

Abstract
Three new modified peptides named grassystatins D–F (1–3) were discovered from a marine cyanobacterium from Guam. Their structures were elucidated using NMR spectroscopy and mass spectrometry (MS). The hallmark structural feature in the peptides is a statine unit which contributes to their aspartic protease inhibitory activity preferentially targeting cathepsins D and E. Grassystatin F (3) was the most potent analogue with IC50 values of 50 and 0.5 nM against cathepsins D and E, respectively. The acidic tumor microenvironment is known to increase the activation of some of the lysosomal proteases associated with tumor metastasis such as cathepsins. Because cathepsin D is a biomarker in aggressive forms of breast cancer and linked to poor prognosis, the effects of cathepsin D inhibition by 1 and 3 on the downstream cellular substrates, cystatin C and PAI-1, were investigated. Furthermore, the functional relevance of targeting cathepsin D substrates was evaluated by examining the effect of 1 and 3 on the migration of MDA-MD-231 cells. Grassystatin F (3) inhibited the cleavage of cystatin C and PAI-1, the activities of their downstream targets cysteine cathepsins and tPA, as well as the migration of the highly aggressive triple negative breast cancer cells, phenocopying the effect of siRNA mediated knockdown of cathepsin D.
Graphical Abstract
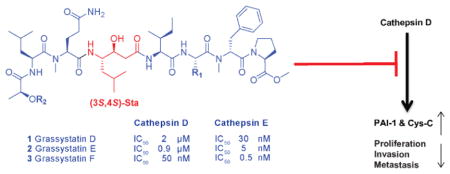
Proteases are involved in the regulation of many physiological processes (e.g. blood coagulation, immune function, cell proliferation, and tissue remodeling) essential to life. Their overexpression and dysregulated activity are linked to many diseases, including chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), neurodegenerative disorders such as Alzheimer’s disease, and cancer.1,2 Due to their implication in the pathogenesis of several diseases, inhibiting proteases is an attractive treatment strategy. Several protease inhibitors have reached the market,2 such as the metalloproteinase inhibitors targeting angiotensin converting enzyme (ACE) for the management of hypertension (e.g., captopril), and the aspartic protease inhibitors targeting HIV protease for the management of AIDS (e.g., ritonavir).2
Proteases have been shown to contribute to cancer progression where the extracellular pH of the tumor microenvironment is often acidic, due to hypoxia and other factors, which plays an important role not only in the expression of some genes but also in the activation of some lysosomal enzymes such as cathepsins with acidic optimal pH for their activity.3 Cathepsin D, a lysosomal aspartic protease, is considered a biomarker in aggressive forms of breast cancer. Its high expression and secretion have been found to correlate with breast cancer tumor aggressiveness, metastasis, and subsequently linked with poor prognosis.4–11 Therefore, novel cancer therapeutics targeting cathepsin D may reduce the metastatic potential and improve the survival rates of breast cancer patients.
We have been exploring marine cyanobacteria which produce modified peptides that have a propensity to inhibit proteases with different selectivity profiles.12–17 Among the wide range of the available cyanobacterial protease inhibitors,12–17 a class of compounds was discovered containing a characteristic statine (γ-amino-β-hydroxy acid) as a pharmacophore for binding and inhibiting aspartic proteases, which was first reported in pepstatin A (4).18,19 Pepstatin A (4) is a natural aspartic protease inhibitor produced by Actinomycetes, which had inspired the design and synthesis of aliskiren, the first orally available renin inhibitor that gained FDA approval in 2007 for the management of hypertension.20–22 Grassystatins A–C, Leu-derived statine-containing compounds isolated from Lyngbya cf. confervoides, are potent cathepsin E inhibitors, which have been shown to reduce antigen presentation by dendritic cells.23 Other cyanobacterial compounds including tasiamides B)24,25 and F,26 which bear a Phe-derived statine core, were isolated from the marine cyanobacteria Symploca sp. and Lyngbya sp., respectively. The tasiamides have been shown to inhibit cathepsins D and E in addition to beta-site amyloid precursor protein cleaving enzyme 1 (BACE1),26 an enzyme involved in the pathogenesis of Alzheimer’s disease. These cyanobacterial secondary metabolites can provide a starting point for the development of therapeutic protease inhibitors, through the design and synthesis of analogues with improved potency and selectivity profiles,27–29 with potential applications in cancer and Alzheimer’s disease. The biological activity of the statine-containing class of cyanobacterial compounds was mostly evaluated in the context of BACE1 inhibition,27 thus their anticancer activities have not been fully investigated.
Our efforts exploring the marine cyanobacteria of Guam, Mariana Islands, have led to the discovery of three related new aspartic protease inhibitors that may have value in the context of cancer. Herein we describe the isolation, structure elucidation, and biological evaluation of the cathepsin D and E inhibitors (1–3, Figure 1) as potential antimetastatic agents targeting breast cancer.
Figure 1.

Grassystatins D–F (1–3) isolated from the marine cyanobacterium VPG 14–61. The differences in structures 2 and 3 compared to 1 are highlighted.
RESULTS AND DISCUSSION
Samples of VPG 14–61 were collected from 7–10 m depth on the reef at Cetti Bay, Guam, and subsequently freeze-dried and extracted with EtOAc:MeOH (1:1). The non-polar extract (1.96 g) was subjected to successive partitioning between solvents of different polarities (EtOAc, BuOH, and H2O). The EtOAc fraction was subjected to silica gel chromatography using a gradient system starting with 30% EtOAc:Hex and ending with 100% MeOH to afford five fractions. The fraction eluting with 1:1 EtOAc:MeOH was further purified by reversed-phase HPLC resulting in the isolation of three compounds named grassystatins D–F (1–3, Figure 1) due to their relatedness to grassystatins A–C (Figure 2).
Figure 2.

Pepstatin A (4) with binding site nomenclature and structurally related cyanobacterial compounds. The structures are color coded to highlight the similarities and differences relative to grassystatins D–F (1–3).
The structures of 1–3 were elucidated using 1D and 2D NMR experiments (1H NMR, 13C NMR, COSY, TOCSY, HSQC, HMBC, and ROESY). The HRESIMS spectrum of 1 in the positive mode exhibited an adduct ion peak at m/z of 981.5626 [M + Na]+ suggesting a molecular formula of C48H78N8O12. 1H and 13C NMR (Table 1) spectra revealed signals indicative of modified peptides: α-protons (δH~ 4–5.5 ppm), carbonyls (δC~ 168–174 ppm), exchangeable protons characteristic of amides (δH~ 7–8 ppm). Also, one O-methyl (δH 3.63 ppm) and two N-methyl (δH 2.91 and 2.92 ppm) signals were apparent. Analysis of 2D NMR data revealed the presence of the following units: O-Me-Pro, N-Me-Phe, Ala, Ile, Sta (4-amino-3-hydroxy-6-methylheptanoic acid), N-Me-Gln, Leu, and lactic acid. The sequence of those units was determined based on correlations observed in both HMBC and ROESY spectra. The HRESIMS data of 2 in the positive mode exhibited an adduct ion peak at m/z of 967.5489 [M + Na]+ suggesting a molecular formula of C47H76N8O12 indicating a CH2 unit less compared to 1. Compound 2 exhibited similarity to 1 in terms of 1H and 13C NMR chemical shifts (Table 2). Further examination of COSY, TOCSY, HSQC, HMBC, and ROESY data revealed the presence of the same units present in 1, except for Gly in place of Ala. The HRESIMS data of 3 in the positive mode exhibited an adduct ion peak at m/z of 1156.6622 [M + Na]+ suggesting a molecular formula of C59H91N9O13. The 1H NMR spectrum and edited HSQC spectrum revealed chemical shifts very similar to 1 (Table 3). The major differences were the presence of an N,N-dimethyl unit (δH/C 2.29/41.22), the deshielded shift in the α-proton of the lactic acid moiety (from δH 3.97 to 4.98), and an additional aromatic group. The combination of COSY and HMBC data revealed the presence of an additional unit: N,N-diMe-Phe, which is connected to the lactic acid unit via an ester linkage. This explains the deshielded shift of the α-proton of the lactic acid moiety (δH 4.98).
Table 1.
NMR Spectroscopic Data for Grassystatin D (1) at 600 MHz (1H), 150 MHz (13C) in DMSO-d6
| C/H no. | δC, typea | δH (J in Hz) | 1H-1H COSY | HMBCb | ROESY | |
|---|---|---|---|---|---|---|
| Pro | 1 | 172.5, C | ||||
| 2 | 59.2, CH | 4.29, dd (8.0, 7.0) | H-3a, H-3b | 3, 4, 5, 7 | H-3a, H-3b, H-4 | |
| 3a | 28.9, CH2 | 2.20, m | H-2, H-3b | 1, 2, 4, 5 | H-2, H-3b, H-4 | |
| 3b | 1.67, m | H-2, H-3a | 1, 2, 4, 5 | H-3a | ||
| 4 | 25.2, CH2 | 1.75, m (2H) | H-3a, H-4, H-5a, H-5b | 2 | H-3b, H-5b | |
| 5a | 46.8, CH2 | 3.35, m | H-4, H-5b | 2, 3, 4 | H-4, H-5b, H-8 | |
| 5b | 3.21, m | H-4, H-5a | 3, 4 | H-4, H-5a, H-8 | ||
| O-Me | 6 | 52.0, CH3 | 3.63, s | 1 | H-3b | |
| N-Me-Phe | 7 | 168.4, C | ||||
| 8 | 55.5, CH | 5.53, dd (10.0, 5.7) | H-9a, H-9b | 7, 9, 10, 16, 17 | H-9a, H-9b, H-5b, H-11/15 | |
| 9a | 34.4, CH2 | 2.99, dd (−14.0, 5.7) | H-8, H-9b | 7, 8, 10, 11/15 | H-8, H-9b | |
| 9b | 2.82, dd (−14.0, 10.0) | H-8, H-9a | 7, 8, 10, 11/15 | H-8, H-9a, H-11/15 | ||
| 10 | 137.9, C | |||||
| 11/15 | 129.9, CH | 7.19, m | H-12/14 | 9, 10, 12/14, 13 | H-8, H-9a, H-9b, H-16, H-19 | |
| 12/14 | 128.2, CH | 7.20, m | H-11/15 | 10, 12/14, 11/15 | H-8, H-9a, H-9b, H-16, H-19 | |
| 13 | 126.4, CH | 7.14, m | 10, 12/14 | |||
| 16 | 30.1, CH3 | 2.91, s | 8, 17 | H-5b, H-8, H-9b, H-11/15, H-18, H-19 | ||
| Ala | 17 | 172.3, C | ||||
| 18 | 44.6, CH | 4.53, qd (7.0, 7.1) | H3-19, NH | 17, 19, 20 | H3-16, H3-19, NH (Ala) | |
| 19 | 16.6, CH3 | 0.69, d (7.0) | H-18 | 17, 18 | H3-16, H-18, NH (Ala) | |
| NH | 8.10, d (7.0) | H-18, H3-19c | 18, 20 | H-18, H3-19, H-21, NH (Ile) | ||
| Ile | 20 | 170.92, C | ||||
| 21 | 56.5, CH | 4.17, dd (9.5, 6.8) | H-22, NH | 20, 22, 23, 24 | H-22, H3-23, H-24, H3-25, NH (Ala), NH (Ile) | |
| 22 | 37.4, CH | 1.61, m | H-21, H3-23, H-24b | 20, 21, 23, 25 | H-21, H-24a, H3-23, NH (Ala), NH (Ile) | |
| 23 | 15.4, CH3 | 0.76, m | H-22 | 21, 22, 24 | H-22, H-24a, H-24b, H-29 | |
| 24a | 24.4, CH2 | 1.36, m | H-24b, H3-25 | 21, 22, 23, 25 | H-22 | |
| 24b | 1.01, m | H-22, H-24a, H3-25 | 21, 22, 23,25 | H-22, NH (Ile) | ||
| 25 | 11.5, CH3 | 0.77, m | H-24a, H-24b | 22, 24 | H-5b, H-21, H-22, NH (Ile), NH (Ala) | |
| NH | 7.74, d (9.5) | H-21 | 20, 21, 22, 26 | H-21, H-22, H-24b, H-27a, H-27b, NH (Ala) | ||
| Sta | 26 | 170.96, C | ||||
| 27a | 40.3, CH2 | 2.17, dd (−14, 9.0) | H-28 | 26, 28, 29 | H-28, NH (Ile) | |
| 27b | 2.10, dd (−14, 3.7) | H-28 | 26, 28, 29 | H-28, H-29, NH (Sta), NH (Ile) | ||
| 28 | 69.7, CH | 3.75, m | H-27a, H-27b, OH | 26, 27, 29 | H-27a, H-27b, H-30b, H-31, OH (Sta), NH (Sta), NH (Ile) | |
| 29 | 51.1, CH | 3.80, m | H-30a, H-30b, NH | 28, 30, 34 | H-27a, H-27b, H-30a, H-30b, H3-33, NH (Sta), NH (Ile) | |
| 30a | 39.8, CH2 | 1.33, m | H-29, H-30b, H-31 | 28, 29 | NH (Sta) | |
| 30b | 1.17, m | H-29, H-30a | 28, 29, 31, 32, 33 | H-28, H-29, H-30a | ||
| 31 | 24.5, CH | 1.43, m | H-30b, H-32, H-33 | 30, 32, 33 | H-28, NH (Sta) | |
| 32 | 23.7, CH3 | 0.83, d (6.6) | H-31 | 30, 31, 32 | H-29, H-30a, H-30b, H-31 | |
| 33 | 21.9, CH3 | 0.79, d (6.6) | H-31 | 30, 31, 33 | H-29, H-30b, H-31 | |
| OH | 4.80, br | H-28 | 27, 28, 29 | H-28 | ||
| NH | 7.12, m | H-29 | 29, 30, 34 | H-29, H-30a, H-31, H-35 | ||
| N-Me-Gln | 34 | 169.7, C | ||||
| 35 | 55.8, CH | 4.87, dd (9.3, 5.9) | H-36a, H-36b | 34, 36, 37, 39, 40 | H-36a, H-36b, NH (Sta) | |
| 36a | 24.1, CH2 | 1.95, m | H-35, H-36b | 34, 35, 37, 38 | H-36b | |
| 36b | 1.79, m | H-35, H-36a | 34, 35, 37, 38 | |||
| 37 | 31.9, CH2 | 1.94, m (2H) | H-37 | 36 | H-35 | |
| 38 | 173.8, C | |||||
| 39 | 30.5, CH3 | 2.92, s | 35, 40 | H-30a, H-31, H3-45, H-35, H-36, H-41, H-42a, NH (Sta), NH (Leu) | ||
| NH2 | ||||||
| Leu | 40 | 172.8, C | ||||
| 41 | 47.0, CH | 4.74, m | H-42a, H-42b, NH | 40, 42, 43, 46 | H-39, H-42a, H-42b, H-43, H3-45, NH (Leu) | |
| 42a | 41.5, CH2 | 1.45, m | H-41, H-42b | 40, 41, 43, 44, 45 | H-42b, NH (Leu) | |
| 42b | 1.33, m | H-41, H-42a | 41, 43, 44, 45 | H-41, H-42a | ||
| 43 | 24.5, CH | 1.57, m | H-42a, H-42b. H3-44, H3-45 | 41, 42, 44, 45 | H-41, H3-44, H3-45,NH (Leu) | |
| 44 | 23.7, CH3 | 0.87, d (6.6) | H-43 | 42, 43, 44 | H-41, H-42a, H-42b, H-43 | |
| 45 | 21.9, CH3 | 0.90, d (6.6) | H-43 | 42, 43, 44 | H-41, H-42a, H-42b, H-43 | |
| NH | 7.64, d (8.5) | H-41 | 40, 41, 42, 46 | H-41, H-42a, H-43, H-47 | ||
| Lactic acid | 46 | 174.4, C | ||||
| 47 | 67.4, CH | 3.96, q (6.6) | H3-48 | 46, 48 | H3-45, NH (Leu) | |
| 48 | 21.5, CH3 | 1.20, d (6.6) | H-47 | 46, 47 | H-47, NH (Leu) | |
| OH | 5.61, br |
Multiplicity derived from APT and HSQC spectra.
