committee. Human muscle samples were taken with the understanding and written consent of each subject and study methodologies conformed to the standards set by the Declaration of Helsinki.
Drug treatments
For the experiments described below, pioglitazone stocks (25 mM in 100% DMSO) were prepared by diluting to 1000X final concentrations and further diluted to 1–25 μM working concentrations in assay media. These concentrations are similar to the plasma pioglitazone concentrations achieved in T2DM individuals receiving 45 mg·day−1 [48] and are lower than that observed in rodent plasma after a single oral dose of 10 mg·kg−1 PIO [34]. Vehicle treatment (VEH) consisted of 100% DMSO, identically diluted in assay media. The pyruvate transport inhibitor UK 5099 (UK) was prepared in an identical manner to PIO and diluted in assay media to 0.05–50 μM. Cell viability was assessed following 24-h treatment with various concentrations of each drug using the RealTime-Glo MT assay (Promega).
14C-pyruvate and 14C-palmitate oxidation assay
We adapted a multiwell assay described by Collins et al. using [2-14C] pyruvate, whereby 14CO2 capture reflects incorporation of the C-2 of pyruvate into the TCA cycle and thus offers a relative index of pyruvate oxidation [49]. Approximately 3 × 105 cells were seeded into each well of a 24-well CulturPlate (Perkin Elmer, Waltham, MA, USA) and allowed to adhere overnight. Culture media was then removed and cells were washed twice with PBS and replaced with a glucose- and serum-free media (SFM; DMEM; Sigma) supplemented with 100 μM sodium pyruvate (Sigma) and sodium bicarbonate (3.7 g·L−1). Treatments were added and the cells were left in a cell culture incubator at 5% CO2, 95% O2 for 2 h. After a 2-h preincubation, the media was refreshed with SFM containing 25 mM HEPES (instead of NaHCO3), 30 μCi [2-14C] sodium pyruvate (Perkin Elmer), and treatments, and the plate was sealed and placed in a 37 °C incubator at ambient air. After 2 h, 400 μL media was removed and added to 2 mL tubes containing 200 μL perchloric acid (1 M). The tube lids, which contained a KOH-saturated filter paper disk for CO2 capture, were rapidly closed and tubes were gently mixed by vortex for 1 h at room temperature. The filter disks were subsequently removed and placed into scintillation vials for counting. An aliquot of unconditioned media was also counted for the determination of specific activity and data were normalized to well protein content (nmol pyruvate·mg protein−1·min−1), assessed using the bicinchoninic acid (BCA) assay (Pierce). Palmitate oxidation was determined as above using 100 μCi U-14C palmitate (Perkin Elmer) and 100 μM palmitate conjugated to BSA (0.3%). Etomoxir (10 μM), an irreversible carnitine palmitoyltransferase inhibitor, was included as a negative control for the palmitate oxidation assay.
Respiration experiments
Whole cell oxygen consumption rate (OCR) was assessed using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA). Cells were seeded (7.5 × 104 H4IIE) the day before the assay and allowed to grow to 80–90% confluence in regular culture media. Media was removed and, after two washes with PBS, replaced with SFM supplemented with 500 μM sodium pyruvate (Sigma), 25 mM HEPES and either VEH, PIO (10 μM), or UK (5 μM). Cells were incubated at room air at 37 °C for 2 h, before being placed into the XF24 Extracellular Flux Analyzer (Seahorse Bioscience) instrument for assessment of oxygen consumption. After assessing the basal pyruvate-stimulated OCR, oilgomycin (4 μM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (4 μM), and rotenone (1 μM) were added at the indicated time-points for the determination of the OCR associated with ATP turnover, maximal uncoupled respiration, and nonmitochondrial respiration, respectively.
Mitochondrial isolation and ATP synthesis
To investigate the direct effect of PIO on mitochondrial metabolism, the rate of ATP synthesis from multiple substrates was examined in isolated mitochondria. Mitochondria were freshly isolated from cultured hepatocytes (~ 1 × 109 cells) or human skeletal muscle (~ 50–100 mg) by differential centrifugation using protocols described previously for cells [50] and tissue [51]. Substrate-specific rates of ATP synthesis were determined by the luminometric method based on firefly luciferase, as described previously by us [51] and others [52]. Isolated mitochondria were incubated with treatments for 15 min before dilution in a luciferin-luciferase reaction buffer containing ADP (600 μM) and substrate (500 μM pyruvate + 2 mM malate, PM; 500 μM glutamate + 2 mM malate, GM; 500 μM succinate, S; 0.7 μM palmitoylcarnitine + 2 mM malate, PCM). The ATP-producing reaction was followed in a 96-well plate for 7 min after which an internal ATP standard (50 pmol) was added to calibrate results which were subsequently normalized to the protein concentration of the mitochondrial preparations (BCA assay; Pierce). The total amount of mitochondrial protein added to each well was approximately 60 ng.
