

Abstract
Accurate modeling of protein ligand binding is an important step in structure-based drug design, is a useful starting point for finding new lead compounds or drug candidates. The ‘Lock and Key’ concept of protein-ligand binding has dominated descriptions of these interactions, and has been effectively translated to computational molecular docking approaches. In turn, molecular docking can reveal key elements in protein-ligand interactions-thereby enabling design of potent small molecule inhibitors directed against specific targets. However, accurate predictions of binding pose and energetic remain challenging problems. The last decade has witnessed more sophisticated molecular docking approaches to modeling protein-ligand binding and energetics. However, the complexities that confront accurate modeling of binding phenomena remain formidable. Subtle recognition and discrimination patterns governed by three-dimensional features and microenvironments of the active site play vital roles in consolidating the key intermolecular interactions that mediates ligand binding. Herein, we briefly review contemporary approaches and suggest that future approaches treat protein-ligand docking problems in the context of a ‘combination lock’ system.
Keywords: Docking, Scoring, Virtual screening, Cavity detection, Pharmacophore, Fragment-based design, Structure-based Drug design, Molecular recognition, Binding energy
Introduction
In 1894, Emil Fischer suggested that the specificity of an enzyme towards its substrate is based on the two components exhibiting complementary geometric shapes that fit perfectly like a ‘key in a lock’. This simple ‘lock and key’ analogy succinctly conceptualized the essence of enzyme substrate interaction where the ‘lock’ describes the enzyme and the ‘key’ describes the substrate or some other small molecule ligand (e.g. a small molecule inhibitor). In such systems, it is a requirement that the ‘key’ (substrate) fit appropriately into the key hole (active site/binding pocket) of the ‘lock’ (enzyme/receptor) for productive biochemistry to take place. Keys that are too small, too large, or with incorrectly positioned notches and grooves, will not fit into the lock (Figure 1).
Figure 1.
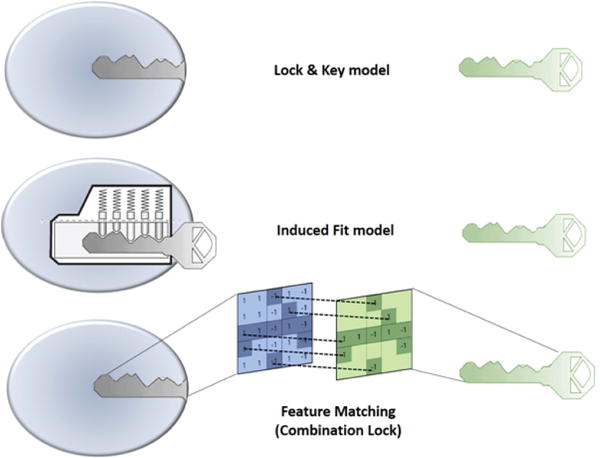
Illustration of ‘Lock and Key’ (top), Induced fit (middle) and Combination Lock (bottom) model of protein-ligand binding interaction.
But, enzymes show conformational flexibility and, on that basis, Daniel Koshland proposed a modification to the ‘lock and key’ model. Koshland’s suggestion was that active sites of enzymes are reshaped during interactions with substrate. This ‘induced fit’ model conceptualizes the ‘lock’ (enzyme) as a dynamic entity and that the ‘key’ (substrate) modulates the shape of the ‘key hole’. This concept paints a picture of an enzyme∷ligand interaction that is more akin to that of a ‘pin tumbler lock’. That is, a device where the pointed teeth and notches on the key allow the pins and wafers in the lock to move up and down until they align with the shear line of the cylindrical grooves of the key. The cylinder moves or rotates within the lock until that fit configuration is reached and the ‘lock’ opens. In an analogous manner, a ‘correct’ substrate aligns with active site residues of the enzyme to induce the appropriate conformational changes required for the desired outcome. ‘Induced fit’ is an attractive hypothesis as it accounts for why certain ligands are not substrates for an enzyme – even though they seemingly satisfy the specific shape requirements to bind to the active site (Figure 1). Computational chemists are now using these basic ideas to model protein-substrate interactions. For reasons of its greater tract ability, the ‘lock and key’ paradigm has, for better or for worse, dominated the philosophical underpinnings of molecular docking approaches. In many respects, ‘induced fit’ approaches are more powerful-albeit more complicated. Below, we review these issues as these apply to molecular docking.
Molecular docking reaches for two major goals. The first is to correctly predict and identify the most favorable binding mode of a given ligand in the active site or binding pocket of a given protein. The second is to correctly rank a family of ligands in accordance to their corresponding experimentally-determined binding affinities [1,2]. The high-throughput version of docking, often referred to as virtual screening or in silico screening, aims to harvest small lists of potential active compounds for downstream experimental testing from a database of millions of compounds [3]. All docking protocols have two essential components: (1) a good positioning algorithm, and (2) a robust ranking or scoring system. Docking requires extensive sampling of conformational space for a ligand in the binding pocket of a protein and thereby generates large numbers of potential poses that orient a ligand within the active site. A good positioning algorithm samples ‘all’ possible binding modes, while the scoring system ranks all the solutions and identifies the most likely ‘binding mode’ of the ligand (Figure 2).
Figure 2.
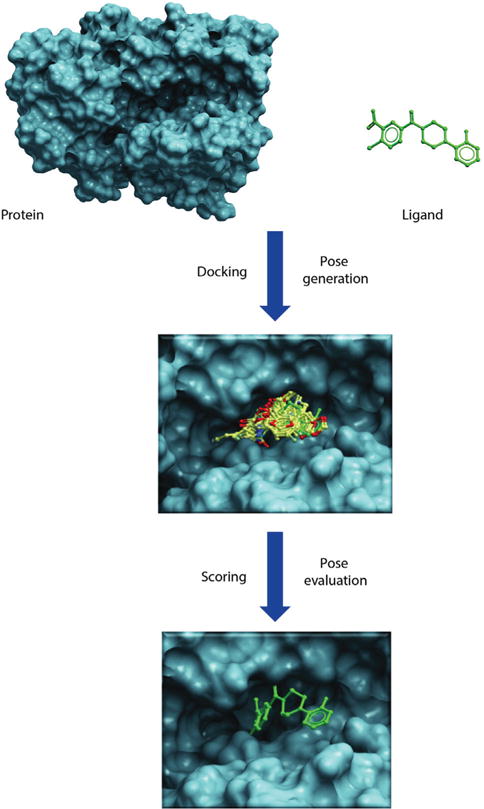
Illustrates docking and scoring scheme as a two-step process. First step involves generation of poses within the binding cavity and second step involves energetic evaluation of poses to find best scoring pose that would mimic the native protein-ligand binding.