Protons showing long-range correlation with indicated carbon.
These couplings were only observed in the TOCSY spectrum.
Table 2.
NMR Spectroscopic Data for Grassystatin E (2) at 600 MHz (1H), 150 MHz (13C) in DMSO-d6
| C/H no. | δCa, type | δH (J in Hz) | 1H-1H COSY | HMBCb | ROESY | |
|---|---|---|---|---|---|---|
| Pro | 1 | 172.5, C | ||||
| 2 | 58.9, CH | 4.25, dd (8.0, 7.0) | H-3a, H-3b | 1, 3 | H-3a | |
| 3a | 28.6, CH2 | 2.14, m | H-2, H-3b | 1, 2, 4, 5 | H-2, H-3b, H-4 | |
| 3b | 1.72, m | H-2, H-3a | 1, 2, 4, 5 | H-3a | ||
| 4 | 25.2, CH2 | 1.78, m (2H) | H-5b | 3, 5 | H-3a, H-5b | |
| 5a | 46.7, CH2 | 3.31, m | H-4, H-5b | 2, 3, 4 | ||
| 5b | 3.23, m | H-4, H-5a | 3, 4 | |||
| O-Me | 6 | 52.1, CH3 | 3.61, s | 1 | ||
| N-Me-Phe | 7 | 168.1, C | ||||
| 8 | 56.1, CH | 5.41, t (7.6) | H-9a, H-9b | 7, 9, 10, 16 | H-9a, H-9b, H-5a, H-5b | |
| 9a | 34.5, CH2 | 3.12, dd (−14.0, 7.6) | H-8, H-9b | 7, 8, 10, 11/15 | H-8, H-9b | |
| 9b | 2.67, dd (−14.0, 7.6) | H-8, H-9a | 7, 8, 10, 11/15 | H-8, H-9a | ||
| 10 | 138.2, C | |||||
| 11/15 | 129.7, CH | 7.19, m | H-9a, H-9b, H-12/14 | 10, 11/15, 13 | H-8, H-9a, H-9b | |
| 12/14 | 128.3, CH | 7.22, m | H-11/15, H-13 | 10, 12/14 | H-8, H-9a, H-9b | |
| 13 | 126.5, CH | 7.14, m | H-12/14 | 11/15 | ||
| 16 | 29.8, CH3 | 2.91, s | 8, 17 | H-8, H-9a, H-9b, H-18a, H-18b | ||
| Gly | 17 | 168.5, C | ||||
| 18a | 40.3, CH | 4.04, dd (−17.1, 5.4) | H-18b, NH | 17, 19 | H-18b, NH (Ala) | |
| 18b | 3.73, dd (−17.1, 5.1) | H-18a, NH | 17, 19 | H-18a, NH (Ala) | ||
| NH | 8.04, m | H-18a, H-18b | H-18a, H-18b, H-20 | |||
| Ile | 19 | 171.5, C | ||||
| 20 | 56.8, CH | 4.21, dd (9.0, 7.0) | H-21, NH | 19, 21, 22, 23 | H-21, H3-24, NH (Gly), NH (Ile) | |
| 21 | 36.9, CH | 1.70, m | H-20, H3-22 | H-20, H-23a, H3-24,NH (Ile) | ||
| 22 | 15.6, CH3 | 0.82, m | H-21 | 20, 21, 23 | H-20, H-21, H-23b | |
| 23a | 24.5, CH2 | 1.41, m | H3-24 | 24 | H-23b | |
| 23b | 1.07, m | H-21, H-23a, H3-24 | 21, 22, 24 | H-23a | ||
| 24 | 11.7, CH3 | 0.79, m | H-23a, H-23b | 21, 23 | H-23a, H-27 | |
| NH | 7.85, d (9.0) | H-20 | 25 | H-26a | ||
| Sta | 25 | 171.26, C | ||||
| 26a | 40.4, CH2 | 2.20, dd (−14, 9.2) | H-26b, H-27 | 25, 27, 28 | H-27, NH (Ile) | |
| 26b | 2.16, dd (−14, 3.8) | H-26a, H-27 | 25, 27 | H-27, NH (Ile) | ||
| 27 | 69.4, CH | 3.78, m | H-26a, H-26b | H-26b, H-29a, H-29b, H3-31 | ||
| 28 | 51.1, CH | 3.80, m | H-29a, H-29b, NH | H-26b, H-29a, H-29b, H3-31, | ||
| 29a | 39.9, CH2 | 1.36, m | H-28, H-29b | 28 | H-29b | |
| 29b | 1.18, m | H-28, H-29a | 28, 30, 31, 32 | H-27, H-29a | ||
| 30 | 24.5, CH | 1.45, m | H-29b, H3-31 | 31, 32 | ||
| 31 | 22.0, CH3 | 0.79, d (6.7) | 29, 30, 32 | H-27 | ||
| 32 | 23.8, CH3 | 0.83, d (6.7) | H-30 | 29, 30, 31 | ||
| OH | 4.85, br | |||||
| NH | 7.16, m | H-28 | 33 | OH | ||
| N-Me-Gln | 33 | 169.8, C | ||||
| 34 | 55.6, CH | 4.90, dd (9.4, 5.9) | H-35a, H-35b | 33, 35, 36, 38, 39 | H-36a, NH (Sta) | |
| 35a | 24.0, CH2 | 1.97, m | H-34, H-35b | 34, 36, 37 | H-4 | |
| 35b | 1.82, m | H-34, H-35a | 33, 34, 36, 37 | H-34, H-35a | ||
| 36 | 31.9, CH2 | 1.95, m (2H) | 34, 35, 37 | H-34 | ||
| 37 | 173.7, C | |||||
| 38 | 30.3, CH3 | 2.91, s | 34, 39 | H-36, H-40, H-41a, H-41b, H3-44 | ||
| NH2 | ||||||
| Leu | 39 | 172.7, C | ||||
| 40 | 46.8, CH | 4.75, m | H-41a, H-41b, NH | 39, 41, 42, 45 | H3-38, H-41a, H-41b, H3-44, NH (Leu) | |
| 41a | 41.5, CH2 | 1.45, m | H-40, H-41b | 39, 40, 42, 43, 44 | H-40, H-41b, NH (Leu) | |
| 41b | 1.33, m | H-40, H-41a | 39, 40, 42, 43, 44 | H-41a | ||
| 42 | 24.5, CH | 1.57, m | H-41b, H3-44 | H3-44 | ||
| 43 | 21.9, CH3 | 0.90, d (6.6) | H-42 | 41, 42, 44 | H-40, H-41b, H-42 | |
| 44 | 23.7, CH3 | 0.86, d (6.6) | H-42 | 41, 42, 43 | H-40, H-41a, H-41b, H-42 | |
| NH | 7.66, d (8.4) | H-40 | 45 | H-40, H-41a, H-46, NH (Ile) | ||
| Lactic acid | 45 | 174.3, C | ||||
| 46 | 67.2, CH | 3.97, q (6.6) | H-47 | 45, 47 | H3-47, NH (Leu) | |
| 47 | 21.4, CH3 | 1.21, d (6.6) | H-46 | 45, 46 | H-46, NH (Leu) | |
| OH | 5.4, br |
Multiplicity derived from HSQC spectra.
Protons showing long-range correlation with indicated carbon.
These couplings were only observed in the TOCSY spectrum.
Table 3.