Pyruvate dehydrogenase activation status
PDH activity was determined in cell lysates using an approach based on the methodology of Constantin et al. [24]. Briefly, H4IIE cells were treated for 2 h with VEH, PIO (10 μM), or DCA (5 mM) in serum- and glucose-free media supplemented with 1 mM sodium pyruvate and 2 mM glutamine in 60 mm dishes. Cells (~ 6 × 106) were washed briefly with PBS, rapidly scraped into PBS and two aliquots were spun in a microcentrifuge for 10 s. One resulting pellet was resuspended in homogenization buffer A (50 mM Tris/HCl, pH 7.8; 200 mM sucrose, 5 mM MgCl2, 5 mM EGTA, 0.1% Triton-X100; pH 7.8) and immediately assayed for active PDH by adding 35 μL lysate to 500 μL reaction buffer (100 mM Tris/HCl, 0.5 mM EDTA, 1 mM MgCl2, 1 mM pyruvate, 1 mM NAD+, 1mM thiamine pyrophosphate, and 0.5 mM Coenzyme A) and removing 150 μL every minute to a fresh vial containing 25 μL of 1 M perchloric acid. Samples were neutralized (6.25 μL 0.25 M KHCO3) and assayed for acetyl-CoA concentration by radioenzymatic assay [53]. For the determination of total PDH, the second cell pellet was resuspended in homogenization buffer B, which was similar to buffer A except for the absence of Triton-X100 and the addition of 10 mM glucose, and freeze-thawed twice prior to assay. Complete catalytic conversion of the enzyme to its active form was achieved by preincubating samples in a buffer containing 5 mM DCA and 2 U·mL−1 hexokinase, to inhibit the PDH kinases, and 100 μM CaCl2, 10 mM MgCl2, and 500 μM spermine, to activate the PDH phosphatases [54,55]. Triton-X100 was added immediately prior to assay, which subsequently followed the protocol described above. PDH activity is expressed as the rate of formation of acetyl-CoA normalized to the protein content of the cell lysate.
Hepatic glucose production
Hepatocellular glucose production was determined as previously described [46] with modifications [47]. Briefly, H4IIE cells were incubated overnight (15 h) in glucose- and serum-free DMEM supplemented with pyruvate (2 mM) plus lactate (20 mM) precursors, and containing either VEH, or various concentrations of PIO, UK, or insulin. For some experiments, glutamine (2–20 mM) or alanine (20 mM) was added, as indicated in figure legends. Glucose release into the media was subsequently measured using the Amplex Red Glucose Assay Kit (Invitrogen), and calibrated using a standard curve generated by spiking in glucose to assay media incubated overnight in the absence of cells. The rate of glucose production was normalized to the protein content of each well.
MPC1 and MPC2 silencing
Silencing of MPC1 and MPC2 mRNA and protein levels in H4IIE cells was performed using 100 nM Dharmacon SmartPool siRNA [MPC1 (BRP44L) siRNA cat#: L-093029-01-000; MPC2 (BRP44) siRNA cat# L-041059-01-0005), introduced via Neon electroporation. Subsequent experiments were performed 24–36 h following electroporation. Knockdown of each protein was subsequently confirmed on western blots using antibodies directed toward MPC1 (Sigma, catalog number: HPA045119) or MPC2 (Cell signaling Technology, catalog number: 46141).
Gene expression analysis
Following the HGP experiments, RNA was extracted and real-time quantitative PCR (qRT-PCR) was performed to assess the expression of rate-limiting HGP genes phosphoenolpyruvate carboxykinase (Pepck), fructose 1,6-bisphosphatase (Fbp1), and glucose 6-phosphatase (G6pc) using Taqman predesigned assays, as described previously [56].
Statistics
Treatment and siRNA effects were analyzed using one- or two-way ANOVA, as appropriate, with Tukey or Bonferroni-corrected t-tests, respectively, performed post-hoc. The number of repeat experiments (n) detailed in each figure legend reflect independent experiments performed using subsequent cell passages, and error bars represent variability between these experiments as standard error of the mean (SEM).
