

Abstract
Kidney stones, especially those that present in childhood/adolescence, may be due to rare inherited disorders such as cystinuria. Early recognition and prompt treatment can help reduce or even prevent the serious long-term complications of these rare stone disorders.
Keywords: Cystinuria, kidney stone, hyperoxaluria, adenine phosphoribosyltransferase (APRT) deficiency, Dent disease, urolithiasis
Kidney stones are one of the most common disorders of the urinary tract, affecting about 10% of the general population (Edvardsson et al., 2013). Although kidney stones are most often diagnosed in older adults, they can occur at any age. A recent study reported that the annual incidence of kidney stones increased 16% in the United States between 1997 and 2012, particularly among adolescents, females, and African Americans (Tasian et al., 2016). During this time period, the risk of kidney stones doubled during childhood for both boys and girls, and the lifetime risk for women increased by 45% (Tasian et al., 2016). The highest rate of increase in kidney stones was among adolescent females, and in any given year, stones were more common among females than males aged 10 to 24 years (Tasian et al., 2016).
Kidney stones that present in childhood are often due to rare inherited metabolic disorders. Although these rare stone disorders can present at any age, onset is usually before the third decade of life. Given the severity and chronicity of these conditions, as well as the associated risk of progressive renal injury, the importance of early diagnosis and appropriate management cannot be overemphasized.
A Renewed Focus on Rare Diseases
A rare disorder is one that affects a very small percentage of a given population. In the United States, a rare disorder is defined as affecting fewer than 200,000 individuals, or approximately 1 in 1,600 individuals (Aronson, 2006). In Europe, it is defined as a disease affecting fewer than 1 in 2,000 individuals (EURORDIS, 2016).
There has been an increasing focus on rare diseases in recent years. The National Human Genome Research Institute recently announced renewed funding for the Centers for Mendelian Genomics, which continues to investigate the genomic basis of rare genetic diseases (National Institutes of Health [NIH], 2016). More relevant to the study of rare kidney stone disorders is the establishment of the Rare Kidney Stone Consortium (RKSC). The Consortium facilitates cooperative exchange of information and resources among investigators, clinicians, patients, and researchers to improve care and outcomes for patients with rare stone diseases (RKSC, 2016).
Rare Kidney Stone Disorders
The inherited rare kidney stone disorders studied by the RKSC include cystinuria, primary hyperoxaluria, adenine phosphoribosyltransferase (APRT) deficiency, and Dent disease (see Table 1). These diseases are characterized by urinary hyperexcretion of insoluble mineral salts, leading to recurrent kidney stones or increased renal calcium deposition (Edvardsson et al., 2013). Other genetic causes of kidney stones are being identified as the ability to genotype more affected individuals becomes faster and less expensive (Braun et al., 2016).
Table 1.
Features of Rare Kidney Stone Disorders
| Genetic Defect | Clinical Features | Pathognomonic Feature | Stone Composition | |
|---|---|---|---|---|
| Cystinuria | Mutations in SLC7A9 or SLC3A1 gene coding for proteins required for tubular cystine reabsorption | Recurrent stones; chronic kidney disease | Hexagonal crystals in urine | Cystine |
| Primary hyperoxaluria | Mutations in gene coding for enzymes of oxalate metabolism (AGT, GRHPR, or HOGA1) | Recurrent radiopaque stones, especially in childhood; nephrocalcinosis, chronic kidney disease | Very high urinary oxalate excretion; oxalate crystal deposition in any biological fluid or tissue | Calcium oxalate |
| APRT deficiency | Mutations in APRT gene coding for protein required for adenine recycling | Radiolucent kidney stones, recurrent UTI, hematuria, red-brown diaper stains, chronic kidney disease | Round, brown crystals with Maltese cross pattern on polarized light microscopy | DHA |
| Dent disease | Mutations in CLCN5 or OCRL1 gene coding for proteins responsible for receptor-mediated endocytosis | Proteinuria, hypercalciuria, chronic kidney disease; oculocerebral syndrome if Lowe syndrome | LMW proteinuria | Calcium oxalate/calcium phosphate |
Notes: AGT = alanine:glyoxylate aminotransferase, APRT = adenine phosphoribosyltransferase, DHA = 2,8-dihydroxyadenine, GRHPR = glyoxylate reductase/hydroxyl pyruvate reductase, HOGA1 = 4-hydroxy-2-oxaloglutarate aldolase, LMW = low-molecular-weight, UTI = urinary tract infection.