NMR Spectroscopic Data for Grassystatin F (3) at 600 MHz (1H), 150 MHz (13C) in DMSO-d6
| C/H no. | δCa, type | δH (J in Hz) | 1H-1H COSY | HMBCb | ROESY | |
|---|---|---|---|---|---|---|
| Pro | 1 | 172.5, C | ||||
| 2 | 59.2, CH | 4.29, t (7.8) | H-3a, H-3b | 1, 3, 4 | H-3a, H-3b, H-4 | |
| 3a | 28.5, CH2 | 2.21, m | H-2, H-3b, H-4 | 1, 4, 5 | H-2, H-3b, H-4 | |
| 3b | 1.67, m | H-2, H-3a | 1, 2, 4 | H-3a | ||
| 4 | 25.1, CH2 | 1.75, m (2H) | H-5a, H-5b | H-3a, H-5b | ||
| 5a | 46.5, CH2 | 3.36, m | H-4, H-5b | 2, 3, 4 | H-5b, H-8 | |
| 5b | 3.22, m | H-4, H-5a | 3, 4 | H-4 | ||
| O-Me | 6 | 51.8, CH3 | 3.63, s | 1 | ||
| N-Me-Phe | 7 | 168.3, C | ||||
| 8 | 55.1, CH | 5.54, dd (10.1, 5.6) | H-9a, H-9b | 7, 9, 10, 16, 17 | H-5a, H-9a, H-9b | |
| 9a | 34.1, CH2 | 2.99, dd (−14.0, 5.6) | H-8, H-9b | 7, 8, 10, 11/15 | H-9, H-9b | |
| 9b | 2.82, dd (−14.0, 10.0) | H-8, H-9a | 7, 8, 10, 11/15 | H-9a | ||
| 10 | 137.8, C | |||||
| 11/15 | 129.5, CH | 7.21, m | H-12/14 | 9, 10, 12/14, 13, 11/15 | ||
| 12/14 | 127.9, CH | 7.20, m | H-11/15, H-13 | 10, 12/14, 13, 11/15 | H-8, H-9a, H-9b, H3-16, H3-19 | |
| 13 | 126.4, CH | 7.16, m | H-12/14 | 11/15 | ||
| 16 | 29.7, CH3 | 2.91, s | 8, 17 | H-8, H-12/14, H3-19 | ||
| Ala | 17 | 172.3, C | ||||
| 18 | 44.3, CH | 4.53, qd (7.0, 7.1) | H3-19, NH | 17, 19, 20 | H3-16, H3-19, NH (Ala) | |
| 19 | 16.3, CH3 | 0.71, d (7.0) | H-18 | 17, 18 | H-18, NH (Ala) | |
| NH | 8.11, d (7.1) | H-18, H3-19c | 18, 19, 20 | H-18, H3-19, H-21, NH (Ile) | ||
| Ile | 20 | 170.90, C | ||||
| 21 | 56.1, CH | 4.18, dd (9.4, 7.0) | H-22, NH | 20, 22, 23, 24 | H-22, H3-23, H-24a, H-24b,NH (Ile), NH (Ala) | |
| 22 | 37.1, CH | 1.62, m | H-21, H3-23 | H-21, H3-23, H-24a, H-24b, NH (Ile), NH (Ala) | ||
| 23 | 15.1, CH3 | 0.76, m | H-22 | 21, 22, 24 | H-22, H-24a, H-24b | |
| 24a | 24.1, CH2 | 1.36, m | H-24b, H3-25 | H-24b | ||
| 24b | 1.01, m | H-22, H-24a, H3-25 | 22, 23, 25 | H-24a | ||
| 25 | 11.3, CH3 | 0.77, m | H-24a, H-24b | 22, 24 | H-21, NH (Ile), NH (Ala) | |
| NH | 7.76, d (9.4) | H-21 | 21, 26 | H-21, H-22, H-24b, H-27a, H-27b, NH (Ala) | ||
| Sta | 26 | 170.97, C | ||||
| 27a | 40.1, CH2 | 2.17, dd (−14, 9.0) | H-27b, H-28 | 26, 28, 29 | H-28, H-29, NH (Ile) | |
| 27b | 2.11, dd (−14, 3.7) | H-27a, H-28 | 26, 28 | H-28, H-29, H-36b, NH (Ile) | ||
| 28 | 69.4, CH | 3.75, m | H-27a, H-27b | H-27a, H-27b, H-30b, H-43, NH (Ile) | ||
| 29 | 50.7, CH | 3.80, m | H-30a, H-30b, NH | H-27a, H-27b, H-30b, H3-45, NH (Sta), NH (Ile) | ||
| 30a | 39.6, CH2 | 1.35, m | H-29, H-30b, H-31 | 29, 31, 32, 33 | H-30b | |
| 30b | 1.18, m | H-29, H-30a | 29, 31, 32, 33 | H-28, H-30a | ||
| 31 | 24.1, CH | 1.65, m | H-30a, H3-32, H3-33 | H3-32, H3-33, | ||
| 32 | 23.5, CH3 | 0.87, d (6.2) | H-31 | 30, 31, 33 | H-30a, H-31, H-41 | |
| 33 | 21.5, CH3 | 0.86, d (6.2) | H-31 | 30, 31, 32 | H-42a | |
| OH | 4.74, br | |||||
| NH | 7.07, d (9.0) | H-29 | 34 | H-35 | ||
| N-Me-Gln | 34 | 169.8, C | ||||
| 35 | 55.5, CH | 4.88, dd (9.5, 6.0) | H-36a, H-36b | 34, 36, 37, 39, 40 | H-36a, H-36b, H-37, NH (Sta) | |
| 36a | 24.0, CH2 | 1.96, m | H-35, H-36b | 37, 38 | H-36b | |
| 36b | 1.80, m | H-35, H-36a | H-36a | |||
| 37 | 31.2, CH2 | 1.94, m (2H) | H-36a, H-36b | 35, 36, 38 | H-35 | |
| 38 | 173.8, C | |||||
| 39 | 30.3, CH3 | 2.90, s | 35, 40 | H-36b, H-37, H-41, H-42b | ||
| NH2 | ||||||
| Leu | 40 | 172.6, C | ||||
| 41 | 47.3, CH | 4.70, m | H-42a, H-42b, NH | 40, 42 | H3-33, H3-39, H-42b, NH (Leu) | |
| 42a | 40.2, CH2 | 1.48, m | H-41, H-42b | 40, 41, 43, 44, 45 | H-42b | |
| 42b | 1.33, m | H-41, H-42a | 43, 44, 45 | H-42a | ||
| 43 | 24.3, CH | 1.44, m | H3-44, H3-45 | H3-44 | ||
| 44 | 23.4, CH3 | 0.83, d (6.6) | H-43 | 42, 43, 45 | H-30b, H-42b, H-43 | |
| 45 | 21.7, CH3 | 0.79, d (6.6) | H-43 | 42, 43, 44 | H-29, H-30b, H-43 | |
| NH | 8.24, d (8.0) | H-41 | 41, 46 | H-41, H-42a, H-47 | ||
| Lactic acid | 46 | 169.9, C | ||||
| 47 | 69.3, CH | 4.98,q (6.7) | H-48 | 48, 49 | H3-48, NH (Leu) | |
| 48 | 17.7, CH3 | 1.31,d (6.7) | H-47 | 46, 47 | H-47, NH (Leu) | |
| N,N-diMe-Phe | 49 | 170.2, C | ||||
| 50 | 67.9, CH | 3.49, t (7.6) | H-51a, H-51b | 49, 51, 52, 58/59 | H-51a, H-51b, H3-58/59 | |
| 51a | 34.8, CH2 | 2.95, dd (−14.0, 6.7) | H-50, H-51b | 49, 50, 52, 53/57 | H-50, H3-58/59 | |
| 51b | 2.85, dd (−14.0, 6.7) | H-50, H-51a | 49, 50, 52, 53/57 | H-50, H3-58/59 | ||
| 52 | 138.7, C | |||||
| 53/57 | 129.1, CH | 7.23, m | 51, 55, 53/57 | H-50, H-51a, H-51b | ||
| 54/56 | 128.2, CH | 7.25, m | H-55 | 52, 54/56 | ||
| 55 | 126.2, CH | 7.18, m | H-54/56 | 53/57 | H-37 | |
| 58/59 | 41.2, CH3 | 2.29, s | 49, 50, 58/59 | H3-48, H-50, H-51a, H-51b |
Multiplicity derived from HSQC spectra.
Protons showing long-range correlation with indicated carbon.
These couplings were only observed in the TOCSY spectrum.
The key structural feature in these compounds (1–3) is the statine (γ-amino-β-hydroxy acid) unit which is present in pepstatin A (4), a potent aspartic protease inhibitor isolated from Actinomycetes.18,19 Compared with other structurally related cyanobacterial secondary metabolites (Figure 2), compounds 1–3 are hybrids between the grassystatins A–C23 and the tasiamides.24–26, 30,31 Compounds 1–3 share the Leu-statine unit of grassystatins A–C23 and have closely related residues flanking the statine moiety to those of the tasiamides.24–26, 30,31 The structure of 2 resembles the structure of grassystatin C in terms of the amino acid units and sequence; however, it differs in the last residue where lactic acid replaces the 2-hydroxy-3-methylpentanoic acid (Hmpa) unit present in grassystatin C. Tasiamide E30 and tasiamide31 cyanobacterial compounds that lack the statine core, were found to exhibit structural similarity to grassystatins D–F (1–3). Compared to tasiamide E, 2 has a similar backbone with an additional Leu-derived statine unit being incorporated between the Ile and N-Me-Gln residues in addition to the presence of lactic acid that replaces the last unit in tasiamide E. On the other hand, tasiamides B24,25 and F26, other tasiamide analogues bearing the Phe-derived statine core [4-amino-3-hydroxy-5-phenylpentanoic acid, Ahppa], appear to confer structural similarity to the grassystatins D and E (1 and 2) backbone with minor differences. The major difference is the replacement of the Phe-derived statine in tasiamides B and F with the Leu-derived statine core. Minor modifications were apparent at the P3′, P2′, and P3 positions (Figure 2).
To assign the absolute configurations of the stereocenters present in grassystatins D–F (1–3), portions of 1–3 (100 μg) were hydrolyzed using 6 N HCl (110 ºC, 24 h) and subjected to enantioselective HPLC-MS analysis. By comparison with authentic standards, the analysis revealed retention times corresponding to L-Pro, N-Me-D-Phe, L-Ala, L-Ile, N-Me-L-Glu, L-Leu, N,N-diMe-L-Phe, and L-lactic acid. The configuration of lactic acid as L was further confirmed by chiral-phase HPLC analysis under different conditions using CuSO4 and by co-injecting the hydrolyzate with L- and D-lactic acid standards. To establish the configuration of the statine unit, a portion of the acid hydrolyzate was derivatized with L-FDLA and subjected to modified Marfey’s analysis. Two peaks corresponding to (3S,4S)-Sta-L-FDLA and (3R,4S)-Sta-L-FDLA were noted, due to epimerization at C-3 resulting from potential dehydration/rehydration, thus revealing the S configuration at C-29 in grassystatins D and F (1 and 3) and C-28 in grassystatin E (2). The relative configuration of the statine unit was assigned based on the coupling constants of the α-methylene signals (H2-27 in grassystatins D and F (1 and 3) and H2-26 in grassystatin E (2)) to the adjacent hydroxy methine (H-28 in 1 and 3; H-27 in 2).32 The deshielded α-methylene signal (H-27a in 1 and 3) shows a large coupling constant to H-28 (J = 9.0 Hz), whereas, the shielded α-methylene signal (H-27b in 1 and 3) shows a small coupling constant to H-28 (J = 3.7 Hz), suggesting that the configuration of the statine core is 3S,4S. Similar coupling constants were also observed in grassystatin E (2) (H-26a; δH 2.20, J = 9.2 Hz) and (H-26b; δH 2.16, J = 3.8 Hz), further supporting the 3S,4S configuration. This 3S,4S configuration is the same as the configuration of the statine unit in grassystatins A–C.23
The similarity of 1–3 to the other statine-containing cyanobacterial compounds (Figure 2) suggests that these compounds might act as protease inhibitors. In particular, the presence of the Leu-derived statine core as in grassystatins A–C23 suggests that these compounds might exhibit aspartic protease inhibitory activity but not BACE1 inhibitory activity as shown in the Phe-derived statine containing tasiamides B24,25 and F.26 To evaluate the antiproteolytic activity of grassystatins D–F (1–3) against aspartic proteases, compounds 1–3 were tested in vitro against cathepsins D and E, which are known to be sensitive to grassystatins A–C.23 All three compounds significantly inhibited both enzymes with grassystatin F (3) being the most potent analogue (Figure 3); however, they were more potent and more selective towards cathepsin E inhibition (Table 4). Pepstatin A (4), on the other hand, did not discriminate between cathepsins D and E, but rather was equipotent against both (Table 4). . Compared to other Leu-derived statine containing analogues, grassystatins D and E (1 and 2) were less potent than grassystatins A and B in inhibiting both enzymes; however, they exhibited comparable potency to grassystatin C in terms of cathepsin D inhibition. In addition, compounds 1 and 2 were slightly more potent than grassystatin C in inhibiting cathepsin E. Compared to tasiamides B and F, grassystatin F (3) exhibited similar cathepsin D inhibitory activity, whereas grassystatins D and E (1 and 2) were ~30-fold less potent (Table 4). Similar to grassystatins D and E (1 and 2), tasiamides B and F terminate with lactic acid; however, the major difference is the replacement of Leu-statine in grassystatins with Phe-statine in tasiamides B and F. On the other hand, grassystatin F (3) terminates with a N,N-diMe-Phe moiety. These data suggest that the Phe-statine unit is important for cathepsin D inhibitory activity and the presence of the terminal N,N-diMe-Phe moiety improves the activity dramatically.
Figure 3.

Dose response curves for grassystatins D–F (1–3) and pepstatin A (4) against A) cathepsin D and B) cathepsin E. The dose-response is presented as % fold inhibition against solvent control (DMSO) and normalized IC50 values are given.
Table 4.
IC50 Values of Grassystatins D–F (1–3), Pepstatin A (4), and Other AnaloguesAgainst Aspartic Proteases
| Cathepsin D | Cathepsin E | Selectivity ratioa | |
|---|---|---|---|
| Grassystatin D (1) | 2000 nM | 30 nM | 66.6 |
| Grassystatin E (2) | 900 nM | 5 nM | 180 |
| Grassystatin F (3) | 50 nM | 0.5 nM | 100 |
| Pepstatin A (4) | 0.35 nM | 0.3 nM | 1.1 |
| Grassystatin A | 26 nM | 0.9 nM | 28.8 |
| Grassystatin B | 7.3 nM | 0.4 nM | 18.3 |
| Grassystatin C | 1600 nM | 43 nM | 37.2 |
| Tasiamide B | 50 nM | 9 nM | 5.5 |
| Tasiamide F | 57 nM | 23 nM | 2.4 |
IC50 CatD/ IC50 CatE.
In order to examine the ability of these compounds to inhibit the cellular enzymes, lysates collected from the triple negative MDA-MB-231 breast cancer cell line, known to express cathepsins D and E, were treated with compounds 1 and 3 in the presence of the reaction buffer and cathepsin D/E fluorogenic substrate. Compounds 1 and 3 successfully inhibited cellular cathepsins D/E; however, their aspartic protease inhibitory activity appeared to be less potent (Figure 4A) compared to the experiments carried out using purified cathepsin D and E enzymes. The IC50 values of grassystatins D and F (1 and 3) were ~5.5 μM and ~5.5 nM respectively, revealing ~183-fold (grassystatin D; 1) and ~11-fold (grassystatin F; 3) reduction in the inhibitory activity against the more sensitive cathepsin E. The reduction in potency could be partly explained by the potential presence of other proteases in MDA-MB-231 lysates, which might be able to cleave the substrate but are not inhibited by the compounds or whose inhibition requires higher compound concentrations.
Figure 4.

The cellular inhibitory activities of grassystatins D and F (1 and 3) and pepstatin A (4) against cathepsin D/E proteases using cell lysates and intact live cells. A) MDA-MB-231 lysates were treated with grassystatins (1 and 3) or pepstatin A (4) in the presence of cathepsin D/E fluorogenic substrate. B) MDA-MB-231 cells were treated with pepstatin A (4), grassystatin D (1) or F (3) for 4 h, cells were lysed, and the protease inhibitory activity was analyzed. C) MDA-MB-231 cells were treated with pepstatin A (4) or grassystatin F (3) for 4 h, cells were trypsinized, collected by centrifugation then lysed, and the protease inhibitory activity was analyzed. D) MDA-MB-231 cells were seeded in 12-well plates in duplicate, after 24 h the medium was replaced with serum free medium buffered with 50 mM HEPES, pH 6.6 and incubated with either DMSO, pepstatin A (4), grassystatins D or F (1 or 3) for 3 days at 37 ºC. The activity of secreted cathepsins D/E was quantified by incubating the 3-day conditioned media (CM) in assay buffer (37 °C, pH 3.5) in the presence of fluorogenic substrate. The asterisks denote significance of P < 0.05 relative to solvent control using two-tailed unpaired t test (* denotes P ≤ 0.05, ** denotes P ≤ 0.01, *** denotes P ≤ 0.001, and **** denotes P ≤ 0.0001).