As simple as the process may sound, both components are themselves complex problems that pose significant challenges [4,5]. Positioning requires exhaustive exploration of accessible conformational space and binding orientations within the active site so as to extensively map interactions between active site residues and ligand. This requires that the process for generating binding modes respect a fine balance between speed and accuracy. That is, the process must not miss valuable solutions while maintaining sufficient computational efficiency to triage nonsensical binding modes. The ability to correctly score and rank the binding modes generated for a ligand presents an even bigger challenge. In cases where a number of different ligands are being interrogated, the scoring function aims to generate a rank list that corresponds to the binding affinity. This is a challenging task as many scoring functions fail to accurately predict binding affinity and often simply report a score which may or may not be at all congruent with experimentally measured binding affinities [6].
Considering the vast conformational sampling space that must often be negotiated in docking experiments, it is not computationally feasible to explore all the degrees of translational and rotational freedom of the ligand along with the internal conformational degree of freedom for protein-ligand complex. Therefore, docking experiments are typically coarse-grained so that only a restricted sampling space is covered, and a limited number of the possible binding modes are sampled. To optimize docking and scoring functions, several methods have recently been developed to add layers of sophistication to simple ‘key into lock’ ideas.
Defining the ‘Lock’
The identification and mapping of a binding site from crystal structure data can reveal key elements in protein-ligand binding [7]. Such knowledge is indispensable for docking and rational drug design since, in the majority of cases, receptor-drug interactions are specific in nature. However, this is not as trivial an undertaking as it may initially seem. The first requirement for any successful docking simulation is to define an active site or binding pocket as this is a critical step in structure-based drug design, and provides a starting point for finding new lead compounds or drug candidates [8]. A broad suite of cavity detection methods has been developed to address these issues in docking and virtual screening simulations [9,10].
The success of docking and structure-based design of a drug molecule for a specific target site depended largely on the quality of information regarding active site architecture because it is the size and shape of active site or binding cavity that dictates the three-dimensional geometry of ligands that will bind within. Pocket architecture also governs the directional and non-directional intermolecular interactions that mediate protein-ligand binding. Thus, clear definition of a binding pocket surface, coupled with identification of protein∷ligand interaction sites, provides a feature set for ligand orientation within a binding substructure. A target protein may have several pockets or cavities for a ligand to bind. Some might be deeply buried in the protein interior, while some might be displayed on the protein surface. However, the precise architecture of these pockets may not be absolutely clear from standard inspection of structural data as these cavities and protrusions are frequently interconnected via small and narrow channels, or are interspersed with numerous holes or voids [9]. The shape and size of binding pockets are also potentially subject to significant variations brought on by rotation of amino acid side-chains, backbone movements, loop motions, and/or ligand-induced conformational changes [9]. Fundamental uncertainties of this nature conspire to make identification of optimal dock solutions more difficult.
After defining the binding site surface, the next crucial step is to locate the interaction sites or “hot spots” within the binding site [11,12]. The primary goal of interaction mapping is to understand the chemical microenvironment of binding so that interaction points can be used to constrain pose possibilities and thereby restrict sampling space to a manageable size. Thus, binding site mapping is a critical step as it defines ‘lock’ parameters and sets the constraints for positioning the ligand in the defined binding region. In addition to preparing the active site for docking, the physicochemical properties and/or interaction can be represented as fields that can be mapped and visualized, interactively, in three dimensions. Using interaction maps, the spatial distributions of properties such as charge, hydrophobicity, etc. can be qualitatively analyzed [12–15]. Points of interaction between the ligand and active site might be elucidated and assessed qualitatively and, in some cases, semi-quantitatively. The importance of mapping interacting features is a critical endeavor since the number of ‘hot spots’ and their contributions to the larger binding process are essential for hypothesis generation. Quality interaction mapping also facilitates the docking process by defining a set of constraints that can be quantified in terms of how many, and which, interaction points might be matched by a ligand or a library of compounds. However, the harsh reality is that, even after defining the binding region for docking and extracting interaction sites, the docking process remains fraught with uncertainties that stem from the inherently dynamic physicochemical properties of the protein-ligand system.
Protein flexibility
Proteins leverage their intrinsic conformational flexibilities to carry out a wide range of biochemical processes in catalysis, protein-protein interaction and functional regulation [16]. In many cases, subtle motions in domains, flexibilities in the protein main chain, or re-orientation of side chains, changes the shape and size of the ligand binding envelope [17]. Ligand binding itself can also effect a change in the topography of binding pocket by inducing loop movements and other conformational shifts. These range from hinge movements of entire domains, to small side-chain rearrangements in residues of the binding pocket [18,19], and even structural transitions that involve opening/closing of otherwise rigid structural elements of the protein about flexible joints. For these reasons, it is always useful to compare holo- and apo-structures of a protein of interest whenever possible. Although most contemporary docking approaches treat ligands as flexible, it remains a challenging task to incorporate protein flexibility into the docking regime. A thorough analysis of side chain flexibility may provide invaluable insights for improving docking run and for optimizing protein-ligand interactions. Despite some recent advancements in considering protein side-chain flexibility in optimizing simulation of protein-ligand interactions, protein flexibility remains one of the most important factors in improvement of methods for docking ligands to their flexible protein partner [20].
Considering the role of water
H2O molecules play myriad roles in biological structure and functions. The importance of structured water molecules in biological systems cannot be overstated given their critical roles in modulating protein–ligand interactions, and these considerations take center stage in the context of drug design and discovery [21]. When a structured water molecule is displaced by a ligand and banished to “bulk” solvent, the act of displacement increases system entropy and helps drive ligand binding. That is, ligand binding is thermodynamically more favorable if the ligand displaces a tightly bound water molecule by replicating its interaction with protein [22]. For protein-ligand complexes, many water molecules are retained in the active site and contribute to the energetics of protein∷ligand interactions independent of entropic considerations. For example, waters can bridge protein and ligand and license what would otherwise represent unfavorable interactions between two chemically incompatible groups (e.g. two bases). Water molecules can also alter the “shape” and microenvironment of the active site by tightly associating with specific residues and thereby present a steric and electrostatic binding pocket profile that is different to the one presented by an anhydrous active site [23,24]. These varied functional involvements of water define yet another set of important considerations that must be respected in quality docking experiments and in rational design of high affinity lead molecules. Accessible surface areas of water molecules, the hydrogen bonds that involve water, the conservation and/or displacement of water, as well as the interaction energetics of water molecules are some of the factors that must be considered in docking simulations. The reality is that contemporary state-of-the art docking algorithms, and the scoring functions that accompany them, do not adequately consider all the explicit and implicit contributions of water molecules to the binding equation. Nonetheless, several docking routines include methods for identifying relevant water molecules and including those contributions in pose generation and in calculating free energies of ligand binding [25].