Patients with rare stone diseases are more likely than other types of stone formers to develop chronic kidney disease, which can progress to end stage renal disease (ESRD) (Rule, Krambeck, & Lieske, 2011). Failure to recognize these diseases may lead to diagnostic delay and preventable renal injury (Edvardsson et al., 2013).
Inherited rare stone diseases should always be considered in the differential diagnosis of stones that occur in children (Edvardsson et al., 2013). Recurrent stones, particularly in pre-pubertal children, should elicit workup for inborn errors of metabolism. All children with reduced kidney function with or without stone disease should undergo metabolic screening for cystinuria, primary hyperoxaluria, APRT deficiency, and Dent disease (Edvardsson et al., 2013).
Diagnostic workup should include a stone analysis, urine chemistry (preferably using 24-hour urine collection), and microscopic examination of urine (Edvardsson et al., 2013). A high index of suspicion along with early diagnosis and treatment may reduce or even prevent the serious long-term complications of these rare stone disorders (Edvardsson et al., 2013).
Cystinuria
Cystinuria is an autosomal recessive disorder in which the kidney, due to a genetic defect in the cystine transporter, is unable to reabsorb cystine in the proximal tubule, resulting in urinary hyperexcretion of amino acids cystine, ornithine, lysine, and arginine (COLA). Of these, only cystine is relatively insoluble at normal urinary pH, leading to stone formation when cystine concentration rises above the solubility limit (Biyani & Cartledge, 2006; Knoll, Zollner, Wendth-Nordahl, Michel, & Alken, 2005; Sumorok & Goldfarb, 2013).
Epidemiology of Cystinuria
Although cystinuria is a rare disorder, it is the most common of the rare stone disorders, accounting for 6% to 8% of stones in children and 1% of stones in adults (Biyani & Cartledge, 2006; Prot-Bertoye et al., 2015). The prevalence of cystinuria is estimated to be 1 in 7,000, globally (Claes & Jackson, 2012), but varies greatly by population. For example, the prevalence of cystinuria is as low as 1 in 100,000 in Sweden and as high as 3% to 4% of the population in Turkey, Oman, and Iran (Al-Marhoon et al., 2015; Celiksoy et al., 2015; Claes & Jackson, 2012; Mohammadjafari et al., 2014; Segal & Thier, 1995). In the United States, the estimated prevalence of cystinuria is about 1 in 10,000, equating to approximately 33,000 cases (NIH, 2014). Given that there are more than 15,000 practicing urology professionals (urologists, urology nurses, and urology physician assistants) in the U.S. (American Urological Association [AUA], 2015; Hilton, 2013; Quallich, 2011), it is likely that every urology practitioner will see at least one cystinuria patient in his or her current practice.
Clinical Presentation Of Cystinuria
Acute stage of cystinuria
Although stones may present at any age, stone presentation in cystinuria most commonly occurs within the first two decades of life, with approximately 50% of cystinuric patients developing their first stone in the first decade of life and 25% to 40% during their teenage years (Biyani & Cartledge, 2006; Edvardsson et al., 2013). About 75% of these patients will present with bilateral stones (Biyani & Cartledge, 2006). Males are generally more severely affected than females, with a larger number of stones (Edvardsson et al., 2013). A small percentage of patients do not form stones. On average, untreated patients experience one new stone every year and undergo a surgical procedure to remove the stones every three years. By middle age, the average cystinuria patient will undergo seven surgical procedures (Barbey et al., 2000).
Chronic kidney disease from kidney stones
Up to 70% of patients with cystinuria may develop some form of chronic kidney disease (CKD), which may lead to end stage renal disease (ESRD) (Barbey et al., 2000). In a retrospective study that collected data from 442 patients with cystinuria, 27% of patients had an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73m2, indicating some degree of CKD. Hypertension was also common in this study, with 28.6% of patients having high blood pressure (Prot-Bertoye et al., 2015). Bilateral stones may also increase the risk for acute kidney injury, particularly in children. Indeed, young children with acute renal failure due to ureteral stones should be tested for cystinuria (Nalcacioglu et al., 2013). Compared with calcium oxalate stone formers, patients with cystinuria are more likely to have abnormal serum creatinine levels and are at higher risk for nephrectomy (Assimos, Leslie, Ng, Streem, & Hart, 2002).