The antiproteolytic activity of compounds 1 and 3 against cathepsins D and E was also evaluated by treating intact live MDA-MB-231 cells; thus, providing an additional level of information with respect to their cellular permeability. The experiment was performed with and without a trypsinization step (before cell lysis) to assess the effect of surface bound compounds on enzyme activity. The trypsinization step involves the addition of trypsin to remove non-internalized, surface bound compound before lysing the cells to account for the activity of only the compound which has penetrated the cells.33 In MDA-MB-231 cells, grassystatin F (3) was found to be more potent compared to pepstatin A (4) with IC50 values roughly between 0.1–1 μM (without trypsinization) (Figure 4B) and between 1–10 μM (with trypsinization) (Figure 4C). It is apparent that grassystatin F (3) exhibited roughly ~10–100 (without or with trypsinization) higher IC50 values against cathepsin D and ~1000–10,000 (without or with trypsinization) IC50 against cathepsin E compared to experiments carried out using purified enzymes. Compared to experiments where lysates were used as the source of enzymes (Figure 4A), 3 was roughly 100–1000 fold (without or with trypsinization) less potent in inhibiting the cellular enzymes. On the other hand, grassystatin D (1) was found to be inactive. The differences in potency could be linked to the reduction in cellular permeability as lower potency was noted when a trypsinization step was incorporated, suggesting that the compound may have remained bound to the cell surface. The cellular studies of 1 and 3 showed that grassystatin F (3) is able to penetrate the cells slightly better than pepstatin A (4), which was previously reported to have poor permeability,34 and inhibit the cellular proteases whereas grassystatin D (1) is not. The differences between 1 and 3 in the ability to penetrate the cell membrane, and ultimately affect the potency towards the inhibition of aspartic proteases, could be attributed to the presence of the N,N-diMe-Phe moiety at its N-terminus, making it less polar compared to grassystatin D (1), which terminates with a lactic acid unit with a free hydroxy group.
Because the lysosomal aspartic protease cathepsin D is known to be secreted by breast cancer cells and is activated in acidic environments to cleave substrates involved in tumor progression, we investigated the effects of compounds 1 and 3 on the activity of secreted cathepsins D/E in MDA-MB-231 cells. The activity of secreted cathepsins D/E was assessed at pH 6.6 in 3 day conditioned media (CM) treated with grassystatins D and F (1 and 3), pepstatin A (4), or solvent control (Figure 4D) in the presence of fluorogenic substrate. The proteolytic activity of secreted cathepsins D/E was monitored by measuring the increase in the fluorescence signal from the fluorescently labeled substrate. Our data show that grassystatins D and F (1 and 3) inhibited the activities of secreted cathepsins D/E with IC50 values between 0.1–1 μM. This inhibitory activity of grassystatin F (3) is similar to that against cathepsin D/E proteases using non-trypsinized MDA-MB-231 cells (Figure 4B); however, grassystatin D (1) was found to be more potent against secreted cathepsins. Pepstatin A (4) was the most potent with IC50 values around 0.1 nM (Figure 4D). Our data suggest that compounds 1, 3, and 4 are better inhibitors of secreted cathepsin D/E proteases than cellular cathepsins D/E, which might in turn be beneficial; however, the relevance of targeting intracellular versus secreted cathepsins in the context of breast cancer remains to be investigated.
To rationalize the differences in potency between grassystatins D and F (1 and 3) with respect to their inhibitory activity against cathepsin D, molecular docking experiments were carried out using AutoDock Vina.35 The crystal structure of cathepsin D bound to pepstatin A (1LYB)36 was used as a starting point. Also, pepstatin A (4) was successfully redocked into cathepsin D (Figure S1) before attempting to dock grassystatins D and F (1 and 3). Compounds 1 and 3 were then docked into cathepsin D (1LYB) and the interactions between each compound and the residues in the enzyme’s binding pocket were examined (Figure 5). In addition to the statine core at P1-P1’, which is the pharmacophore for inhibition, residues at P2 and the N-terminal unit of the inhibitor are known to be critical for activity.23 It has been previously reported that cathepsin D favors hydrophobic residues at the P2 position compared to cathepsin E which prefers polar residues instead.37 In the structures of both 1 and 3, the P2 position has the polar N-Me-Gln residue, thus explaining in part the reduced potency towards cathepsin D inhibition compared to pepstatin A (4) which has the hydrophobic Val unit at P2. This is consistent with the docked structures of grassystatins A and C in cathepsin D,23 where the polar residues Asn in grassystatin A and N-Me-Gln in grassystatin C replace the hydrophobic Val moiety in pepstatin A (4), thus being less potent inhibitors against cathepsin D.
Figure 5.

Docked structures of A) grassystatin F (3) and B) grassystatin D (1) in cathepsin D (1LYB) using Autodock Vina.
The main difference between grassystatins D and F (1 and 3) structures is the presence of an additional N,N-diMe-Phe unit in 3 at its N-terminus. Docked structures of 1 and 3 provided some insight to the structural basis underlying the differences in potency between both compounds against cathepsin D. In grassystatin F (3), the N,N-diMe-Phe unit was found to be in close proximity to the polar residues (Tyr-10, Gln-14, Thr-125, and Lys-130) within the cathepsin D binding pocket (Figure 5A), suggesting that the N,N-diMe-Phe moiety may help in anchoring the inhibitor within the binding cleft of the enzyme. This interaction was absent in the docked structure of grassystatin D (1) (Figure 5B) as its structure terminates with lactic acid. A similar observation was reported in the docked structures of grassystatins A and C into cathepsin D.23 Grassystatin C exhibited lower potency compared to grassystatin A, mainly due to the absence of the terminal N,N-diMe-Val in grassystatin C. The docked structure of grassystatin A in cathepsin D reveals the presence of several polar residues in close proximity to the terminal N,N-diMe-Val in grassystatin A, including Tyr-10, Gln-14, Thr-125, Lys-130, Gln-258, and Gln-260.23 It has been previously reported that the basic nitrogen in N,N-diMe containing ligands, which can act as either a proton donor or acceptor, was used to improve the binding affinity of compounds interacting with Asp residue in the active site of the H1 receptor 38 compared to histamine which has a primary amine. This N,N-diMe moiety is present in several clinically effective H1 antagonists39 and was found to be crucial for improved inhibitor binding.
As cathepsin D is a biomarker of invasive breast cancer and correlates with poor prognosis,4,5,8 and in order to choose a cell-based system to further examine the effect of our cathepsin D inhibitors (1–3) in the context of targeting breast cancer, conditioned media (CM) was prepared from several breast cancer cell lines (ER+: MCF7; triple negative: MDA-MB-231 and MDA-MB-436; HER2+: BT474) and assessed for the level of secreted cathepsin D as well as cystatin C (Cys-C) and plasminogen activator inhibitor-1 (PAI-1), two cellular substrates of cathepsin D, using Western blot and ELISA (Figure 6).
Figure 6.

The secretion level of cathepsin D, PAI-1, and cystatin C in conditioned media (CM) prepared from breast cancer cells. 24 h serum free CM (pH 7.4) collected from four breast cancer cell lines (MCF7, MDA-MB-231, MDA-MB-436, and BT474) were centrifuged to remove detached cells, concentrated and analyzed by A) Western blot, B) PAI-1 ELISA, and C) Cys-C ELISA.
Cathepsin D secretion was found to be highest in the triple negative breast cancer cells MDA-MB-231 and MDA-MB-436, which are known to be more aggressive and invasive than the estrogen receptor positive MCF7 cell line. No secreted cathepsin D was detected in the HER2+ BT474 cell line. Likewise, the serine protease inhibitor, PAI-1 was secreted in MCF7 and both MDA-MB-231 and MDA-MB-436, but was undetectable in BT474 cells, with the highest levels detected in the triple negative cell lines. The cysteine cathepsin inhibitor, Cys-C, was secreted into the media of all breast cancer cells tested with the highest levels detected in MDA-MB-231 cells. The secretion level of these biomarkers measured using ELISA (Figure 6B, 6C) further supported the results obtained by Western blot (Figure 6A). The high secretion levels of PAI-1 and Cys-C could be partly due to less degradation of both substrates by cathepsin D, which may not be very active under conditions where the CM has been collected (pH 7.4) since cathepsin D activity is optimal at pH 4.5–5. Also, several studies support a role of PAI-1, independent from its protease inhibitory activity, in promoting tumorigenesis, which might explain in part the high levels detected in the aggressive breast cancer cell lines.40–43
As grassystatins A–C (1–3) are more selective towards cathepsin E inhibition, it is imperative to use a model system that highly expresses cathepsin D compared to cathepsin E. We assessed the expression levels of cathepsins D and E in MDA-MB-231 cells using RT-qPCR (Figure S2), and our data revealed ~1400 fold higher expression of cathepsin D relative to cathepsin E. Therefore, based on secretion and expression levels of cathepsin D, which correlated with the aggressiveness of breast cancer cells, and the presence of both substrates, the triple negative MDA-MB-231 cell line was selected to carry out further experiments investigating the effects of the cathepsin D inhibitors, grassystatins D and F (1 and 3) and pepstatin A (4), on cellular targets of cathepsin D: PAI-1 and Cys-C.
Cathepsin D has been shown to enhance the cleavage of Cys-C under acidic pH.44 To test this hypothesis and to investigate the effect of cathepsin D inhibitors on the cleavage of Cys-C, recombinant human Cys-C (14 kDa) was incubated with cathepsin D (enzyme to substrate ratio 1:5) at 37 °C in the presence and absence of 1 μM and 5 μM cathepsin D inhibitors (pepstatin A (4), grassystatins D (1) and F (3)) (Figure 7A). The reaction was analyzed by SDS-PAGE under reducing conditions and silver stained. Incubating cathepsin D with Cys-C resulted in the proteolysis of Cys-C into cleavage products with a size of about 4 kDa compared to incubating Cys-C alone; however, this cleavage was reduced/prevented by the addition of cathepsin D inhibitors to the reaction (Figure 7A). At 5 μM final concentration, the 4 kDa fragment had disappeared in pepstatin A (4) treated samples and the intensity of the band was reduced with grassystatin F (3) treatment, which was not the case in grassystatin D (1) treated samples. At lower concentration (1 μM), the effect of the inhibitors on the 4 kDa band was less prominent. It has been reported in previous studies that the proteolytically active 51 kDa and the mature forms (34 and 14 kDa) of human cathepsin D cleaved Cys-C to 10 and 5 fragments, respectively,44,45 where the main cleaved peptide bonds identified are Phe-Phe, Leu-Tyr, Tyr-Leu, and Phe-Tyr.46
Figure 7.
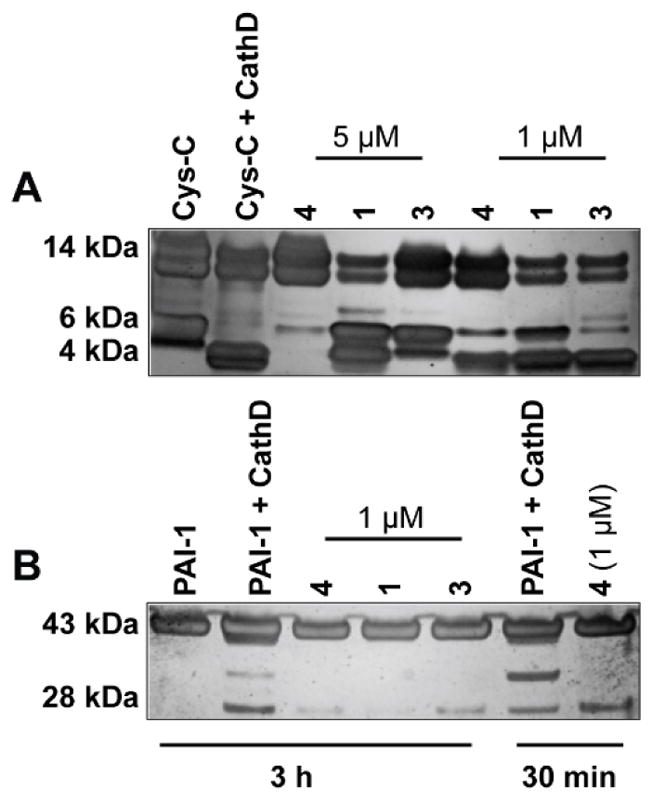
Cleavage of Cys-C and PAI-1 by cathepsin D in vitro. A) Incubation of recombinant human Cys-C with cathepsin D (enzyme to substrate ratio 1:5) in 100 mM NaCl, 100 mM sodium acetate (pH 4.0) at 37 °C for 3 h in the presence and absence of cathepsin D inhibitors (1 and 5 μM). B) Incubation of active PAI-1 with cathepsin D (enzyme to substrate ratio 1:5) in 100 mM NaCl, 100 mM sodium acetate (pH 4.0) at 37 °C for 3 h in the presence and absence of cathepsin D inhibitors (1 μM). Fragments were separated on SDS-PAGE under reducing conditions and silver stained.
