

Ataxia telangiectasia (AT) is a rare autosomal-recessive disorder caused by mutations in the AT-mutated (ATM) gene (chromosome 11q 22-23) with absent or aberrant ATM protein kinase.1 It is usually characterized by childhood-onset cerebellar ataxia, oculocutaneous telangiectasia, oculomotor apraxia, immunodeficiency, increased malignancy risk, and early mortality (classical AT).1 Recently it has become apparent that a spectrum of milder adult-onset phenotypes exist (variant AT), associated with residual expression of functional ATM protein.2,3 These patients demonstrate a predominance of extrapyramidal features, later age at ataxia onset, slower progression, and an extended lifespan. We report the case of a woman first diagnosed with AT at the unusually late age of 60.
Case report
A 60-year-old woman was referred to the Sheffield, UK, Ataxia Clinic with progressive gait deterioration over a period of 8 years. The patient had a vague childhood history of choreoathetosis but remained mobile and stable without considerable gait difficulties until age 52 years. She retained the ability to walk with walking aids. On examination, she had signs of cerebellar dysfunction including gaze-evoked nystagmus, limb ataxia, dysarthria, and intention tremor. In addition, she had some evidence of facial but no ocular telangiectasia (figure 1). There was no evidence of oculomotor apraxia. She did not have frequent infections and there was no history of cancer in her or her family. There was no family history of any neurologic problem. MRI demonstrated cerebellar atrophy (figure 2). Neurophysiology showed a predominantly sensory neuropathy. The α-fetoprotein levels, which are raised in classical AT, were within the upper limit of normal on repeated testing, 15 KU/L (range 0–15 KU/L). She had slightly reduced level of immunoglobulins, with immunoglobulin G being 5.24 g/L (normal 6–16), immunoglobulin M 0.31 g/L (normal 0.5–2), and normal immunoglobulin A. Genetic testing for Friedreich and spinocerebellar ataxias types 1, 2, 3, 6, and 7 were negative. Screening for celiac disease and vitamin E levels was normal. In view of the possible early onset of ataxia, the α-fetoprotein being at the upper limit of normal, low immunoglobulins, and the presence of facial telangiectasia, DNA was sent for further genetic analysis. This revealed compound heterozygosity for hypomorphic ATM mutations producing residual levels of normal (splice mutation c.1066-6 T>G [IV510-6 T>G]) and mutant ATM protein with kinase activity (missense mutation c.7271 T>G p.Val2424Gly), confirming a diagnosis of AT. Serum levels of aprataxin and senataxin protein were within normal parameters.
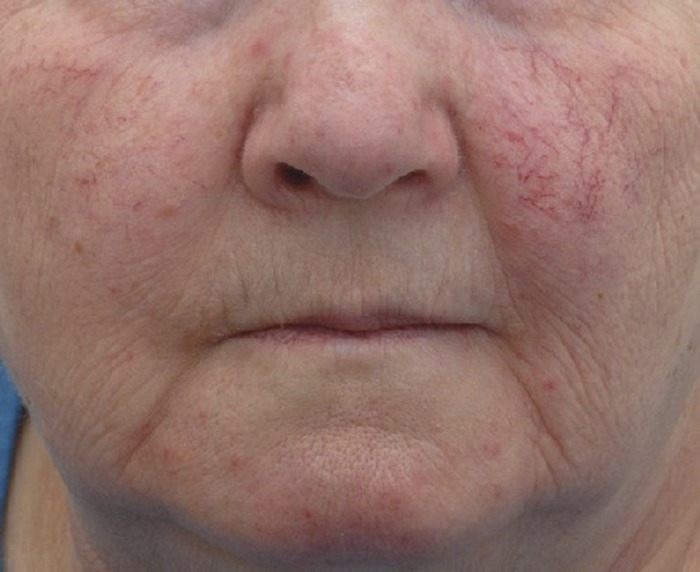
Facial telangiectasia
Figure 1. Evidence of facial but no ocular telangiectasia in the patient. This served as an etiologic clue along with reduced immunoglobulins and an α-fetoprotein level at the upper limit of normal.