Protonation and ionization states of binding site residues
In addition to managing issues associated with protein flexibility and solvent, both the computational intensities and uncertainties of the docking problem are compounded for protein∷ligand systems with variable ionization states, and contributions of metals and counter ions [26]. Protein ligand interactions are sensitive to subtle changes in microenvironment of the binding site. Change in pH, buffer, ionic strength, and temperature conditions under which the data are collected also affect the microenvironment of an active site [27]. Protonation states of active site residues are typically not well-assigned, even in high resolution X-ray crystal structures, and therefore present little information to prepare the structure for docking [28]. Moreover, protein crystals are typically solvent rich (30–70%)-values that often include the crystallization buffer [29]. The accompanying ions and solvent molecules are distributed throughout the protein molecule in accord with the electrostatic properties of the solvent-accessible pockets. Altering ambient pH often alters the ionization states of residues and thereby influences the shape and electrostatic properties of the binding pocket, and ultimately the set of ligand-binding solutions [30]. Multiplicity of protonation states in ligand–protein complexes is an often overlooked aspect in protein structure preparation as emphasized by the fact that current modeling techniques frequently ignore the possibility of multiple protonation states.
There is recent progress on this front, however. New algorithms such as the computational titration protocol implemented in Hydropathic Interaction (HINT) seek to identify and optimize all possible protonation states so that rational models with atomic details can be constructed and applied to model ligand-binding energetic [26,30,31]. By modeling all ionizable residues in the binding pocket, and calculating all the possible protonation states of residues and functional groups within the active site, the computational-titration methodology realistically samples the dynamic behavior of labile H-atoms in the active site microenvironment. In particular, an important aspect of the active site microenvironment that is often ignored is the dielectric constant within the active site [32,33]. While comprehensive estimations of polarizability and binding energies are computationally expensive endeavors, simplified models that use macroscopic dielectric models, either uniform or distance-dependent, are being productively applied to descriptions of binding site microenvironments [34,35]. The message is that accurate prediction of binding free energies requires that pH, ionization and entropic contributions be taken into account in docking and virtual screening experiments.
Entropy
Entropic considerations, as well as the contributions of hydrophobicity, in ligand binding cannot be overstated but are often poorly characterized and poorly quantified [36,37]. Entropy and hydrophobicity are difficult to measure and therefore difficult to computationally model. It is for this reason that these parameters are sacrificed in favor of computational efficiency. Most approaches consider enthalpic and entropic contributions separately and sum these interactions to a cumulative score [38]. However, protein-ligand binding is a concerted event, and entropy and hydrophobicity are thermodynamic quantities which cannot be accurately described by a simple summation. Solvation and desolvation effects that involve hydrophobic interactions are significant factors in protein∷ligand interactions but are particularly difficult to model computationally. But, the effort is worthwhile. Docking simulations that adequately consider the entropic, solvation/desolvation, and thermodynamic components of a binding reaction yield information whether the binding is enthalpy- or entropy driven and provide vital insights into the free-energy changes in the system [39–43].
Finding the right ‘key’
Once the ‘lock’ is defined (i.e boundary and interacting features within the binding pocket are delineated) the next core issue is to find a suitable key for the lock. To accomplish this task, the first step is fitting the ligand (key) into the binding pocket (key hole) and finding the best fit. That effort involves sampling different ligand conformations and orientations within the binding pocket and measuring the fitness of different alternative poses to identify the most favorable fit. Thus, docking approaches share two components: (i) a search algorithm that generates a sufficient set of different poses so that it exhaustively samples nearly all possible conformations and orientations for a ligand, and (ii) a scoring algorithm which evaluates the generated poses, approximates their binding energies, and identifies an optimal binding pose(s). Several different search algorithms have evolved over the past decades that were based on a variety of computational approaches [44–47]. Interestingly, the evolution of computational docking approaches offers interesting parallels to the evolution of thought from ‘lock and key’ to ‘induced fit’ hypotheses. Several approaches, with different degrees of sophistication, evolved from ‘rigid body’ considerations to ‘flexible ligand’ docking methods, and are still evolving into ever more sophisticated and computationally intensive ‘flexible-ligand and flexible receptor’ methods [48–51]. In rigid body approaches both the receptor and ligand are treated as static units and search algorithm tries to orient a rigid ligand within a rigid binding pocket [52–54]. Flexible-ligand methods treat the receptor (protein) as a rigid entity, but impart flexibility to the ligand and explore different conformations in systematic or random stochastic manners [48–51,55]. By contrast, ‘flexible-ligand and flexible-receptor’ approaches treat both receptor and ligand as flexible entities [56–59]. Despite the significant progress made in flexible protein-ligand docking, significant improvement is still needed.
One of the earliest docking approaches involved systematic search logic [60,61]. However, the search becomes ever more complex with increasing ligand flexibility as the number of degree of freedom of the ligand molecule obviously increases. Such an approach was implemented in methods where ligand and binding pocket were considered to be rigid and ligand was fitted using shape complementarity as determined by point complementarity or distance geometry approaches [62,63]. In such docking methods, the shape of both the receptor site and the ligand is interrogated based on criteria of shape and pharmacophoric points. Orientations are generated through various alignment procedures in order to maximize the pharmacophoric constraints and shape complementarity. However, it is not feasible to exhaustively explore available conformational space, and an acceptable balance has to be struck between speed and accuracy so that as many binding modes can be explored as is feasible. Fragment-based approaches that involve either incremental construction of ligand in the binding pocket, or by simply placing and joining the fragment, circumvent problems associated with combinatorial explosion of conformers generated by the previous approaches [64–66].
Stochastic methods involving random sampling of conformational space of ligand in the binding pocket are also being widely applied in many docking algorithms. Algorithms using Monte Carlo sampling, coupled with Metropolis criterion, are applied to exhaustively interrogate the conformational space [67]. Simulated annealing protocols, combined with grid-based energy evaluations, can be coupled with such an approach to overcome high conformational energy barriers in the sampling regime [68]. Another such stochastic approach that has been successfully implemented in docking algorithm is the genetic algorithm-based sampling of conformational space [69–71]. In this approach, multi-conformers referred as chromosomes are evaluated, crossed and mutated and the best possible solution is selected based on a fitness function. The ultimate solution is represented by the best scored conformation of the total conformers after a suitable number of generations. GOLD (Genetic Optimization for Ligand Docking) is the most widely used algorithm of this type for flexible molecular docking [72].