Acknowledgments
This work was supported in part by National Institute of Health Grant KO1DK098314.
Abbreviations
- FBP
fructose 1,6-biphosphatase
- G6P
glucose-6-phosphatase
- HGP
hepatocellular glucose production
- MPC
mitochondrial pyruvate carrier
- PC
pyruvate carboxylase
- PEPCK
phosphoenolpyruvate carboxykinase
- PIO
pioglitazone
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- T2DM
type II diabetes mellitus
- TZD
thiazolidinedione
- UK
UK5099
- VEH
vehicle
Footnotes
Author contributions
CS designed, performed, and analyzed the experiments and wrote the paper. GD and GC performed experiments. RD, MA, and LN conceived the study. LN designed the experiments, analyzed the data, and wrote the paper. All authors reviewed the results and approved a final version of the manuscript.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Rangwala SM, Lazar MA. Peroxisome proliferator-activated receptor gamma in diabetes and metabolism. Trends Pharmacol Sci. 2004;25:331–336. doi: 10.1016/j.tips.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, Clement SC, Henry RR, Hodis HN, Kitabchi AE, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011;364:1104–1115. doi: 10.1056/NEJMoa1010949. [DOI] [PubMed] [Google Scholar]
- 3.Eldor R, DeFronzo RA, Abdul-Ghani M. In vivo actions of peroxisome proliferator-activated receptors: glycemic control, insulin sensitivity, and insulin secretion. Diabetes Care. 2013;36(Suppl 2):S162–S174. doi: 10.2337/dcS13-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miyazaki Y, Mahankali A, Matsuda M, Glass L, Mahankali S, Ferrannini E, Cusi K, Mandarino LJ, DeFronzo RA. Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care. 2001;24:710–719. doi: 10.2337/diacare.24.4.710. [DOI] [PubMed] [Google Scholar]
- 5.Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 6.Rogue A, Spire C, Brun M, Claude N, Guillouzo A. Gene expression changes induced by PPAR gamma agonists in animal and human liver. PPAR Res. 2010;2010:325183. doi: 10.1155/2010/325183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E, Gavrilova O, Reitman ML. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest. 2000;106:1221–1228. doi: 10.1172/JCI11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazaki Y, DeFronzo RA. Rosiglitazone and pioglitazone similarly improve insulin sensitivity and secretion, glucose tolerance and adipocytokines in type 2 diabetic patients. Diabetes Obes Metab. 2008;10:1204–1211. doi: 10.1111/j.1463-1326.2008.00880.x. [DOI] [PubMed] [Google Scholar]
- 9.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Dello Russo C. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 10.Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhausl W, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–1059. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Munoz-Yague T, Solis-Herruzo JA. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol. 2013;11:88. doi: 10.1186/1741-7007-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanz MN, Sanchez-Martin C, Detaille D, Vial G, Rigoulet M, El-Mir MY, Rodriguez-Villanueva G. Acute mitochondrial actions of glitazones on the liver: a crucial parameter for their antidiabetic properties. Cell Physiol Biochem. 2011;28:899–910. doi: 10.1159/000335804. [DOI] [PubMed] [Google Scholar]
- 13.Bova MP, Tam D, McMahon G, Mattson MN. Troglitazone induces a rapid drop of mitochondrial membrane potential in liver HepG2 cells. Toxicol Lett. 2005;155:41–50. doi: 10.1016/j.toxlet.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab. 2004;286:E252–E260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 15.Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)–relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS ONE. 2013;8:e61551. doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 18.Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, et al. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab. 2015;22:669–681. doi: 10.1016/j.cmet.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, Burgess SC, Finck BN. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 2015;22:682–694. doi: 10.1016/j.cmet.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendes-Mourao J, Halestrap AP, Crisp DM, Pogson CI. The involvement of mitochondrial pyruvate transport in the pathways of gluconeogenesis from serine and alanine in isolated rat and mouse liver cells. FEBS Lett. 1975;53:29–32. doi: 10.1016/0014-5793(75)80674-0. [DOI] [PubMed] [Google Scholar]