Brain MRI
Figure 2. Brain coronal T2 (A) and sagittal T1 (B) MRI of the patient demonstrates cerebellar atrophy. This is a nonspecific finding seen in a number of progressive cerebellar disorders.
DISCUSSION
This patient is an example of the late age at onset at which variant AT may present, having developed a cerebellar syndrome in the fifth decade. A median onset of ataxia at 27 years, with age at death of 37 years, in patients possessing residual ATM kinase activity have previously been reported.2,3 Examples of survival past the fifth decade exist in rare case studies.4
Recent studies demonstrate the strong correlation between ATM kinase activity and phenotypic severity across the AT disease spectrum.3 Patients with classical AT, possessing biallelic null ATM mutations, present in early infancy with a progressive and debilitating cerebellar ataxia. Wheelchair use is near invariable by late childhood. Oculomotor apraxia, oculocutaneous telangiectasia, immunodeficiency, recurrent pulmonary infection, endocrinopathy, and acute radiation sensitivity manifest early in the disease course.1 Life expectancy is reduced to the mid-20s due to respiratory failure and malignancy (primarily hematologic).4
In variant AT, the presence of hypomorphic ATM mutations (missense or leaky-splice) attenuates the clinical phenotype by conferring residual levels of functional ATM protein kinase.1 These heterogeneous patients are characterized by one or more of the following: later age at clinical onset, slower progression, and an extended lifespan compared with classical AT. Extrapyramidal movement disorders rather than cerebellar signs dominate the presentation and often predate ataxia, which may or may not emerge in a slowly progressive form in adulthood.3 Oculocutaneous telangiectasia and other characteristic features are typically mild or absent.2 MRI cerebellar morphology is often normal. The α-fetoprotein, while raised, may still be within normal ranges. Importantly, risk of malignancy as an adult—while lower than in classical AT—remains elevated compared with the healthy population.5 Solid tumors predominate, and occur later than in classical AT. The burden of malignancy in heterozygous carriers of ATM mutations, comprising 0.5%–1% of the UK population, may also be elevated.6
The current case highlights the difficulty the diagnosis of variant AT represents, in which presentation is mild, of marked late onset, and initially noncerebellar. Indeed, ataxia and telangiectasia may be absent.2 Early diagnostic genetic testing in suspicious cases is often required, and should be considered in view of the high risk of malignancy.
STUDY FUNDING
No targeted funding reported.
DISCLOSURES
The authors report no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
Correspondence to: m.hadjivassiliou@sheffield.ac.uk
Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
Footnotes
Correspondence to: m.hadjivassiliou@sheffield.ac.uk
REFERENCES
- 1.Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair. 2004;3:1187–1196. doi: 10.1016/j.dnarep.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 2.Verhagen MM, Abdo WF, Willemsen MA. Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology. 2009;73:430–437. doi: 10.1212/WNL.0b013e3181af33bd. [DOI] [PubMed] [Google Scholar]
- 3.Verhagen MM, Last JI, Hogervorst FB. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia-telangiectasia: a genotype-phenotype study. Hum Mutat. 2012;33:561–571. doi: 10.1002/humu.22016. [DOI] [PubMed] [Google Scholar]
- 4.Dork T, Bendix-Waltes R, Wegner RD, Stumm M. Slow progression of ataxia-telangiectasia with double missense and in frame splice mutations. Am J Med Genet A. 2004;126A:272–277. doi: 10.1002/ajmg.a.20601. [DOI] [PubMed] [Google Scholar]
- 5.Micol R, Ben Slama L, Suarez F. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J Allergy Clin Immunol. 2011;128:382–389. doi: 10.1016/j.jaci.2011.03.052. [DOI] [PubMed] [Google Scholar]
- 6.Thompson D, Duedal S, Kirner J. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]