In contrast to systematic and stochastic approaches, molecular dynamics-based and heuristic tabu searches are also implemented to explore the sample space [73,74]. However, molecular dynamics is computationally expensive which restricts its use in docking. To circumvent the problem of exhaustive sampling, tabu search approaches are adopted where a list of already explored conformations is maintained and only unexplored spaces are sampled [75]. This avoids reinvestigating space already sampled by associating previously sampled conformations with a degree of penalty. Apart from these deterministic approaches, hybrid consensus logic combine features from other two approaches [76,77]. Although these approaches can exhaustively generate and sample all possible conformations within the active site, it remains a fact that the success of any docking program is measured by how well it reproduces experiment.
The success of whole molecule docking, de novo construction of molecules into a target site, or screening large virtual combinatorial libraries is ultimately dependent on the accuracy of the scoring function that ranks the compounds. Ligand orientations can be evaluated on the fly as the ligand or fragment is positioned within the cavity, or all the generated poses can be scored in the end. The scoring methods that are used in high throughput settings i.e. that deal with thousands of diverse compounds, can be evaluated by how well the corresponding relative binding affinities can be predicted. That need has spurred development of multiple methods which can be subdivided in four major approaches: force field-based methods, semi-empirical approaches, empirical scoring methods, knowledge-based potentials, and consensus scoring functions that are a combination of multiple scoring functions [78–80].
Force field-based methods
Force field-based scoring methods generally use a molecular mechanics force field. This parameter contains terms for intramolecular forces (e.g. bond, angle and dihedral terms) between atoms bonded to each other, plus energy terms for intermolecular forces that describe the forces between non-bonded atoms (e.g. Van der Waals and Coulombic terms). There are also a number of widely and successfully applied molecular mechanics-based scoring functions [81–84]. Their popularity in virtual screening programs is a reflection of their simplicity. Though faster and simpler, these functions are not ideal for simulating biomolecular interactions as those methods were developed for calculating gas phase enthalpy of binding. Thus, this class of scoring approaches has many drawbacks, primarily that these ignore hydrophobic interactions, and solvation and entropic effects.
Empirical scoring methods
Empirical scoring methods offer an alternative approach to pure molecular mechanics-based force field scoring methods [85]. The principle is that the binding free energy of a non-covalent protein-ligand complex can be factorized into a sum of localized and chemically intuitive interactions. The terms accounting for different contributions such as hydrogen bonds, hydrophobic interactions, entropic effects are normalized by weighting factors derived from regression analyses of data from training sets comprised of well characterized protein-ligand complexes. Based on the assumption of additivity, the binding affinity is estimated as a sum of interactions multiplied by weighting factors and solved by equation of the type (1):
| (1) |
Where fi is a simple geometrical function of the ligand (rl) and receptor (rp) coordinates [6]. However, accuracy of these methods depends upon the quality of the experimental binding data and of the crystallographic structural data of the training set.
Semi-empirical approaches
Semi-empirical scoring functions combine the above two approaches and incorporate empirical, or empirically calibrated, energetic terms for interactions that cannot be computed by pure molecular mechanics-based methods. Thus, implicit binding energy terms such as hydrogen bonding, solvent effects, hydrophobicity and entropic terms are included in the scoring functions. In contrast to force field-based scoring functions, semi-empirical scoring terms also more accurately estimate binding energies by accounting for entropic and solvation effects known to significantly affect biological interactions in aqueous medium [86–89].
Knowledge-based scoring
Knowledge-based scoring functions [90] are rule-based regimes where rules are derived from the analysis of structural data of known and well characterized receptor-ligand interactions. The exponential growth and availability of protein-ligand crystal structures is enabling derivation and formulation of rule sets based on frequencies of chemical interactions. Scoring functions of this type seek to capture the knowledge about protein-ligand binding that is implicitly stored in the protein data bank by means of statistical analysis of structural data. That is, potentials are obtained by statistical analysis of atom-pairing frequencies observed in crystal structures of protein-ligand complexes [91]. Again, the accuracy of knowledge-based scoring function depends on the quality of experimental data, as it incorporates structural knowledge without considering inconsistencies in experimental and structural data.
Consensus scoring
Although multiple approaches have been implemented for derivation of a robust scoring function, none of the scoring functions are ideal. Invariably, various approximations are made to strike a balance between speed and accuracy. Taking into consideration the limitations of anyone scoring function, the concept of consensus scoring evolved from the base premise that a combination of different scoring functions will buffer inherent weaknesses in individual functions and offer better performance [92]. A consensus between a set of scoring functions can be reached either by averaging the rank assigned by each scoring function, or averaging the score value calculated by different functions. Ideally, the best scoring function should be able to discriminate between native and non-native binding modes and be able to calculate the actual free energy of binding.
Combination Lock and Key
Traditional docking approaches largely operate on ‘lock and key’ concepts, and this philosophy has enjoyed some successes in estimating the native binding poses of small molecule ligands. A variety of sophisticated approaches have come on-line in recent years that consider conformational flexibility for both ligand and protein [93]. However, the fact remains that both ‘lock and key’ and ‘induced fit’ approaches provide a simplistic views of ligand-binding phenomena that in actuality represent intricate molecular recognition/interaction processes. For this reason, we prefer to view protein-ligand recognition and binding reactions in terms of a ‘combination lock’ system (Figure 1). In this scenario, a tandem combination of complementary features provided by both the protein and the ligand match as in case of a ‘combination lock’. Upon satisfying a suitable combination of features a binding event then ensues. For matching to occur, both feature variables on protein and ligand fine-tune and adapt in a search for the best complementarity. That is, the better the feature matching the tighter the binding. The questions then come to: (i) what are these features, (ii) how are these features encoded in the three-dimensional structure, and (iii) how is the three-dimensional feature code decoded by binding partners? The features could be geometric properties based on the three-dimensional structure of the molecule (e.g. shape, size, volume, surface area, etc.) and/or physicochemical features described by intrinsic electronic properties of a molecule (e.g. electrostatic, hydropathic and van der Waals energetic components). While the energy-based features are more dynamic in nature, and manifest themselves in three-dimensional interaction fields, the geometry-based properties are static in character. It is the sum of pharmacophoric chemical features (e.g. hydrogen bond donor/acceptors, aromatic centers, etc.), geometric features, and intrinsic electronic features of the molecules that define unique interaction fingerprints. The spatial arrangement of these various properties is a particularly discriminating property as electronic, hydropathic and van der Waals energetic properties have varying intensities in three-dimensional space and thereby form unique fields the strength of which vary from point to point and are distance dependent. The patterning of these feature sets in three-dimensional space forms the essence of molecular recognition.