The serine protease inhibitor, PAI-1, is another substrate of cathepsin D and a regulator of plasminogen activation, reported to be involved in the extracellular matrix remodeling, adhesion, migration and metastasis.40,41 Cathepsin D has been shown in previous studies to increase the proteolysis of PAI-1.47,48 To investigate the effects of our cathepsin D inhibitors on the cleavage of PAI-1, recombinant active PAI-1 (43 kDa) was incubated with cathepsin D (enzyme to substrate ratio 1:5) at 37 °C in the presence and absence of 1 μM cathepsin D inhibitors (pepstatin A (4), grassystatins D (1) and F (3)) (Figure 7B). The reaction was analyzed by SDS-PAGE under reducing conditions, followed by silver staining of the gel. Incubation of PAI-1 with cathepsin D resulted in the proteolysis of PAI-1 into several cleavage products compared to the incubation of PAI-1 alone; however, this cleavage was prevented/reduced by the addition of cathepsin D inhibitors to the reaction, supporting the previous studies that PAI-1 is indeed a substrate of cathepsin D.
To further support the in vitro data and to investigate the molecular mechanism by which cathepsin D inhibitors elicit their cellular effects, the effect on Cys-C degradation was evaluated using the MDA-MB-231 cell line. It has been previously reported that under acidic conditions secreted cathepsin D degrades the extracellular Cys-C in MCF7.44 We therefore investigated the effect of pH on the level of secreted cellular Cys-C in MDA-MB-231 cells. A reduction in the level of extracellular Cys-C was noted at pH 6.6 compared to pH 7.4 due to the fact that cathepsin D is more active at acidic pH. This is a direct result of the degradation by cathepsin D, and possibly by other unknown factors (Figure 8A). This degradation, therefore, might be blocked with cathepsin D inhibitors. In order to examine the effects of 1 and 3, the concentration of Cys-C in a 3-day serum free CM-pH 6.6 collected from MDA-MB-231 cells treated with either grassystatins D and F (1 and 3), pepstatin A (4), or DMSO was quantified by ELISA (Figure 8B). The concentration of the secreted Cys-C quantified by ELISA was found to be slightly but not significantly higher (8% increase) in 1 and 3 treated CM at the highest concentration (100 μM) compared to the control suggesting potential inhibition of Cys-C cleavage owing to the cathepsin D inhibitory activity of the compounds (1 and 3) (Figure 8B). The non-significant change noted in the concentration of Cys-C could be due to the complexity of the cellular system, compared to the in vitro experiment, with the presence of several other proteases that might compete with cathepsin D for Cys-C cleavage and in that case a higher concentration of cathepsin D is required for the cleavage. In support of that, a previous proteomic analysis revealed that Cys-C is a substrate of matrix metalloproteinase 2 (MMP2), which cleaves and subsequently inactivates Cys-C,49 thus highlighting the complexity of the proteolytic network that operates in the cellular system and tumor microenvironment.
Figure 8.

The effect of cathepsin D inhibition on the degradation and activity of extracellular Cys-C and cysteine cathepsins, respectively. MDA-MB-231 cells were seeded in 12-well plates in duplicate, after 24 h the medium was replaced with serum free medium buffered with 50 mM HEPES, pH 7.4 or pH 6.6 and incubated with either DMSO or pepstatin A (4), grassystatins D and F (1 and 3) for 3 days at 37 ºC. A, B) The concentration of Cys-C in the 3-day CM was quantified by human Cys-C ELISA. C, D) The activity of cysteine cathepsins was quantified by incubating the 3-day CM in assay buffer (37 °C, pH 5.5) in the presence of fluorogenic substrate. The asterisks denote significance of P < 0.05 relative to solvent control using two-tailed unpaired t test (* denotes P ≤ 0.05, ** denotes P ≤ 0.01, *** denotes P ≤ 0.001, and **** denotes P ≤ 0.0001).
Because Cys-C is an inhibitor of cysteine cathepsins, its cleavage by cathepsin D has been shown to increase the proteolytic activity of cysteine cathepsins44 such as cathepsins B and L which play an important role in tumor progression.50 To test this hypothesis, we investigated the proteolytic activity of cysteine cathepsins in MDA-MB-231 cells using 3-day (acidic and neutral) CM. At pH 6.6, the proteolytic activity of cysteine cathepsins was 3-fold higher than the activity measured at pH 7.4 (Figure 8C), suggesting a potential role of cathepsin D, being activated under acidic conditions, degrading Cys-C and ultimately resulting in enhancement of the proteolytic activity of cysteine cathepsins. In order to assess the effects of cathepsin D inhibitors (1 and 3) on the proteolytic activity of cysteine cathepsins, the 3-day CM collected from MDA-MB-231 cells treated with or without grassystatins D (1), F (3), and pepstatin A (4) at acidic pH (6.6) was incubated in an assay buffer (pH 5.5) in the presence of a fluorogenic substrate. The proteolytic activity was monitored by measuring the increase in fluorescence. Our data (Figure 8D) show that pepstatin A (4) was the most potent compared to the other inhibitors (1 and 3) with ~50% inhibition of cysteine cathepsin activity at 10 and 1 μM. Grassystatin F (3) was found to inhibit the activity of cysteine cathepsins at 10 μM with ~35% inhibition, whereas grassystatin D (1) was found to be inactive. E-64, a direct inhibitor of cysteine cathepsins, was used as a positive control and found to be more potent than the cathepsin D inhibitors tested with ~60% inhibition of activity. To rule out the potential effect of cell viability on the activity of cysteine cathepsins, the effects of cathepsin D (1, 3, and 4) and cysteine cathepsin (E-64) inhibitors on the viability of MDA-MB-231 cells were measured under similar conditions. There was no reduction in cell viability by the compounds tested suggesting that the inhibition of cysteine cathepsin activity is potentially due to the indirect effect of cathepsin D inhibition (in case of 1, 3, and 4) rather than reduced cell number.
To complement our in vitro PAI-1 degradation results, we aimed to investigate the effects of cathepsin D inhibitors on PAI-1 using a cell-based system. Similar to Cys-C, we investigated the effect of an acidic environment on the level of extracellular PAI-1 secreted by MDA-MB-231 cells. Our data show ~50% decrease in the concentration of secreted PAI-1 at pH 6.6 compared to pH 7.4 (Figure 9A), likely due to the degradation of PAI-1 by cathepsin D as reported in a previous study.47 Next we examined the effects of our inhibitors on the concentration of total PAI-1 (active, latent “inactive”, complexed with tissue and urokinase plasminogen activators (tPA and uPA)), in the 3-day CM collected from MDA-MB-231 cells treated with either inhibitors (1, 3, and 4) or solvent control at pH 6.6, using PAI-1 human ELISA (Figure 9B). Grassystatin F (3) resulted in ~73% increase in the concentration of secreted PAI-1 compared to the solvent control only at the highest concentration tested (100 μM), whereas grassystatin D (1) did not have a significant effect at the same concentration tested. On the other hand, pepstatin A (4) had an opposite effect compared to 1 and 3. Our data show that 4 decreased the concentration of secreted PAI-1 in a dose-dependent manner, suggesting that pepstatin A (4) and grassystatins (1 and 3) might act on PAI-1 differently where 4 might have an effect on the secretion, which remains to be investigated.
Figure 9.

The effect of cathepsin D inhibition on the degradation and activity of extracellular PAI-1 and tPA respectively. MDA-MB-231 cells were seeded in 12-well plates in duplicate, after 24 h the medium was replaced with serum free medium buffered with 50 mM HEPES, pH 7.4 or pH 6.6 and incubated with either DMSO or pepstatin A (4), grassystatins D and F (1 and 3) for 3 days at 37 ºC. A, B) The concentration of PAI-1 in the 3-day CM was quantified by PAI-1 human ELISA kit. C, D) The activity of tPA was quantified by tPA human chromogenic activity assay. The asterisks denote significance of P < 0.05 relative to solvent control using two-tailed unpaired t test (* denotes P ≤ 0.05, ** denotes P ≤ 0.01, *** denotes P ≤ 0.001, and **** denotes P ≤ 0.0001).
The proteolysis of PAI-1 by cathepsin D has been proposed and reported to enhance the proteolytic activities of secreted plasminogen activators (PA) and in particular tPA,47 potentially leading to tumor progression, invasion and metastasis. To evaluate the effects of cathepsin D and its inhibitors on activities of secreted PA, the tPA activity in the 3-day CM of MDA-MB-231 cells was first measured at two different pH levels (6.6 and 7.4), using tPA human chromogenic activity assay (Figure 9C). Also, the tPA activities were assessed at pH 6.6 in the 3-day CM treated with pepstatin A (4), grassystatins D and F (1 and 3) or solvent control (Figure 9D). Consistent with the proteolytic activities of cysteine cathepsins at acidic pH (Figure 8A), tPA activity was shown to be higher at pH 6.6 compared to pH 7.4, suggesting probably low levels of PAI-1 due to potential degradation via cathepsin D. Compared to the control, pepstatin A (4), reduced the activity of tPA with highest reduction in activity achieved at 100 and 10 μM final concentration, in which the tPA activity was found to be ~34 and 36%, respectively. Treatment with grassystatin F (3) resulted in ~30% inhibition of tPA activity whereas grassystatin D (1) was found to be inactive. Additionally, cell viability was measured in parallel under similar conditions to eliminate the potential effect of cytotoxicity on tPA activity. None of the compounds 1, 3, and 4 affected cell viability, suggesting that the inhibition of tPA activity is potentially due to their inhibition of cathepsin D. There are still discrepancies between the studies regarding the role of tPA in cancer. Unlike uPA, clinical studies in breast cancer have shown that tPA level is a marker of good prognosis.51,52 However, several other studies have shown that tPA has a role in promoting tumor progression through various mechanisms.53–57
As cathepsin D is known to correlate with tumor aggressiveness, invasion and metastasis in breast cancer,4–11 we assessed the effects of the aspartic protease inhibitors grassystatins D and F (1 and 3), and pepstatin A (4) on the migration of MDA-MB-231 cells. Pepstatin A (4) and grassystatin F (3) at 5 μM significantly inhibited the migration of the highly invasive triple negative breast cancer cells (MDA-MB-231) after 48 h by 48% and 57%, respectively, whereas grassystatin D (1) had no effect on migration (Figure 10A). The role of cathepsin D in promoting migration of MDA-MB-231 cells was validated by siRNA knockdown of cathepsin D. Our data showed that 20 nM siRNA resulted in ~50% inhibition in migration compared to the negative control (Figures 10B, S3), phenocopying the effect of grassystatin F (3) and validating that cathepsin D is the functionally relevant target.
Figure 10.

The effect of cathepsin D inhibitors and siCTSD on the migration of MDA-MB-231 cells. A) MDA-MB-231 cells were incubated for 48 h in the presence of 5 μM cathepsin D inhibitors (grassystatins D and F (1 and 3) and pepstatin A (4)) and the effect was compared to the solvent control. B) MDA-MB-231 cells were incubated in the presence of 20 nM siCTSD and the effect was compared to the negative control. The graph represents number of migrated cells in each treatment group. The asterisks denote significance of P < 0.05 relative to solvent control using two-tailed unpaired t test (* denotes P ≤ 0.05, ** denotes P ≤ 0.01, *** denotes P ≤ 0.001, and **** denotes P ≤ 0.0001).
Because cathepsin E is involved in antigen processing and induction of proinflammatory responses, we previously investigated similar Leu-derived statine containing compounds, grassystatins A–C as cathepsin E inhibitors in immune cells.23 As compounds 1–3 also had the ability to inhibit cathepsin D, we investigated their therapeutic potential for inhibiting tumor progression and metastasis in the highly invasive MDA-MB-231 breast cancer cell line. Cathepsin D is secreted as pro-cathepsin D (52 kDa), and under acidic conditions it is self-activated into cathepsin D, which then may cleave some substrates contributing to cancer progression (Figure 11).44 Several mechanisms by which cathepsin D mediates its tumorigenic/invasive effects were hypothesized and investigated (Figure 11).44,47 Cathepsin D has been shown to increase the proteolysis of plasminogen activator inhibitor-1 (PAI-1), a regulator of plasminogen activators uPA and tPA which convert plasminogen to plasmin, a serine protease that plays an important role in cancer progression through its various functions, one of which is the cleavage of the CUB domain-containing protein 1 (CDCP1).58,59 Another mechanism involves the degradation of Cys-C, a potent extracellular inhibitor of cysteine cathepsins60–61 and has been shown to inhibit growth and metastasis of tumor cells.62–63 Cysteine cathepsins such as B and L are often dysregulated in cancer cells and therefore, have been implicated in proliferation, invasion, and metastasis.60,64 Prior studies have shown that under acidic conditions secreted pro-cathepsin D activates and converts secreted pro-cathepsin B into a mature form.65 Also, among the different secreted cysteine cathepsins which are detected as proenzymes, cathepsin B was detected as a mature enzyme in the CM of MCF7 under acidic pH.44 Moreover, it has been reported that activated cathepsin B can activate and convert pro-uPA to uPA which has the ability to convert plasminogen into plasmin, ultimately resulting in the degradation of the ECM components.66 This highlights the complexity of the protease network which overall contributes to tumor aggressiveness and metastasis. Several reports have suggested the therapeutic utility of Cys-C and cysteine cathepsin inhibitors to target cancer 2,62,67 and here we show that targeting cathepsin D with potent cathepsin D/E inhibitors may potentially have therapeutic utility in reducing the migration of the highly invasive breast cancer cells. The discrepancies in the IC50 values obtained in each assay could be due to the complexity of the protease network and the proteolytic activation cascade as we were investigating the effect of our aspartic protease inhibitors on different levels of downstream cellular substrates, which might be regulated differently with the influence of various endogenous factors/inhibitors and timing.