- 21.Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci U S A. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hildyard JC, Ammala C, Dukes ID, Thomson SA, Halestrap AP. Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim Biophys Acta. 2005;1707:221–230. doi: 10.1016/j.bbabio.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Yang C, Harrison C, Jin ES, Chuang DT, Sherry AD, Malloy CR, Merritt ME, DeBerardinis RJ. Simultaneous steady-state and dynamic 13C NMR can differentiate alternative routes of pyruvate metabolism in living cancer cells. J Biol Chem. 2014;289:6212–6224. doi: 10.1074/jbc.M113.543637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Anal Biochem. 1991;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- 25.Raman P, Foster SE, Stokes MC, Strenge JK, Judd RL. Effect of troglitazone (Rezulin) on fructose 2,6-bisphosphate concentration and glucose metabolism in isolated rat hepatocytes. Life Sci. 1998;62:Pl89–Pl94. doi: 10.1016/s0024-3205(97)01177-6. [DOI] [PubMed] [Google Scholar]
- 26.Raman P, Judd RL. Role of glucose and insulin in thiazolidinedione-induced alterations in hepatic gluconeogenesis. Eur J Pharmacol. 2000;409:19–29. doi: 10.1016/s0014-2999(00)00806-2. [DOI] [PubMed] [Google Scholar]
- 27.Gastaldelli A, Miyazaki Y, Mahankali A, Berria R, Pettiti M, Buzzigoli E, Ferrannini E, DeFronzo RA. The effect of pioglitazone on the liver: role of adiponectin. Diabetes Care. 2006;29:2275–2281. doi: 10.2337/dc05-2445. [DOI] [PubMed] [Google Scholar]
- 28.Bogacka I, Xie H, Bray GA, Smith SR. The effect of pioglitazone on peroxisome proliferator-activated receptor-gamma target genes related to lipid storage in vivo. Diabetes Care. 2004;27:1660–1667. doi: 10.2337/diacare.27.7.1660. [DOI] [PubMed] [Google Scholar]
- 29.Furnsinn C, Brunmair B, Neschen S, Roden M, Waldhausl W. Troglitazone directly inhibits CO(2) production from glucose and palmitate in isolated rat skeletal muscle. J Pharmacol Exp Ther. 2000;293:487–493. [PubMed] [Google Scholar]
- 30.Lembert N, Joos HC, Idahl LA, Ammon HP, Wahl MA. Methyl pyruvate initiates membrane depolarization and insulin release by metabolic factors other than ATP. Biochem J. 2001;354:345–350. doi: 10.1042/0264-6021:3540345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin ES, Moreno KX, Wang JX, Fidelino L, Merritt ME, Sherry AD, Malloy CR. Metabolism of hyperpolarized [1- C]pyruvate through alternate pathways in rat liver. NMR Biomed. 2016;29:466–474. doi: 10.1002/nbm.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MK, Olefsky JM. Acute effects of troglitazone on in vivo insulin action in normal rats. Metabolism. 1995;44:1166–1169. doi: 10.1016/0026-0495(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 33.Fulgencio JP, Kohl C, Girard J, Pegorier JP. Troglitazone inhibits fatty acid oxidation and esterification, and gluconeogenesis in isolated hepatocytes from starved rats. Diabetes. 1996;45:1556–1562. doi: 10.2337/diab.45.11.1556. [DOI] [PubMed] [Google Scholar]
- 34.Fujita Y, Yamada Y, Kusama M, Yamauchi T, Kamon J, Kadowaki T, Iga T. Sex differences in the pharmacokinetics of pioglitazone in rats. Comp Biochem Physiol C Toxicol Pharmacol. 2003;136:85–94. doi: 10.1016/s1532-0456(03)00194-7. [DOI] [PubMed] [Google Scholar]
- 35.Halestrap AP, Scott RD, Thomas AP. Mitochondrial pyruvate transport and its hormonal regulation. Int J Biochem. 1980;11:97–105. doi: 10.1016/0020-711x(80)90241-4. [DOI] [PubMed] [Google Scholar]
- 36.Wimhurst JM, Manchester KL. Some aspects of the kinetics of rat liver pyruvate carboxylase. Biochem J. 1970;120:79–93. doi: 10.1042/bj1200079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Portenhauser R, Wieland O. Regulation of pyruvate dehydrogenase in mitochondria of rat liver. Eur J Biochem. 1972;31:308–314. doi: 10.1111/j.1432-1033.1972.tb02534.x. [DOI] [PubMed] [Google Scholar]