Using the ‘combination lock’ concept, the essential challenge in developing the next generation of robust and predictive docking model is to accurately derive the critical interaction features and map their arrangement in three-dimensional space. These encoded features and properties must first be extracted to define exclusive ‘interaction fingerprints’ for both a ligand binding substructure on the receptor and for the ligand. These unique features and ‘interaction fingerprints’ can be stored as mathematical representations in two- or three-dimensional matrices. Subsequently, machine learning and feature matching algorithms can extract the relevant features and simulate the corresponding protein-ligand binding interactions [94,95]. Features extracted from physical-chemical properties and energies will have broad applicability in deriving target-focused docking and scoring in addition to developing regimes for generating target-focused libraries in silico (Figure 3).
Figure 3.
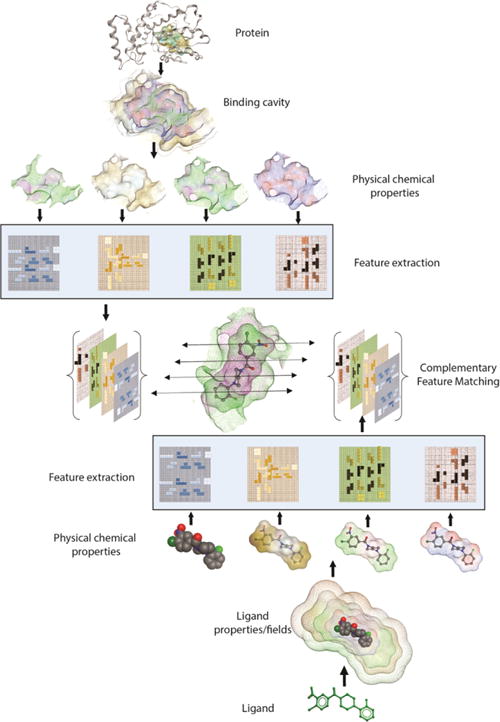
Schematic of ‘Combination Lock’ hypothesis based on feature matching. Protein and Ligand’s physicochemical properties are mapped and relevant binding features are extracted. Matching the combination of best complementary features between protein and ligand ensues optimal fit.
The availability of substantially more protein-ligand complex data and robust machine learning algorithms suggests that feature matching methodology may now be even more effective approach to predict and characterize protein-ligand binding. Recently, a combination of structure-based QSAR approach was implemented to generate descriptive and predictive models for phosphodiesterase-4 inhibitors [96]. This approach applies machine learning methodology to describes protein-ligand binding based on matching of ligand pharmacophore feature pairs with those of the target binding pocket. The method takes advantage of structure of binding pocket to derive feature sets or descriptors which is used as a reference for matching and makes it unique and target specific. Similar feature sets are generated for ligands followed by generation of structure-based pharmacophore key (SBPPK) from the protein-ligand complex based on their feature matching patterns with the binding pocket. Once the feature pairs are generated for both the receptor and ligands machine learning methods can be employed to determine pattern matches to build descriptive and predictive models of protein-ligand interactions. The method was successfully applied to study the SAR (Structure Activity Relationship) of 35 PDE-4 inhibitors. In another similar approach, atom based Interaction Fingerprint (IF) were applied to describe the patterns of ligand pharmacophores that interacted with proteins in complex [97]. These fingerprints are calculated from the distance of pairs of ligand pharmacophore features that interact with protein atoms delineating important geometrical patterns of ligand pharmacophores. From a physicochemical and pharmacological perspective, the detected patterns of ligand features would facilitate an understanding of the structure-activity relationship of the protein-ligand interactions. The method further allows a comparison of the interaction patterns of a target with those of several other targets and facilitates in sillico screening against other homologous proteins. Some of these approaches are applied as a pre-screen and to filter large databases of small molecules before they are actually docked into the protein binding pocket. This database filtering procedure was applied to virtually screen HIV protease inhibitors from ZINC database [98]. The method involved identification of binding site topology and generating site interaction points based on physicochemical property. The resultant functional/interaction properties are saved as a receptor site’s distance matrix. Similar to receptor site distance matrix, functional interaction points are located in small molecule ligand and a similar topological matrix is generated. The methodology can be seen as a comparison and matching of the ligand’s distance matrices with receptor’s matrices. Overlay and matching of receptor and ligand site matrices with each complementary pair, describes ligand’s functionalities mapped onto receptor’s binding pocket. Similar matrices can be generated for small molecules and large databases can be screened as comparing the matrices is a simple matter of matching each molecule’s distance matrix with the one generated from the protein’s binding pocket. The high proportion of known active compounds recovered in the top ranks along with target specificity signifies a promising future for the feature matching approaches for virtual screening. Such hybrid QSAR, machine learning approach that take into account ligand features as well have been applied and benchmarked against traditional rigid body docking methods and affords similar or better enrichment ratios in virtual screening [99–102]. We suggest that ‘combination lock’-driven approaches better capture the complex inter-relationships between feature properties of interacting biomolecules, and that implementation of such approaches will herald significant progress in our ability to model protein-ligand binding events with superior accuracy.
Conclusion
A primary aim of structure-based drug design is to adequately describe the binding interactions between a drug and its target. Traditionally, and perhaps in a tired analogy, protein-ligand binding is treated as a ‘Lock and Key’ system. Although pioneering studies in flexible docking and free energy calculation are making significant progress towards improving the accuracy of docking and virtual screening regimes these technologies remain complex, are time consuming and, for a variety of reasons, still suffer errors. Paradigm shifts in docking and scoring regimes are being driven by the evolution of artificial intelligence and machine learning algorithms for pose scoring and evaluation. With the availability of experimental binding data from bioactivity databases the molecular docking field is witnessing the emergence of hybrid approaches that combine ligand-based and structure-based approaches. Some of the current methods extend ligand-based machine learning strategies and principles in the direction of structure-based approaches. Based on feature extraction and correlation with crystallographic and bioactivity data, robust predictive models can now be generated complementing structure-based approach. Such hybrid ‘Combination Lock’ approaches are evolving technology and albeit with number of limitations, holds great promise for future progress in drug discovery and development.
Acknowledgments
This work was supported by grants GM44530, GM112591 from the National Institutes of Health and BE- 0017 from the Robert A. Welch Foundation (VAB). We also extend our thanks to The Laboratory for Molecular Simulation and High Performance Research Computing (HPRC) at Texas A&M University for providing software, support, and computer time.