Figure 11.
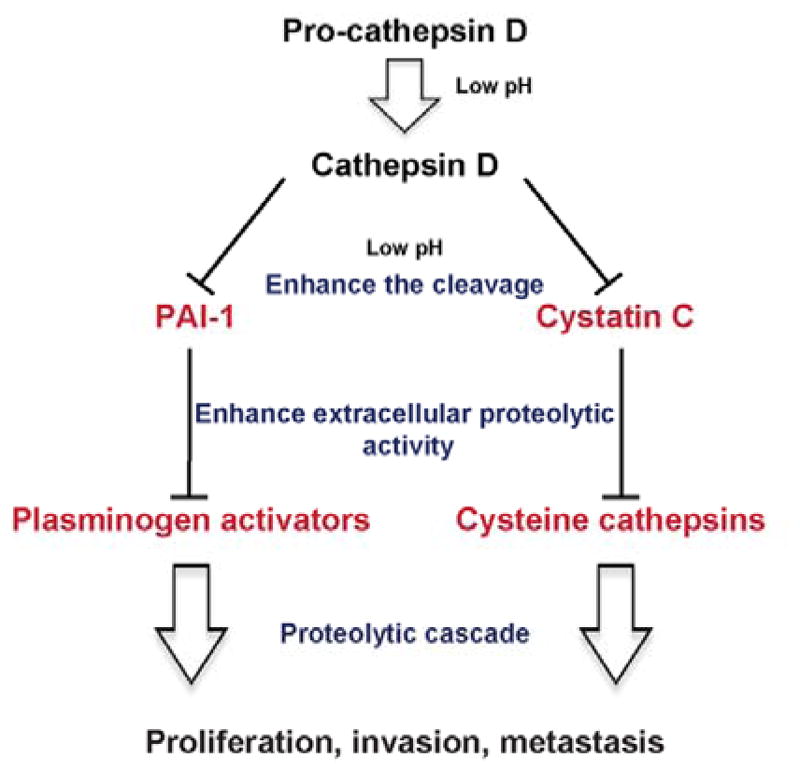
Proposed mechanisms by which cathepsin D contributes to tumor aggressiveness and metastasis via its effect on PAI-1 and Cys-C.
In conclusion, our chemical investigations of a cyanobacterium from Guam led to the discovery of grassystatins D–F (1–3) which are dual inhibitors of cathepsins D and E with preference for cathepsin E. This may represent an issue with respect to their selectivity profile that remains to be investigated; however, it was previously established that the grassystatin scaffold can be used to modulate selectivity profiles.27 Phe-statine containing compounds are much better inhibitors of cathepsin D, especially if they are derivatized with the N,N-diMe unit. Therefore, future studies would be directed towards the development of more selective compounds with enhanced potency against cathepsin D. Our data provide a proof-of-concept study of the therapeutic potential of this structural scaffold of cyanobacterial compounds, and we believe that grassystatins D–F (1–3) can serve as potentially useful probes to aid in understanding the role of cathepsin D in cancer and starting points to develop agents for combination therapy.
Experimental section
General Experimental Procedure
The optical rotation was measured using a Perkin-Elmer 341 polarimeter. The 1H, 13C and 2D NMR spectra were obtained in DMSO-d6 using Agilent VNMRS-600 MHz, 5-mm cold probe spectrometer. The spectra were referenced using the residual solvent signal [δH/C 2.5/39.5 (DMSO-d6)]. The HRESIMS data were obtained in the positive mode using Agilent LC-TOF mass spectrometer equipped with APCI/ESI multimode ion source detector. LCMS data were obtained using API 3200 (Applied Biosystems) equipped with an HPLC system (Shimadzu). Biological Material. Samples of VPG 14–61 were collected from 7–10 m depth at Cetti Bay, Guam on June 6, 2014. 16S rRNA gene sequence has been deposited in GenBank (accession #MG098886). This specimen is related to poorly resolved, polyphyletic genera of cyanobacteria described as Leptolyngbya and Phormidium (Oscillatoriales). Voucher specimens are retained at the Smithsonian Marine Station at Ft. Pierce.
Extraction and Isolation
The freeze dried sample of cyanobacterium (9.78 g) was subjected to both non-polar extraction using EtOAc–MeOH (1:1) and polar extraction using EtOH–H2O (1:1). The non-polar extract (1.96 g) was later partitioned between EtOAc and H2O. The H2O fraction was combined with the polar extract and further partitioned between BuOH and H2O. The EtOAc fraction (184.3 mg) was concentrated and subjected to silica gel chromatography using the following gradient (30% EtOAc–Hex, 100% EtOAc, 10% MeOH–EtOAc, 1:1 EtOAc–MeOH, and finally 100% MeOH). The fraction that eluted with 1:1 EtOAc–MeOH (52.7 mg) was then applied to a C18 SPE cartridge and elution initiated with H2O followed by aqueous solutions containing 50, 75, and 100% MeOH. The material eluting with 50% MeOH–H2O was then purified by semi-preparative reversed-phase HPLC [Synergi Hydro 4u-RP, 250 × 10.0 mm; flow rate, 2.0 mL/min; PDA detection 200–800 nm] using a linear MeOH–H2O gradient (60–100% MeOH over 10 min, 100% MeOH for 20 min) to afford 1 (5.3 mg), 2 (0.9 mg), and 3 (1.3 mg) at tR 13.8 min, 13.5 min, and 15.5 min, respectively. Compound 3 was further purified by HPLC [Luna 5u Phenyl-Hexyl, 250 × 10.0 mm; flow rate, 2.0 mL/min; PDA detection 200–800 nm] using a linear MeOH–H2O gradient (70–100% MeOH over 5 min, 100% MeOH for 20 min) to afford 3 (0.9 mg) at tR 13.6 min.
Grassystatin D (1)
Colorless amorphous solid; [α]20D −32 (c 0.235, MeOH); 1H and 13C NMR data (DMSO-d6), Table 1; HRESIMS m/z 981.5626 [M + Na]+ (calcd for C48H78N8O12Na).
Grassystatin E (2)
Colorless amorphous solid; [α]20D −24 (c 0.05, MeOH); 1H and 13C NMR data (DMSO-d6) Table 2; HRESIMS m/z 967.5489 [M + Na]+ (calcd for C47H76N8O12Na).
Grassystatin F (3)
Colorless amorphous solid; [α]20D −19 (c 0.04, MeOH); 1H and 13C NMR data (DMSO-d6) Table 3; HRESIMS m/z 1156.6622 [M + Na]+ (calcd for C59H91N9O13Na).
Acid Hydrolysis and Chiral Amino Acid Analysis by LC–MS
A sample of compounds 1–3 (100 μg each) was hydrolyzed with 6 N HCl (0.5 mL) at 110 ºC for 24 h. The hydrolyzate was concentrated to dryness, reconstituted in 100 μL of H2O, and then analyzed by chiral-phase HPLC [Chirobiotic TAG (250 × 4.6 mm), Supelco; solvent: MeOH–10 mM NH4OAc (40:60, pH 5.48); flow rate 0.5 mL/min; detection by ESIMS in positive ion mode MRM scan)]. L-Pro, N-Me-D-Phe, L-Ala, L-Ile, N-Me-L-Glu, L-Leu, and N,N-diMe-L-Phe eluted at tR 13.9, 44.1, 8.3, 8.9, 6.6, 9.0, and 122.6 min, respectively. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: L-Pro (13.9; 116→70), D-Pro (36.3), N-Me-L-Phe (24.1; 180.1→134.1), N-Me-D-Phe (44.1), L-Ala (8.3; 90→44), D-Ala (15.0), L-Ile (8.9; 132→86), L-allo-Ile (9.0), D-Ile (23.7), D-allo-Ile (20.3), N-Me-L-Glu (6.6; 162→98), N-Me-D-Glu (16.8), L-Leu (9.0; 132→86), D-Leu (19.7), N,N-diMe-L-Phe (122.6; 194.1→148.1), N,N-diMe-D-Phe (116). The compound-dependent MS parameters were as follows: Pro: DP 32.4, EP 4, CE 21.8, CXP 2.8; N-Me-Phe: DP 29, EP 4, CE 20, CXP 3; Ala: DP 26, EP 3, CE 19, CXP 3; Ile: DP 32, EP 7, CE 17, CXP 3; N-Me-Glu: DP 32, EP 7, CE 17, CXP 3; Leu: DP 32, EP 7, CE 17, CXP 3; N,N-diMe-Phe: DP 33, EP 4, CE 20, CXP 3 . The source and gas-dependent MS parameters were as follows: CUR 50, CAD medium, IS 5500, TEM 750, GS1 65, GS2 65. In order to separate Ile isomers, the mobile phase was changed to MeOH–10 mM NH4OAc (90:10, pH 5.55). The acid hydrolyzates of 1–3 showed a peak corresponding to L-Ile (tR 12.7). The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: L-Ile (12.7; 132→86), L-allo-Ile (13.4), D-Ile (58.6), D-allo-Ile (48.0). The L-lactic acid was detected in the negative ion mode [Chirobiotic TAG (250 × 4.6 mm), Supelco; solvent: MeOH–10 mM NH4OAc (40:60, pH 5.51); flow rate 0.5 mL/min; detection by ESIMS in negative ion mode MRM scan)]. The MS parameters used were as follows: DP −26, EP −3, CE −18, CXP −3, CUR 30, CAD High, IS −4500, TEM 750, GS1 65, GS2 65. L-Lactic acid from the hydrolyzate eluted at tR 6.9 min. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: L-lactic acid (6.9; 89.1→43.3), D-lactic acid (7.5).
The lactic acid unit in the hydrolyzate was also examined under different HPLC conditions to confirm this assignment [Chirex 3126 (D)-penicillamine (250 × 4.6 mm) 5 micron (Phenomenex); solvent: 2 mM CuSO4; flow rate 1 mL/min; detection by UV (254 nm)]. L-Lactic acid from the hydrolyzate eluted at tR 27.5 min. The retention times (tR, min) of the authentic standards were as follows: L-lactic acid (27.5), D-lactic acid (33.3). The assignment was confirmed by a co-injection.
Modified Marfey’s Analysis to Determine the Configuration of Statine Units
Samples of 1–3 (35 μg) were subjected to acid hydrolysis at 110 ºC for 24 h using 6 N HCl. The hydrolyzates were derivatized with L-FDLA through the addition of 10 μL of 1 M NaHCO3 followed by 50 μL of 1% acetone solution of L-FDLA. The contents were mixed and heated over a hot plate at 30–40 ºC for 1 h with frequent mixing. The reaction mixture was then cooled at room temperature, acidified with 2N HCl (5 μL), dried, and re-suspended in 1:1 MeCN–H2O. Aliquots were analyzed by reversed-phase HPLC [Alltima HP C18 HL (250 × 4.6 mm), 5 μm, Alltech; flow rate 0.5 mL/min; detection by ESIMS in negative ion mode (MRM scan, 468→408)] using a linear gradient of MeOH in H2O (both containing 0.1% HCOOH, 40–100% MeOH over 50 min). The MS parameters used were as follows: DP −32, EP −7, CE −33, CXP −1, CUR 40, CAD High, IS −4500, TEM 750, GS1 65, GS2 65. Two peaks corresponding to (3S,4S)-Sta-L-FDLA and (3R,4S)-Sta- L-FDLA (tR 37.3, 37.5) were observed in the three samples. The retention times (tR, min; MRM ion pair, parent→product) of the authentic amino acids were as follows: (3S,4S)-Sta-L-FDLA (37.3; 468→408), (3R,4S)-Sta-L-FDLA (37.5), (3S,4S)-Sta-D-FDLA (47.2) [corresponding to (3R,4R)-Sta-L-FDLA], (3R,4S)-Sta-D-FDLA (47.6) [corresponding to (3S,4R)-Sta-L-FDLA].
Molecular Docking
AutoDock Vina 1.035 was used to carry out molecular docking experiments of 1 and 3. The compounds were docked into the crystal structure of cathepsin D bound to pepstatin A (PDB ID: 1LYB).36 The 3D structures of 1 and 3 were obtained and then energetically minimized using Chem3D Pro 12.0 software [Cambridge Corporation]. For receptor preparation, H2O molecules were removed and polar hydrogens were added using Pymol and Autodock tools. Autodock tools software was then used to generate receptor grid. In structures of 1 and 3, all the bonds were considered rotatable except the amide bonds and the rings. Pymol software was then used to examine and analyze the interactions of docked structures of 1 and 3 into cathepsin D. Pepstatin A (4) was also docked into cathepsin D and was compared with the X-ray structure of pepstatin A bound to cathepsin D (Figure S1).
Cell Culture
MDA-MB-231 cells were propagated and maintained in Dulbecco’s modified Eagle medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT) and 1% antibiotic-antimycotic (Invitrogen) at 37 °C humidified air and 5% CO2.
Cell Viability Assay
MDA-MB-231 cells were seeded in 96-well plates (12,000 cells/well). After 24 h incubation, the cells were treated with different concentrations of compound 1–3, pepstatin A (4, Enzo Life Sciences), or solvent control (DMSO). Following 48 h of incubation, cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide according to the manufacturer’s instructions (Promega).
To measure cell viability using the same conditions as preparation of CM, MDA-MB-231 cells were seeded and after 24 h incubation, the media was replaced with serum free media (DMEM, without sodium bicarbonate, buffered with 50 mM HEPES, pH 6.6 adjusted using 1N HCl) and treated with compounds 1–3, pepstatin A (4), E-64, or solvent control. Cell viability was measured after 72 h using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide according to the manufacturer’s instructions (Promega).
