

A 62-year-old previously healthy man was diagnosed with a deep vein thrombosis, a suspicious nevus, and macrocytic anemia. Otherwise, his hematogram and coagulation indices were normal. His vitamin B12 level was 52 pg/mL (reference 211–911 pg/mL), iron level was 58 mcg/dL (reference 59–158 mcg/dL), and folate level was normal. His nevus pathology revealed an in situ melanoma. Assuming his anemia and hypercoagulable state were related to newfound melanoma, and given his lack of neurologic complaints, his physician regarded the vitamin B12 level as incidental and asymptomatic. He went home on anticoagulation and iron supplementation.
He returned 1 month later, still anemic, with unrelated small-bowel obstruction. He underwent emergent lysis of adhesions without bowel resection, receiving 30 minutes of inhaled nitrous oxide (N2O) during anesthesia. Postoperatively, he recovered physically, but developed dysgusia and anorexia, and was discharged home on hospital day 5.
Two months later, he again returned, now unable to walk. Shortly after surgery, he developed numbness and burning in his feet, which quickly ascended to his knees. The patient had progressive difficulties with balance and falls, requiring a walker, then becoming nonambulatory. His daughter also observed progressive confusion, requiring assistance with all activities of daily living.
His general medical and cranial nerve examinations were unremarkable. Mental status examination was notable for bradyphrenia, disorientation to time, and mild naming difficulties. His strength was decreased in distal arms and throughout his legs to 4/5. Reflexes were absent throughout and there was no Babinski sign. He demonstrated patchy loss of pinprick and temperature sensation throughout, with vibratory and joint position sense absent to his elbows and hips in respective limbs. He had pseudoathetosis and sensory ataxia in both arms and trunk. His hemoglobin was 8 g/dL, and mean corpuscular volume was 106 femtoliters. Vitamin B12 was undetectable, homocysteine was 221 μmol/L (reference ⩽15 μmol/L), and methylmalonic acid (MMA) was 107 nmol/mL (reference ⩽0.4 nmol/mL). Intrinsic factor antibodies were present, but not parietal cell antibodies. The remainder of his neuropathy laboratory evaluation was normal, including studies for diabetes, thyroid, syphilis, and paraproteinemia. MRI of the spinal cord demonstrated abnormal signal within the posterior column from C2 to C7 and from T9 to the conus medullaris (figure 1).
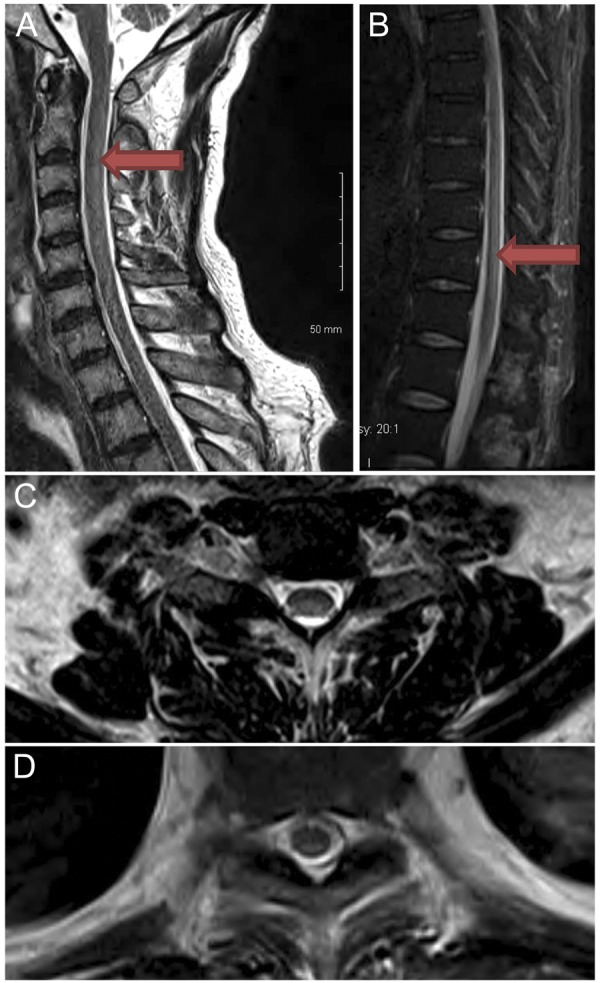
Abnormal signal in the cervical and thoracic spine
Figure 1. T2-weighted sagittal image of cervical (A) and thoracic (B) spine demonstrates subtle hyperintensity posteriorly, C2–C7, and T9 through conus. Axial T2 image at C5–C6 (C) and T9 (D) demonstrates hyperintensity dorsally.
He received aggressive parenteral B12 repletion and underwent inpatient rehabilitation, demonstrating significant improvement. At 4-month follow-up, he had a normal mental status examination, near normal strength with minimal weakness in hip and knee flexors bilaterally, and greatly improved large- and small-fiber sensory modalities. He ambulated unassisted and was independent in all activities of daily living. Vitamin B12 level was >2,000 pg/mL, homocysteine was 11 μmol/L, and MMA was 0.22 nmol/mL. Repeat MRI of the spine demonstrated resolution of the abnormalities seen before.