References
- 1.Lengauer T, Rarey M. Computational methods for biomolecular docking. Curr Opin in Struct Biol. 1996;6:402–406. doi: 10.1016/s0959-440x(96)80061-3. [DOI] [PubMed] [Google Scholar]
- 2.Vajda S, Guarnieri F. Characterization of protein-ligand interaction sites using experimental and computational methods. Curr Opin Drug Discov Devel. 2006;9:354–362. [PubMed] [Google Scholar]
- 3.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 4.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: Current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 5.Huang SY, Zhou XQ. Advances and Challenges in Protein-Ligand Docking. Int J Mol Sci. 2010;11:3016–3034. doi: 10.3390/ijms11083016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spyrakis F, Cozzini P, Kellogg GE. Docking and scoring in drug discovery Burger’s Medicinal Chemistry and Drug Discovery. 7th. John Wiley & Sons, Inc; New Jersey, USA: 2009. [Google Scholar]
- 7.Kleywegt GJ, Jones TA. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr D Biol Crystallogr. 1994;50:178–185. doi: 10.1107/S0907444993011333. [DOI] [PubMed] [Google Scholar]
- 8.Campbell SJ, Gold ND, Jackson RM, Westhead DR. Ligand binding: functional site location, similarity and docking. Curr Opin Struct Biol. 2003;13:389–395. doi: 10.1016/s0959-440x(03)00075-7. [DOI] [PubMed] [Google Scholar]
- 9.Tripathi A, Kellogg GE. A novel and efficient tool for locating and characterizing protein cavities and binding sites. Proteins. 2010;78:825–842. doi: 10.1002/prot.22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vajda S, Guarnieri F. Characterization of protein-ligand interaction sites using experimental and computational methods. Curr Opin Drug Discov Devel. 2006;9:354–362. [PubMed] [Google Scholar]
- 11.Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hotspots in protein-protein and protein-ligand interfaces. Bioinformatics. 2006;22:1335–1342. doi: 10.1093/bioinformatics/btl079. [DOI] [PubMed] [Google Scholar]
- 12.Tripathi A, Surface JA, Kellogg GE. Using active site mapping and receptor-based pharmacophore tools: prelude to docking and de novo/fragment-based ligand design. Methods Mol Biol. 2011;716:39–54. doi: 10.1007/978-1-61779-012-6_3. [DOI] [PubMed] [Google Scholar]
- 13.RosenfieId RE, Swanson JSM, Meyer EF, CarrelI JHL, Murray-Rust P. Mapping the atomic environment of functional groups: turning 3D scatter plots into pseudo-density contours. J Mol Graph. 1984;2:43–46. [Google Scholar]
- 14.Barillari C, Marcou G, Rognan D. Hot-spots-guided receptor-based pharmacophores (HS-Pharm): a knowledge-based approach to identify ligand anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J Chem Inf Model. 2008;48:1396–1410. doi: 10.1021/ci800064z. [DOI] [PubMed] [Google Scholar]
- 15.Landon MR, Lancia DR, Jr, Yu J, Thiel SC, Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J Med Chem. 2007;50:1231–1240. doi: 10.1021/jm061134b. [DOI] [PubMed] [Google Scholar]
- 16.Leach AR. Ligand docking to proteins with discrete side-chain flexibility. J Mol Biol. 1994;235:345–356. doi: 10.1016/s0022-2836(05)80038-5. [DOI] [PubMed] [Google Scholar]
- 17.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heaslet H, Rosenfeld R, Giffin M, Lin YC, Tam K, et al. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Crystallogr Sect D Biol Crystallogr. 2007;63:866–875. doi: 10.1107/S0907444907029125. [DOI] [PubMed] [Google Scholar]
- 19.Tripathi A, Nile AH, Bankaitis VA. Sec14-like phosphatidylinositol-transfer proteins and diversification of phosphoinositide signalling outcomes. Biochem Soci Trans. 2014;42:1383–1388. doi: 10.1042/BST20140187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat Chem. 2014;6:575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed MH, Amadasi A, Bayden AS, Cashman DJ, Cozzini P, et al. Understanding Water and Its Many Roles in Biological Structure: Ways to Exploit a Resource for Drug Discovery. Methods in Pharmacology and Toxicology. Computer-Aided Drug Discovery. 2016:85–110. [Google Scholar]
- 22.Rarey M, Kramer B, Lengauer T. The particle concept: placing discrete water molecules during protein-ligand docking predictions. Proteins. 1999;34:17–28. [PubMed] [Google Scholar]
- 23.Cozzini P, Fornabaio M, Marabotti A, Abraham DJ, Kellogg GE, et al. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J Med Chem. 2002;45:2469–2483. doi: 10.1021/jm0200299. [DOI] [PubMed] [Google Scholar]
- 24.Fornabaio M, Spyrakis F, Mozzarelli A, Cozzini P, Abraham DJ, et al. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 3. The free energy contribution of structural water molecules in HIV-1 protease complexes. J Med Chem. 2004;47:4507–4516. doi: 10.1021/jm030596b. [DOI] [PubMed] [Google Scholar]
- 25.Verdonk ML, Chessari G, Cole JC, Hartshorn MJ, Murray CW, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48:6504–6515. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 26.Tripathi A, Fornabaio M, Spyrakis F, Mozzarelli A, Cozzini P, et al. Complexity in modeling and understanding protonation states: computational titration of HIV-1-protease-inhibitor complexes. Chem Biodivers. 2007;4:2564–2577. doi: 10.1002/cbdv.200790210. [DOI] [PubMed] [Google Scholar]
- 27.Antosiewicz J, Mc Cammon JA, Gilson MK. Prediction of pH-dependent properties of Proteins. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 28.Park MS, Gao C, Stern HA. Estimating binding affinities by docking/scoring methods using variable protonation states. Proteins. 2011;79:304–314. doi: 10.1002/prot.22883. [DOI] [PubMed] [Google Scholar]
- 29.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 30.Fornabaio M, Cozzini P, Mozzarelli A, Abraham DJ, Kellogg GE. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 2. Computational titration and pH effects in molecular models of neuraminidase-inhibitor complexes. J Med Chem. 2003;46:4487–4500. doi: 10.1021/jm0302593. [DOI] [PubMed] [Google Scholar]
- 31.Kellogg GE, Fornabaio M, Chen DL, Abraham DJ, Spyrakis F, et al. Tools for building a comprehensive modeling system for virtual screening under real biological conditions: The Computational Titration algorithm. J Mol Graph Model. 2006;24:434–439. doi: 10.1016/j.jmgm.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Krishtalik LI, Kuznetsov AM, Mertz EL. Electrostatics of proteins: description in terms of two dielectric constants simultaneously. Proteins. 1997;28:174–182. [PubMed] [Google Scholar]