In Vitro Cathepsins D and E Inhibition Assay
To assess the aspartic protease inhibitory activity of 1–3, in vitro cathepsin D and E inhibition assays were carried out. In brief, cathepsin D (Enzo Life Sciences) and cathepsin E (R&D systems) were freshly prepared in the assay buffer [100 mM NaOAc/100 mM NaCl (pH 3.5)]. The enzyme solution was added in each well such that the final concentration is 2 μg/mL for cathepsin D and 0.05 μg/mL for cathepsin E. This was followed by the addition of various concentrations (half log dilutions starting from 100 μM final concentration) of test compounds (1–3 and pepstatin A 4) dissolved in DMSO. The plate was then incubated at room temperature for 15 min. Subsequently, substrate solution Mca-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-(Dnp)-D-Arg-NH2 [(Enzo Life Sciences); Mca = (7-methoxycoumarin-4-yl)acetyl; Dnp = 2,4,-dinitrophenyl] prepared in DMSO was then added such that the final concentration is 10 μM. The enzyme activities were monitored by measuring the increase in fluorescence signal from the fluorescently labeled substrate every 5 min for 120 min (Ex 320 nm, Em 405 nm) using a SpectraMax M5 plate reader (Molecular Devices).
Inhibition of Cellular Cathepsins D and E
To examine the inhibitory activity of grassystatin D and F (1, 3) and pepstatin A (4, Enzo Life Sciences) against cellular cathepsins (D and E), kinetic assays were carried out using lysates collected from MDA-MB-231. Briefly, the cells were lysed with PhosphoSafe buffer (EMD Millipore) and 50 μL of the lysate was incubated for 15 min with different concentrations (10 fold dilutions starting from 100 μM final concentration) of test compounds, solvent control (DMSO) and assay buffer (50 mM NaOAc, pH 4.0). Following the incubation period, the substrate solution Mca-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-(Dnp)-D-Arg-NH2 [(Enzo Life Sciences); Mca = (7-methoxycoumarin-4-yl)acetyl; Dnp = 2,4,-dinitrophenyl] prepared in DMSO was then added such that the final concentration is 10 μM. The total volume of the reaction was 100 μL. The enzyme activities were monitored by measuring the increase in fluorescence signal from the fluorescently labeled substrate every 30 sec for 30 min (Ex 320 nm, Em 405 nm) using a SpectraMax M5 plate reader.
To assess the cell permeability of compounds (1, 3) and pepstatin A (4, Enzo Life Sciences), intact live MDA-MB-231 cells were treated directly with test compounds and the inhibitory activity was measured. In brief, the cells were seeded in 24-well plate (150000 cells/well) and when reached 80% confluency, they were treated with various concentrations of test compounds and solvent control (DMSO). Following 4 h incubation, the medium was removed, the cells were washed with PBS and either lysed with PhosphoSafe buffer or trypsinized for 10 min (to remove non-internalized, surface bound compound). After trypsinization, the cells were collected by centrifugation, re-suspended in the lysis buffer and the lysate was kept on ice for 60 min followed by centrifugation (10 min, 4 °C) to remove cell debris. 50 μL of the lysate was incubated with assay buffer (50 mM NaOAc, pH 4.0) in the presence of substrate solution Mca-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-(Dnp)-D-Arg-NH2 (10 μM final concentration). The total volume of the reaction was 100 μL. The enzyme activities were monitored at 37 °C by measuring the increase in fluorescence signal from the fluorescently labeled substrate every 30 sec for 30 min (Ex 320 nm, Em 405 nm) using a SpectraMax M5 plate reader.
Inhibition of Secreted Cathepsins D and E
MDA-MB-231 cells were seeded in 12-well plates in duplicate (150000 cells/well) and 24 h later the media was replaced with serum free DMEM (without NaHCO3 buffered with 50 mM HEPES buffer and the pH was adjusted to 6.6 using 1N HCl), and treated with either grassystatins D and F (1 and 3), pepstatin A (4, Enzo Life Sciences) or DMSO. After 3 days the conditioned media were harvested, centrifuged to remove detached cells and 50 μL of CM were added in black 96-well plate in the presence of 50 μL assay buffer (100 mM NaOAc/100 mM NaCl (pH 3.5)) containing the fluorogenic substrate Mca-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-(Dnp)-D-Arg-NH2 [(Enzo Life Sciences); Mca = (7-methoxycoumarin-4-yl)acetyl; Dnp = 2,4,-dinitrophenyl] prepared in DMSO (10 μM final concentration). The total volume of the reaction was 100 μL. The secreted cathepsins D/E proteolytic activity was monitored every 5 min for 2 h (Ex 320 nm, Em 405 nm) using a SpectraMax M5 plate reader.
Measurement of Level of Secreted Cathepsin D, Cystatin C, and PAI-1
The breast cancer cell lines (MDA-MB-231, MCF7, MDA-MB-436, and BT474) were cultured in 10 cm dishes. When the cells reached 75% confluency, the media were replaced with serum free DMEM. After 24 h, the media were collected, centrifuged to remove detached cells, and concentrated using Amicon centrifugal filter units (Millipore). The protein concentration in samples was measured using BCA protein assay kit (Pierce) and 25 μg of proteins were separated by SDS-PAGE (NuPAGE 4–12% Bis-Tris mini gels, Invitrogen), transferred to polyvinylidene difluoride (PVDF) membranes, probed with antibodies and detected with the SuperSignal West Femto Maximum Sensitivity Substrate (Pierce). Anti-cathepsin D and anti-cystatin C antibodies were obtained from Abcam. PAI-1 antibody was obtained from BD Biosciences. The secondary anti-rabbit and anti-mouse antibodies were obtained from Cell-Signaling Technology. For ELISA experiments, cystatin C ELISA was performed according to the manufacturer’s instructions using Quantikine Human Cystatin C Immunoassay kit (DSCTC0) from R&D systems. PAI-1 ELISA was carried out according to the manufacturer’s instructions using PAI-1 (SERPINE1) Human ELISA Kit from Abcam (ab108891).
RNA Isolation and Reverse Transcription
RNA was isolated using RNeasy mini kit (Qiagen). cDNA synthesis was carried out using SuperScript II Reverse Transcriptase and Oligo(dT) (Invitrogen).
Real Time Quantitative Polymerase Chain Reaction (qPCR)
The reaction solution (25 μL) was prepared using 1 μL aliquot of cDNA, 12.5 μL of TaqMan gene expression assay mix, 1.25 μL of 20X TaqMan gene expression assay mix, and 10.25 μL of RNase-free water. ABI 7300 sequence detection system was used to carry out the experiment. The thermocycler program used was: 2 min at 50 °C, 10 min at 95 °C, and 15 s at 95 °C (40 cycles), and 1 min at 60 °C. CTSD (Hs00157205_m1) and CTSE (Hs00157213_m1) were used as a target genes and GAPDH (Hs02758991_g1) was used as endogenous control.
Cleavage of Cystatin C by Cathepsin D In Vitro
Recombinant Human Cystatin C (PHP0044; LifeTechnologies) and cathepsin D from human liver (Sigma) were incubated in enzyme to substrate ratio (1:5) for 3 h, pH 4.0, 37 °C. Briefly, to the assay buffer (100 mM NaCl, 100 mM sodium acetate, pH 4.0) 0.4 μL of 25 μg/50μL cathepsin D was added to give a final concentration of 2 μg/mL and 10 μL of 10 μg/100 μL cystatin C was added to give a final concentration of 10 μg/mL. The mixture was then treated with cathepsin D inhibitors (1 and 5 μM final concentration). The total volume of the mixture was 100 μL. The reaction was then incubated for 3 h at 37 °C. After the incubation period, the reaction was stopped by freezing. Each sample (20 μL) was then boiled for 5 min in 5 μL sample buffer containing 2% mercaptoethanol. Cystatin C fragments were then separated by SDS-PAGE (NuPAGE 4–12% Bis-Tris mini gels, Invitrogen) and stained using silver stain kit (Pierce).
Degradation of Extracellular Cystatin C by Secreted Cathepsin D
MDA-MB-231 cells were seeded in 12-well plates in duplicate (150000 cells/well) and 24 h later the media was replaced with serum free DMEM (without NaHCO3 buffered with 50 mM HEPES buffer and the pH was adjusted to 6.6 using 1N HCl), and treated with either grassystatins D and F (1 and 3), pepstatin A (4) or DMSO. After 3 days the conditioned media were harvested, centrifuged to remove detached cells, and analyzed by cystatin C ELISA from R&D according to the manufacturer’s instructions.
Quantification of Cysteine Cathepsins
MDA-MB-231 cells were seeded in 12-well plates in duplicate (150000 cells/well) and 24 h later the media was replaced with serum free DMEM (without NaHCO3 buffered with 50 mM HEPES buffer and the pH was adjusted to 6.6 using 1N HCl), and treated with either grassystatins D and F (1 and 3), pepstatin A (4) or DMSO. After 3 days the conditioned media were harvested, centrifuged to remove detached cells and 50 μL of CM were added in black 96-well plate in the presence of 50 μL assay buffer (100 mM sodium acetate buffer (pH 5.5), containing 4 mM DTT, and 2 mM EDTA) and the fluorogenic substrate Z-Phe-Arg-AMC (40 μM). The cysteine cathepsins proteolytic activity was monitored every 5 min for 2 h (Ex 350 nm, Em 460 nm) using a SpectraMax M5 plate reader. The cysteine cathepsin inhibitor N-[N-(L-3-trans-carboxyirane-2-carbonyl)-L-leucyl]-agmatine (E-64; 10 μM; Sigma) was used as a positive control.
Cleavage of PAI-1 by Cathepsin D
Recombinant active PAI-1 (43 kDa) and cathepsin D from human liver were purchased from Sigma. Cathepsin D and PAI-1 were incubated in enzyme to substrate ratio (1:5) for 3 h, pH 4.0, 37 °C. Briefly, to the assay buffer (100 mM NaCl, 100 mM sodium acetate (pH 4.0) 0.4 μL of 25μg/50μL cathepsin D was added to give a final concentration of 2 μg/mL and 4 μL of 25μg/100μL PAI-1 was added to give a final concentration of 10 μg/mL. The mixture was then treated with cathepsin D inhibitors (1 μM final concentration). The total volume of the mixture was 100 μL. The reaction was then incubated for 3 h at 37 °C. After the incubation period, the reaction was stopped by freezing. Each sample (20 μM) was then boiled for 5 min in 5 μL sample buffer containing 2% mercaptoethanol. PAI-1 fragments were then separated by SDS-PAGE (NuPAGE 4–12% Bis-Tris mini gels, Invitrogen) and stained using a silver stain kit (Pierce).
Activity of tPA and Concentration of PAI-1 in the CM
MDA-MB-231 cells were seeded in 12-well plates in duplicate (150000 cells/well) and 24 h later the media was replaced with serum free DMEM (without NaHCO3 buffered with 50 mM HEPES buffer and the pH was adjusted to 6.6 using 1N HCl), and treated with either grassystatins D and F (1, 3) pepstatin A (4) or DMSO. After 3 days the conditioned media (CM) were harvested, centrifuged to remove detached cells, and analyzed by tPA human chromogenic activity assay (Abcam) according to the manufacturer’s instructions. Briefly, the plate was incubated at 37 °C in a humidified incubator for increasing periods of time and the optical density was measured at 405 nm every hour for up to 8 h using a SpectraMax M5 plate reader. The same CM was also analyzed for PAI-1 concentration using PAI-1 (SERPINE1) Human ELISA Kit from Abcam (ab108891) according to the manufacturer’s instructions.
Transwell Migration Assays
The migration assays were performed using BD falcon cell culture inserts (353097) where the bottom of the insert is a polyethylene terephthalate membrane (PET) having 8 μm pore size. MDA-MB-231 cells (5000 cells/well) were suspended in complete medium (DMEM containing 10% FBS), treated with compound, and 500 μL were seeded into the inserts. On the other side of the membrane, 750 μL of complete medium treated with the same concentration of the compound were seeded. The plate was then incubated for 48 h. Following the incubation period, non-migrated cells were removed using cotton swabs and kimwipes. Cells that were migrated to the underside of the membrane were stained with crystal violet and counted under the microscope. The assays were performed in triplicate and the experiments were repeated twice.
Cathepsin D (CTSD) Knockdown
MDA-MB-231 cells (300,000 cells/well) were seeded in 6-well plate. After 24 h, the cells were transfected for 48 h with 20 nM CTSD siRNA (s137; Life Technologies) using silentFect reagent according to the manufacturer’s instructions (BIO-RAD). RT-qPCR was performed to determine the extent of CTSD knockdown.
Supplementary Material
Acknowledgments
The research was supported by the National Institutes of Health grant R01CA172310. We are very grateful to Thomas Sauvage and Larissa H. dos Santos for assistance sequencing the cyanobacterium. This is Smithsonian Marine Station contribution #xxx.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publication website at DOI: Docking figure for pepstatin A (Figure S1), expression levels of cathepsins D and E in MDA-MB-231 cells (Figure S2), cathepsin D knockdown in MDA-MB-231 cells (Figure S3), and NMR spectra (1H NMR, COSY, TOCSY, HSQC, HMBC, ROESY) for compounds 1–3 and (13C NMR and APT) for compound 1.