DISCUSSION
Vitamin B12 (cobalamin) is an essential cofactor in 2 major biochemical pathways (figure 2), contributing to carbohydrate and lipid synthesis and to myelin sheath maintenance.1 Even marginally low levels of vitamin B12 may cause neurologic symptoms with elevated MMA and homocysteine, whose measurements provide a more sensitive gauge for functional deficiency than vitamin B12 alone.1
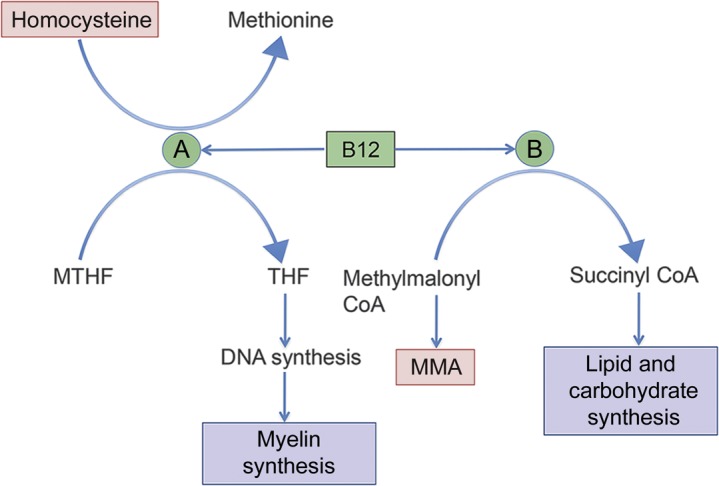
Biochemical reactions requiring vitamin B12 as a cofactor
Figure 2. Methionine synthetase (A) transmethylates methyl-tetrahydrofolate (MTHF) and homocysteine to methionine and tetrahydrofolate (THF), important for DNA and myelin synthesis. Methylmalonyl CoA-mutase (B) converts methylmalonyl CoA to succinyl CoA, which enters the Krebs cycle to contribute to lipid and carbohydrate synthesis. Items in red are increased with vitamin B12 deficiency.
The consequences of cobalamin deficiency are various, and some presentations are more common than others. Neurologically, decreased activity of methionine synthetase functionally impairs myelin,2 causing neuropathy, dorsal column dysfunction, and cognitive decline. Hematologically, impaired DNA synthesis causes anemia, and methyltetrahydrofolate accumulation within immature red blood cells causes macrocytosis. Prothrombotic consequences of cobalamin deficiency stem from elevated levels of homocysteine,3 which promotes platelet aggregation, free radical–induced endothelial damage, and vessel lumen narrowing.4
N2O accelerates the development of cobalamin deficiency symptoms by irreversibly inactivating cobalamin, and by decreasing methionine synthetase activity directly.5 Previous reports suggest that the symptoms of early subacute combined degeneration following N2O exposure may be reversible with repletion of vitamin B12.5,6 Recovery to independent ambulation has been reported in as few as 4 weeks with cobalamin therapy, with near-complete symptom resolution thereafter,5 and with imaging improvement on MRI.7
This case highlights anemia and DVT as an uncommon pair of symptoms from cobalamin deficiency, the latter due to secondary hyperhomocysteinemia. This symptom pair, just as with more classical presentations, should prompt evaluation of vitamin B12 levels. Prior to N2O administration, however, even isolated anemia should trigger cobalamin evaluation due to potential morbidity. Similarly in cases of unprovoked DVT, assessment of homocysteine levels may be important, especially if concurrent anemia or neuropathy exists, or if there are risk factors for B12 deficiency. Finally, this case demonstrates the potential reversibility of neurologic symptoms after N2O exposure with early, high-dose, parenteral cobalamin repletion.
STUDY FUNDING
No targeted funding reported.
DISCLOSURES
J.T. Jordan and J. Weiser report no disclosures. P.C. Van Ness serves on the Editorial Board of Archives of Neurology and receives research support from UCB, NeuroPace, and Eisai. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
Correspondence to: jtjordan@partners.org
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
Footnotes
Correspondence to: jtjordan@partners.org
REFERENCES
- 1.Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45:1435–1440. doi: 10.1212/wnl.45.8.1435. [DOI] [PubMed] [Google Scholar]
- 2.Metz J. Cobalamin deficiency and the pathogenesis of nervous system disease. Annu Rev Nutr. 1992;12:59–79. doi: 10.1146/annurev.nu.12.070192.000423. [DOI] [PubMed] [Google Scholar]
- 3.Remacha AF, Souto JC, Ramila E, Perea G, Sarda MP, Fontcuberta J. Enhanced risk of thrombotic disease in patients with acquired vitamin B12 and/or folate deficiency: role of hyperhomocysteinemia. Ann Hematol. 2002;81:616–621. doi: 10.1007/s00277-002-0560-6. [DOI] [PubMed] [Google Scholar]
- 4.Eldibany MM, Caprini JA. Hyperhomocysteinemia and thrombosis: an overview. Arch Pathol Lab Med. 2007;131:872–884. doi: 10.5858/2007-131-872-HATAO. [DOI] [PubMed] [Google Scholar]
- 5.Flippo TS, Holder WD. Neurologic degeneration associated with nitrous oxide anesthesia in patients with vitamin B12 deficiency. Arch Surg. 1993;128:1391–1395. doi: 10.1001/archsurg.1993.01420240099018. [DOI] [PubMed] [Google Scholar]
- 6.Schilling RF. Is nitrous oxide a dangerous anesthetic for vitamin B12-deficient subjects? JAMA. 1986;255:1605–1606. [PubMed] [Google Scholar]
- 7.Singer MA, Lazaridis C, Nations SP, Wolfe GI. Reversible nitrous oxide-induced myeloneuropathy with pernicious anemia: case report and literature review. Muscle Nerve. 2008;37:125–129. doi: 10.1002/mus.20840. [DOI] [PubMed] [Google Scholar]