- 33.Demchuck E, Wade RC. Improving the continuum dielectric approach to calculating pKa’s of ionizable groups in proteins. J Phys Chem. 1996;100:17373–17387. [Google Scholar]
- 34.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, et al. Current status of the AMOEBA polarizable force field. J Phys Chem B. 2010;114:2549–2564. doi: 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 36.Finkelstein AV, Janin J. The price of lost freedom: entropy of bimolecular complex formation. Protein Eng. 1989;3:1–3. doi: 10.1093/protein/3.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Murray CW, Verdonk ML. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J Comput Aided Mol Des. 2002;16:741–753. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
- 38.Salaniwal S, Manas ES, Alvarez JC, Unwalla RJ. Critical evaluation of methods to incorporate entropy loss upon binding in high-throughput docking. Proteins. 2007;66:422–435. doi: 10.1002/prot.21180. [DOI] [PubMed] [Google Scholar]
- 39.Ruvinsky AM, Kozintsev AV. New and fast statistical-thermodynamic method for computation of protein-ligand binding entropy substantially improves docking accuracy. J Comput Chem. 2005;26:1089–1095. doi: 10.1002/jcc.20246. [DOI] [PubMed] [Google Scholar]
- 40.Kellogg GE, Burnett JC, Abraham DJ. Very Empirical Treatment of Solvation and Entropy: a Force Field Derived From Log Po/w. J Comput Aided Mol Des. 2001;15:381–393. doi: 10.1023/a:1011136228678. [DOI] [PubMed] [Google Scholar]
- 41.Kellogg GE, Joshi JS, Abraham DJ. New tools for modeling and understanding hydrophobicity and hydrophobic interactions. Med Chem Res. 1992;1:444–453. [Google Scholar]
- 42.Ruvinsky AM. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11scoring functions. J Comput Chem. 2007;28:1364–1372. doi: 10.1002/jcc.20580. [DOI] [PubMed] [Google Scholar]
- 43.Kongsted J, Ryde U. An improved method to predict the entropy term with the MM/PBSA approach. J Comput Aided Mol Des. 2009;23:63–71. doi: 10.1007/s10822-008-9238-z. [DOI] [PubMed] [Google Scholar]
- 44.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes andchallenges. Curr Pharm Des. 2005;11:323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 45.Blaney J, Dixon J. A good ligand is hard to find: automated docking methods. Perspect Drug Discov Des. 1993;1:301–319. [Google Scholar]
- 46.Taylor RD, Jewsbury PJ, Essex JW. A review of protein-small molecule docking methods. J Comput Aided Mol Des. 2002;16:151–166. doi: 10.1023/a:1020155510718. [DOI] [PubMed] [Google Scholar]
- 47.Muegge I, Rarey I. Small molecule docking and scoring. In: Lipkowitz KB, Boyd DB, editors. Reviews Comput Chem. John Wiley and Sons, Inc; New York: 2001. pp. 1–61. [Google Scholar]
- 48.Oshiro CM, Kuntz ID, Dixon JS. Flexible ligand docking using a genetic algorithm. J Comput Aided Mol Des. 1995;9:113–130. doi: 10.1007/BF00124402. [DOI] [PubMed] [Google Scholar]
- 49.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 50.Taylor RD, Jewsbury PJ, Essex JW. FDS: flexible ligand and receptor docking with a continuum solvent model and soft-core energy function. J Comput Chem. 2003;24:1637–1656. doi: 10.1002/jcc.10295. [DOI] [PubMed] [Google Scholar]
- 51.Jain AN. Surflex: fully automated flexible molecular docking using a molecular similarity-based search engine. J Med Chem. 2003;46:499–511. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 52.Sauton N, Lagorce D, Villoutreix BO, Miteva MA. MS-DOCK: accurate multiple conformation generator and rigid docking protocol for multi-step virtual ligand screening. BMC Bioinformatics. 2008;9:184. doi: 10.1186/1471-2105-9-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng EC, Gschwend DA, Blaney JM, Kuntz ID. Orientational sampling and rigid-body minimization in molecular docking. Proteins. 1993;17:266–278. doi: 10.1002/prot.340170305. [DOI] [PubMed] [Google Scholar]
- 54.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 55.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 56.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mangoni R, Roccatano D, Di Nola A. Docking of flexible ligands to flexible receptors in solution by molecular dynamics simulation. Proteins. 1999;35:153–162. doi: 10.1002/(sici)1097-0134(19990501)35:2<153::aid-prot2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 58.Huang ZN, Wong CF, Wheeler RA. Flexible protein-flexible ligand docking with disrupted velocity simulated annealing. Proteins. 2008;71:440–454. doi: 10.1002/prot.21781. [DOI] [PubMed] [Google Scholar]
- 59.Zhao Y, Sanner MF. FLIPDock: Docking flexible ligands into flexible receptors. Proteins. 2007;68:726–737. doi: 10.1002/prot.21423. [DOI] [PubMed] [Google Scholar]
- 60.Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. doi: 10.1146/annurev.biophys.32.110601.142532. [DOI] [PubMed] [Google Scholar]
- 61.Perola E, Xu K, Kollmeyer TM, Kaufmann SH, Prendergast FG, et al. Successful virtual screening of a chemical database for farnesyl transferase inhibitor leads. J Med Chem. 2000;43:401–408. doi: 10.1021/jm990408a. [DOI] [PubMed] [Google Scholar]
- 62.Jiang F, Kim S. “Soft docking”: matching of molecular surface cubes. J Mol Biol. 1991;219:79–102. doi: 10.1016/0022-2836(91)90859-5. [DOI] [PubMed] [Google Scholar]
- 63.Kuntz I, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 64.Sandak B, Nussinov R, Wolfson HJ. An automated computer vision and robotics-based technique for 3-D flexible biomolecular docking and matching. Comput Appl Biosci. 1995;11:87–99. doi: 10.1093/bioinformatics/11.1.87. [DOI] [PubMed] [Google Scholar]
- 65.DesJarlais R, Sheridan RP, Dixon JS, Kuntz ID, Venkataraghavan R. Docking flexible ligands to macromolecular receptors by molecular shape. J Med Chem. 1986;29:2149–2153. doi: 10.1021/jm00161a004. [DOI] [PubMed] [Google Scholar]
- 66.Sandak B, Nussinov R, Wolfson HJ. A method for biomolecular structural recognition and docking allowing conformational flexibility. J Comput Biol. 1998;5:631–654. doi: 10.1089/cmb.1998.5.631. [DOI] [PubMed] [Google Scholar]
- 67.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller A, Teller E. Equation of State Calculations by Fast Computing Machines. J Chem Phys. 1953;21:1087. [Google Scholar]