References
- 1.Lopez-Otin C, Bond JS. J Biol Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turk B. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 3.Kato Y, Ozawa S, Miyamoto C, Maehata Y, Suzuki A, Maeda T, Baba Y. Cancer Cell Int. 2013;13:1–8. doi: 10.1186/1475-2867-13-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foekens JA, Look MP, Bolt-de Vries J, Meijer-van Gelder ME, van Putten WL, Klijn JG. Br J Cancer. 1999;79:300–307. doi: 10.1038/sj.bjc.6990048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Westley BR, May FE. Br J Cancer. 1999;79:189–190. doi: 10.1038/sj.bjc.6690032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohri SS, Vashishta A, Vetvickova J, Fusek M, Vetvicka V. Int J Biol Macromol. 2007;41:204–209. doi: 10.1016/j.ijbiomac.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Garcia M, Platet N, Liaudet E, Laurent V, Derocq D, Brouillet JP, Rochefort H. Stem Cells. 1996;14:642–650. doi: 10.1002/stem.140642. [DOI] [PubMed] [Google Scholar]
- 8.Rochefort H. Eur J Cancer. 1992;28A:1780–1783. doi: 10.1016/0959-8049(92)90003-k. [DOI] [PubMed] [Google Scholar]
- 9.Rochefort H, Liaudet-Coopman E. Apmis. 1999;107:86–95. doi: 10.1111/j.1699-0463.1999.tb01530.x. [DOI] [PubMed] [Google Scholar]
- 10.Garcia M, Derocq D, Pujol P, Rochefort H. Oncogene. 1990;5:1809–1814. [PubMed] [Google Scholar]
- 11.Berchem G, Glondu M, Gleizes M, Brouillet JP, Vignon F, Garcia M, Liaudet-Coopman E. Oncogene. 2002;21:5951–5955. doi: 10.1038/sj.onc.1205745. [DOI] [PubMed] [Google Scholar]
- 12.Kwan JC, Liu Y, Ratnayake R, Hatano R, Kuribara A, Morimoto C, Ohnuma K, Paul VJ, Ye T, Luesch H. ChemBioChem. 2014;15:799–804. doi: 10.1002/cbic.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller B, Friedman AJ, Choi H, Hogan J, McCammon JA, Hook V, Gerwick WH. J Nat Prod. 2014;77:92–99. doi: 10.1021/np400727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taori K, Matthew S, Rocca JR, Paul VJ, Luesch H. J Nat Prod. 2007;70:1593–1600. doi: 10.1021/np0702436. [DOI] [PubMed] [Google Scholar]
- 15.Kwan JC, Taori K, Paul VJ, Luesch H. Mar Drugs. 2009;7:528–538. doi: 10.3390/md7040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo D, Chen QY, Luesch H. J Org Chem. 2016;81:532–544. doi: 10.1021/acs.joc.5b02386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salvador LA, Taori K, Biggs JS, Jakoncic J, Ostrov DA, Paul VJ, Luesch H. J Med Chem. 2013;56:1276–1290. doi: 10.1021/jm3017305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morishima H, Takita T, Aoyagi T, Takeuchi T, Umezawa H. J Antibiot. 1970;23:263–265. doi: 10.7164/antibiotics.23.263. [DOI] [PubMed] [Google Scholar]
- 19.Umezawa H, Aoyagi T, Morishima H, Matsuzaki M, Hamada M. J Antibiot. 1970;23:259–262. doi: 10.7164/antibiotics.23.259. [DOI] [PubMed] [Google Scholar]
- 20.Rahuel J, Rasetti V, Maibaum J, Rueger H, Goschke R, Cohen NC, Stutz S, Cumin F, Fuhrer W, Wood JM, Grutter MG. Chem Biol. 2000;7:493–504. doi: 10.1016/s1074-5521(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 21.Wood JM, Maibaum J, Rahuel J, Grutter MG, Cohen NC, Rasetti V, Ruger H, Goschke R, Stutz S, Fuhrer W, Schilling W, Rigollier P, Yamaguchi Y, Cumin F, Baum HP, Schnell CR, Herold P, Mah R, Jensen C, O’Brien E, Stanton A, Bedigian MP. Biochem Biophys Res Commun. 2003;308:698–705. doi: 10.1016/s0006-291x(03)01451-7. [DOI] [PubMed] [Google Scholar]
- 22.Webb RL, Schiering N, Sedrani R, Maibaum J. J Med Chem. 2010;53:7490–7520. doi: 10.1021/jm901885s. [DOI] [PubMed] [Google Scholar]
- 23.Kwan JC, Eksioglu EA, Liu C, Paul VJ, Luesch H. J Med Chem. 2009;52:5732–5747. doi: 10.1021/jm9009394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams PG, Yoshida WY, Moore RE, Paul VJ. J Nat Prod. 2003;66:1006–1009. doi: 10.1021/np030114z. [DOI] [PubMed] [Google Scholar]
- 25.Sun T, Zhang W, Zong C, Wang P, Li Y. J Pept Sci. 2010;16:364–374. doi: 10.1002/psc.1254. [DOI] [PubMed] [Google Scholar]
- 26.Al-Awadhi FH, Ratnayake R, Paul VJ, Luesch H. Bioorg Med Chem. 2016;24:3276–3282. doi: 10.1016/j.bmc.2016.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Zhang W, Li L, Salvador LA, Chen T, Chen W, Felsenstein KM, Ladd TB, Price AR, Golde TE, He J, Xu Y, Li Y, Luesch H. J Med Chem. 2012;55:10749–10765. doi: 10.1021/jm301630s. [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Chen W, Xu Y, Ren S, Zhang W, Li Y. Bioorg Med Chem. 2015;23:1963–1974. doi: 10.1016/j.bmc.2015.03.034. [DOI] [PubMed] [Google Scholar]
- 29.Zhang W, Sun T, Ma Z, Li Y. Mar Drugs. 2014;12:2308–2325. doi: 10.3390/md12042308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mevers E, Haeckl FP, Boudreau PD, Byrum T, Dorrestein PC, Valeriote FA, Gerwick WH. J Nat Prod. 2014;77:969–975. doi: 10.1021/np401051z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams PG, Yoshida WY, Moore RE, Paul VJ. J Nat Prod. 2002;65:1336–1339. doi: 10.1021/np020184q. [DOI] [PubMed] [Google Scholar]
- 32.Preciado A, Williams PG. J Org Chem. 2008;73:9228–9234. doi: 10.1021/jo8012429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. J Biol Chem. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 34.Zaidi N, Burster T, Sommandas V, Hermann T, Boehm BO, Driessen C, Voelter W, Kalbacher H. Biochem Biophys Res Commun. 2007;364:243–249. doi: 10.1016/j.bbrc.2007.09.114. [DOI] [PubMed] [Google Scholar]
- 35.Trott O, Olson AJ. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baldwin ET, Bhat TN, Gulnik S, Hosur MV, Sowder RC, Cachau RE, Collins J, Silva AM, Erickson JW. Proc Natl Acad Sci USA. 1993;90:6796–6800. doi: 10.1073/pnas.90.14.6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao-Naik C, Guruprasad K, Batley B, Rapundalo S, Hill J, Blundell T, Kay J, Dunn BM. Proteins. 1995;22:168–181. doi: 10.1002/prot.340220209. [DOI] [PubMed] [Google Scholar]
- 38.Ghoneim OM, Legere JA, Golbraikh A, Tropsha A, Booth RG. Bioorg Med Chem. 2006;14:6640–6658. doi: 10.1016/j.bmc.2006.05.077. [DOI] [PubMed] [Google Scholar]
- 39.Zhang MQ, Leurs R, Timmerman H. In: Burgers Medicinal Chemistry and Drug Discovery. 5. Wolff ME, editor. John Wiley; New York: 1997. pp. 495–559. [Google Scholar]
- 40.Lee CC, Huang TS. J Cancer Mol. 2005;1:25–36. [Google Scholar]
- 41.Wilkins-Port CE, Freytag J, Higgins SP, Higgins PJ. Cell Commun Insights. 2010;3:1–10. doi: 10.4137/cci.s2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmieri D, Lee JW, Juliano RL, Church FC. J Biol Chem. 2002;277:40950–40957. doi: 10.1074/jbc.M202333200. [DOI] [PubMed] [Google Scholar]
- 43.Giacoia EG, Miyake M, Lawton A, Goodison S, Rosser CJ. Mol Cancer Res. 2014;12:322–34. doi: 10.1158/1541-7786.MCR-13-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurent-Matha V, Huesgen PF, Masson O, Derocq D, Prébois C, Gary-Bobo M, Lecaille F, Rebière B, Meurice G, Oréar C, Hollingsworth RE, Abrahamson M, Lalmanach G, Overall CM, Liaudet-Coopman E. FASEB J. 2012;26:5172–5181. doi: 10.1096/fj.12-205229. [DOI] [PubMed] [Google Scholar]
- 45.Lenarcic B, Krasovec M, Ritonja A, Olafsson I, Turk V. FEBS Lett. 1991;280:211–215. doi: 10.1016/0014-5793(91)80295-e. [DOI] [PubMed] [Google Scholar]
- 46.Dunn BM, Hung S. Biochim Biophys Acta. 2000;1477:231–240. doi: 10.1016/s0167-4838(99)00275-7. [DOI] [PubMed] [Google Scholar]
- 47.Maynadier M, Farnoud R, Lamy PJ, Laurent-Matha V, Garcia M, Rochefort H. Int J Oncol. 2013;43:1683–1690. doi: 10.3892/ijo.2013.2095. [DOI] [PubMed] [Google Scholar]
- 48.Simon D, Xu H, Vaughan DE. Biochim Biophys Acta. 1995;1268:143–151. doi: 10.1016/0167-4889(95)00063-x. [DOI] [PubMed] [Google Scholar]
- 49.Dean RA, Butler GS, Hamma-Kourbali Y, Delbe J, Brigstock DR, Courty J, Overall CM. Mol Cell Biol. 2007;27:8454–8465. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nomura T, Katunuma N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J Med Invest. 2005;52:1–9. doi: 10.2152/jmi.52.1. [DOI] [PubMed] [Google Scholar]
- 51.Duffy MJ, O’Grady P, Devaney D, O’Siorain L, Fennelly JJ, Lijnen HR. Cancer Res. 1988;48:1348–1349. [PubMed] [Google Scholar]
- 52.Chappuis PO, Dieterich B, Sciretta V, Lohse C, Bonnefoi H, Remadi S, Sappino AP. J Clin Oncol. 2001;19:2731–2738. doi: 10.1200/JCO.2001.19.10.2731. [DOI] [PubMed] [Google Scholar]
- 53.Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, Mueller BM, Cravatt BF. Proc Natl Acad Sci USA. 2004;101:13756–13761. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu K, Yang J, Tanaka S, Gonias SL, Mars WM, Liu Y. J Biol Chem. 2006;281:2120–2127. doi: 10.1074/jbc.M504988200. [DOI] [PubMed] [Google Scholar]
- 55.Hurtado M, Lozano JJ, Castellanos E, López-Fernández LA, Harshman K, Martínez-A C, Ortiz AR, Thomson TM, Paciucci R. Gut. 2007;56:1266–1274. doi: 10.1136/gut.2006.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ortiz-Zapater E, Peiró S, Roda O, Corominas JM, Aguilar S, Ampurdanés C, Real FX, Navarro P. Am J Pathol. 2007;170:1573–1584. doi: 10.2353/ajpath.2007.060850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Díaz VM, Planaguma J, Thomson TM, Reventos J, Paciucci R. Gastroenterology. 2002;122:806–819. doi: 10.1053/gast.2002.31885. [DOI] [PubMed] [Google Scholar]
- 58.Deryugina EI, Quigley JP. J Biomed Biotechnol. 2012;2012:1–21. doi: 10.1155/2012/564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Law ME, Corsino PE, Jahn SC, Davis BJ, Chen S, Patel B, Pham K, Lu J, Sheppard B, Nørgaard P, Hong J, Higgins P, Kim JS, Luesch H, Law BK. Oncogene. 2013;32:1316–1329. doi: 10.1038/onc.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mohamed MM, Sloane BF. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 61.Wallin H, Bjarnadottir M, Vogel LK, Wasselius J, Ekstrom U, Abrahamson M. Biochimie. 2010;92:1625–1634. doi: 10.1016/j.biochi.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Kopitz C, Anton M, Gansbacher B, Kruger A. Cancer Res. 2005;65:8608–8612. doi: 10.1158/0008-5472.CAN-05-1572. [DOI] [PubMed] [Google Scholar]
- 63.Wegiel B, Jiborn T, Abrahamson M, Helczynski L, Otterbein L, Persson JL, Bjartell A. PLoS One. 2009;4:e7953. doi: 10.1371/journal.pone.0007953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turk V, Kos J, Turk B. Cancer Cell. 2004;5:409–410. doi: 10.1016/s1535-6108(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 65.Van der Stappen JW, Williams AC, Maciewicz RA, Paraskeva C. Int J Cancer. 1996;67:547–554. doi: 10.1002/(SICI)1097-0215(19960807)67:4<547::AID-IJC14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi H, Moniwa N, Sugimura M, Shinohara H, Ohi H, Terao T. Biochim Biophys Acta. 1993;1178:55–62. doi: 10.1016/0167-4889(93)90109-3. [DOI] [PubMed] [Google Scholar]
- 67.Bell-McGuinn KM, Garfall AL, Bogyo M, Hanahan D, Joyce JA. Cancer Res. 2007;67:7378–7385. doi: 10.1158/0008-5472.CAN-07-0602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.