- 38.Halestrap AP. The mitochondrial pyruvate carrier: has it been unearthed at last? Cell Metab. 2012;16:141–143. doi: 10.1016/j.cmet.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Lavoie L, van de Werve G. Hormone-stimulated glucose production from glycogen in hepatocytes from streptozotocin diabetic rats. Metabolism. 1991;40:1031–1036. doi: 10.1016/0026-0495(91)90125-g. [DOI] [PubMed] [Google Scholar]
- 40.Matsuda M, Defronzo RA, Glass L, Consoli A, Giordano M, Bressler P, Delprato S. Glucagon dose-response curve for hepatic glucose production and glucose disposal in type 2 diabetic patients and normal individuals. Metabolism. 2002;51:1111–1119. doi: 10.1053/meta.2002.34700. [DOI] [PubMed] [Google Scholar]
- 41.Vigueira Patrick A, McCommis Kyle S, Schweitzer George G, Remedi Maria S, Chambers Kari T, Fu X, McDonald William G, Cole Serena L, Colca Jerry R, Kletzien Rolf F, et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep. 2014;7:2042–2053. doi: 10.1016/j.celrep.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halestrap AP. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem J. 1975;148:85–96. doi: 10.1042/bj1480085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell. 2014;56:414–424. doi: 10.1016/j.molcel.2014.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowman CE, Zhao L, Hartung T, Wolfgang MJ. Requirement for the mitochondrial pyruvate carrier in mammalian development revealed by a hypomorphic allelic series. Mol Cell Biol. 2016;36:2089–2104. doi: 10.1128/MCB.00166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hectors TL, Vanparys C, Pereira-Fernandes A, Knapen D, Blust R. Mechanistic evaluation of the insulin response in H4IIE hepatoma cells: new endpoints for toxicity testing? Toxicol Lett. 2012;212:180–189. doi: 10.1016/j.toxlet.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 46.de Raemy-Schenk AM, Trouble S, Gaillard P, Page P, Gotteland JP, Scheer A, Lang P, Yeow K. A cellular assay for measuring the modulation of glucose production in H4IIE cells. Assay Drug Dev Technol. 2006;4:525–533. doi: 10.1089/adt.2006.4.525. [DOI] [PubMed] [Google Scholar]
- 47.Norton L, Fourcaudot M, Abdul-Ghani MA, Winnier D, Mehta FF, Jenkinson CP, Defronzo RA. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia. 2011;54:3132–3142. doi: 10.1007/s00125-011-2289-z. [DOI] [PubMed] [Google Scholar]
- 48.Hanefeld M. Pharmacokinetics and clinical efficacy of pioglitazone. Int J Clin Pract Suppl. 2001;121:19–25. [PubMed] [Google Scholar]
- 49.Collins CL, Bode BP, Souba WW, Abcouwer SF. Multiwell 14CO2-capture assay for evaluation of substrate oxidation rates of cells in culture. Biotechniques. 1998;24:803–808. doi: 10.2144/98245st04. [DOI] [PubMed] [Google Scholar]
- 50.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 51.Daniele G, Eldor R, Merovci A, Clarke GD, Xiong J, Tripathy D, Taranova A, Abdul-Ghani M, DeFronzo RA. Chronic reduction of plasma free fatty acid improves mitochondrial function and whole-body insulin sensitivity in obese and type 2 diabetic individuals. Diabetes. 2014;63:2812–2820. doi: 10.2337/db13-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wibom R, Hagenfeldt L, von Dobeln U. Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal Biochem. 2002;311:139–151. doi: 10.1016/s0003-2697(02)00424-4. [DOI] [PubMed] [Google Scholar]
- 53.Cederblad G, Carlin JI, Constantin-Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Anal Biochem. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- 54.Sheu KF, Hu CW, Utter MF. Pyruvate dehydrogenase complex activity in normal and deficient fibroblasts. J Clin Invest. 1981;67:1463–1471. doi: 10.1172/JCI110176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pezzato E, Battaglia V, Brunati AM, Agostinelli E, Toninello A. Ca2+-independent effects of spermine on pyruvate dehydrogenase complex activity in energized rat liver mitochondria incubated in the absence of exogenous Ca2+ and Mg2+ Amino Acids. 2008;36:449. doi: 10.1007/s00726-008-0099-5. [DOI] [PubMed] [Google Scholar]
- 56.Norton L, Chen X, Fourcaudot M, Acharya NK, DeFronzo RA, Heikkinen S. The mechanisms of genome-wide target gene regulation by TCF7L2 in liver cells. Nucleic Acids Res. 2014;42:13646–13661. doi: 10.1093/nar/gku1225. [DOI] [PMC free article] [PubMed] [Google Scholar]