- 68.Hart T, Read RJ. A multiple-start Monte Carlo docking method. Proteins. 1992;13:206–222. doi: 10.1002/prot.340130304. [DOI] [PubMed] [Google Scholar]
- 69.Clark D, Westhead DR. Evolutionary algorithms in computer-aided molecular design. Comput Aided Mol Des. 1996;10:337–358. doi: 10.1007/BF00124503. [DOI] [PubMed] [Google Scholar]
- 70.Judson R. Genetic Algorithms and their use in Chemistry. In: Lipkowitz KB, Boyd DB, editors. Reviews in Computational Chemistry. John Wiley & Sons, Inc; New York: 1997. pp. 1–73. [Google Scholar]
- 71.Morris G, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 72.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 73.Vieth M, Hirst J, Dominy B, Daigler H, Brooks C. Assessing search strategies for flexible docking. J Comput Chem. 1998;19:1623–1631. [Google Scholar]
- 74.Wu G, Robertson DH, Brooks CL, 3rd, Vieth M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm based MD docking algorithm. J Comput Chem. 2003;24:1549–1562. doi: 10.1002/jcc.10306. [DOI] [PubMed] [Google Scholar]
- 75.Baxter C, Murray CW, Clark DE, Westhead DR, Eldridge MD. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins. 1998;33:367–382. [PubMed] [Google Scholar]
- 76.Price M, Jorgensen W. Analysis of Binding Affinities for Celecoxib Analogues with COX-1 and COX-2 from Combined Docking and Monte Carlo Simulations and Insight into the COX-2/COX-1 Selectivity. J Am Chem Soc. 2000;122:9455–9466. [Google Scholar]
- 77.Hoffmann D, Kramer B, Washio T, Steinmetzer T, Rarey M, et al. Two-stage method for protein-ligand docking. J Med Chem. 1999;42:4422–4433. doi: 10.1021/jm991090p. [DOI] [PubMed] [Google Scholar]
- 78.Bohm H, Stahl M. The use of scoring functions in drug discovery applications. In: Lipkowitz KB, Boyd DB, editors. Reviews Comput Chem Wiley-VCH. John Wiley and Sons, Inc; New York: 2002. pp. 41–87. [Google Scholar]
- 79.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 80.Schulz-Gasch T, Stahl M. Scoring functions for protein–ligand interactions: A critical perspective. Drug Discov Today Technol. 2004;1:231–239. doi: 10.1016/j.ddtec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 81.Kaminski G, Jorgensen WL. Performance of the AMBER94, MMFF94, and OPLS-AA Force Fields for Modeling Organic Liquids. J Phys Chem. 1996;100:18010–18013. [Google Scholar]
- 82.Weiner S, Kollman PA, Nguyen DT, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;252:230–252. doi: 10.1002/jcc.540070216. [DOI] [PubMed] [Google Scholar]
- 83.CERTARA. The SYBYL software. Certara; New Jersey, USA: 1995. [Google Scholar]
- 84.Halgren T. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 85.Dinur U, Hagler A. New Approaches to Empirical Force Fields. In: Lipkowitz KB, Boyd DB, editors. Reviews in Computational Chemistry. John Wiley & Sons, Inc; New York: 1991. pp. 99–164. [Google Scholar]
- 86.Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, et al. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- 87.Naim M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, et al. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model. 2007;47:122–133. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]
- 88.Zou X, Yaxiong S, Kuntz ID. Inclusion of Solvation in Ligand Binding Free Energy Calculations Using the Generalized-Born Model. J Am Chem Soc. 1999;121:8033–8043. [Google Scholar]
- 89.Verdonk M, Chessari G, Cole JC, Hartshorn MJ, Murray CW, et al. Modeling water molecules in protein-ligand docking using GOLD. J Med Chem. 2005;48:6504–6515. doi: 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- 90.Koppensteiner W, Sippl MJ. Knowledge based potentials-back to the roots. Biochemistry (Mosc) 1998;63:247–252. [PubMed] [Google Scholar]
- 91.Muegge I. PMF scoring revisited. J Med Chem. 2006;49:5895–5902. doi: 10.1021/jm050038s. [DOI] [PubMed] [Google Scholar]
- 92.Clark R, Strizhev A, Leonard JM, Blake JF, Matthew JB. Consensus scoring for ligand/protein interactions. J Mol Graph Model. 2002;20:281–295. doi: 10.1016/s1093-3263(01)00125-5. [DOI] [PubMed] [Google Scholar]
- 93.Sousa SF, Ribeiro AJM, Coimbra JTS, Neves RPP, Martins SA, et al. Protein-Ligand Docking in the New Millennium – A Retrospective of 10Years in the Field. Curr Med Chem. 2013;20:2296–2314. doi: 10.2174/0929867311320180002. [DOI] [PubMed] [Google Scholar]
- 94.Khamis MA, Gomaa W, Ahmed WF. Machine Learning in Computational Docking. Artif Intell Med. 2015;63:135–152. doi: 10.1016/j.artmed.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 95.Duch W, Swaminathan K, Meller J. Artificial Intelligence Approaches for Rational Drug Design and Discovery. Curr Phar Des. 2007;13:1497–1508. doi: 10.2174/138161207780765954. [DOI] [PubMed] [Google Scholar]
- 96.Dong X, Zheng W. A New Structure-Based QSAR Methods Affords both Descriptive and Predictive Models for Phosphodiesterase-4 Inhibitors. Curr Chem Genom. 2008;2:29–39. doi: 10.2174/1875397300802010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sato T, Honma T, Yokoyama S. Combining Machine Learning and Pharmacophore-Based Interaction Fingerprints for in Silico Screening. J Chem Inf Model. 2010;50:170–185. doi: 10.1021/ci900382e. [DOI] [PubMed] [Google Scholar]
- 98.Takkis K, Garcia-Sosa AT, Sild S. Virtual Screening for HIV Protease Inhibitors Using a Novel Database Filtering Procedure. Mol Inf. 2015;34:485–492. doi: 10.1002/minf.201400170. [DOI] [PubMed] [Google Scholar]
- 99.Ballester PJ, Mitchell JBO. A Machine Learning Approach to Predicting Protein-Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics. 2010;26:1169–1175. doi: 10.1093/bioinformatics/btq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zsoldos Z, Reid D, Simon A, Sadjad BS, Johnson AP. eHiTS: An Innovative Approach to the Docking and Scoring Function Problem. Curr Prot Pep Sci. 2006;7:421–435. doi: 10.2174/138920306778559412. [DOI] [PubMed] [Google Scholar]
- 101.Zsoldos Z, Reid D, Simon A, Sadjad BS, Johnson AP. eHiTS: A New Fast, Exhaustive Flexible Ligand Docking System. J Mol Graph Model. 2007;26:198–212. doi: 10.1016/j.jmgm.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 102.Da C, Kireev D. Structural Protein-Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. J Chem Inf Model. 2014;54:2555–2561. doi: 10.1021/ci500319f. [DOI] [PMC free article] [PubMed] [Google Scholar]