Diagnosis of Cystinuria
The diagnosis of cystinuria is easily made by stone analysis, microscopic examination of the urine, and 24-hour urine testing. Microscopic examination of urine will often show hexagonal crystals, which are pathognomonic of cystinuria (see Figure 1). Analysis of 24-hour urine will show elevated cystine concentrations of greater than 400 mg/L urine (Biyani & Cartledge, 2006).
Figure 1. Hexagonal Crystals in the Urine Are Pathognomonic of Cystinuria.
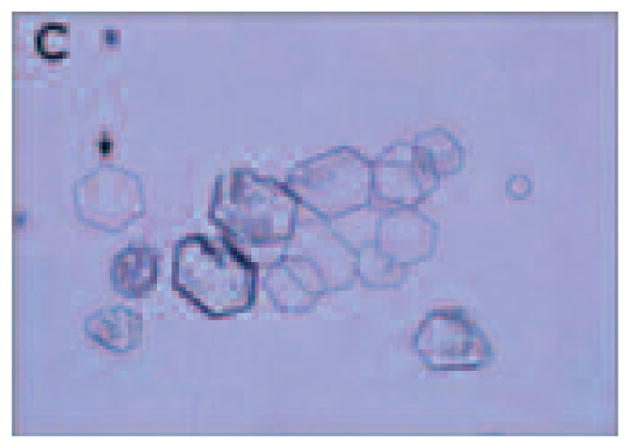
Source: Reprinted with permission from Edvardsson et al., 2013.
Treatment of Cystinuria
The goal of treatment in cystinuria is to prevent recurrence of stones by decreasing urinary cystine concentrations to below the solubility limit (< 250 mg/L) or increasing the solubility of cystine. According to AUA guidelines for the management of kidney stones, the first approach to treatment of cystinuria is a conservative program that includes initiation of therapeutic lifestyle changes involving increased fluid intake and restriction of sodium and protein, as well as urinary alkalinization therapy (Pearle et al., 2014). If conservative therapy fails to reduce urinary cystine concentrations to less than 250 mg/L or stones recur despite therapy, cystine-binding thiol drugs are the next step in treatment (Pearle et al., 2014).
Therapeutic Lifestyle Changes and Urinary Alkalinization
Fluid intake should be increased to achieve a urine volume of at least 2.5 liters daily (Pearle et al., 2014). The general goal of fluid intake is to reduce the concentration of cystine to less than 250 mg/L at pH 7, and patients should be counseled on the importance of maintaining high fluid intake. For patients with cystinuria, achieving this goal may require much higher levels of fluid intake (up to 5 L/day) than for other types of stone formers (Pearle et al., 2014).
Clinicians should counsel patients with cystinuria to limit sodium intake with the goal of decreasing urine cystine excretion (Lindell, Denneberg, Edholm, & Jeppsson, 1995; Pearle et al., 2014; Rodriguez, Santos, Málaga, & Martínez, 1995). Ideally, sodium intake should be limited to 2,300 mg or less (Pearle et al., 2014).
A low-protein/vegetarian diet reduces cystine excretion because foods of animal origin are rich in cystine and methionine, which is metabolized to cystine (Pearle et al., 2014). Reducing animal protein intake also helps with urinary alkalinization. In a study of seven patients with cystinuria, urinary cystine excretion decreased significantly when patients were maintained on a low-protein diet (9% protein) versus a higher-protein diet (27% protein) (Rodman et al., 1984).
At urine pH 7.5, the solubility of cystine increases to about 500 mg/L (Barbey et al., 2000), hence the importance of urine alkalinization therapy in patients with cystinuria. Urine alkalinization therapy with potassium citrate is recommended for patients with cystine stones to raise urinary pH to an optimal level (Pearle et al., 2014). This may require dosing throughout the day (Pearle et al., 2014). Potassium citrate is preferred over sodium bicarbonate because sodium bicarbonate increases sodium load and cystine excretion (Fjellstedt, Denneberg, Jeppsson, & Tiselius, 2001). However, potassium citrate should not be used in patients with severe renal impairment (Fjellstedt et al., 2001).
Following initiation of therapeutic lifestyle changes and urine alkalinization therapy, 24-hour urine should be collected again after three to six months and analyzed. If urinary cystine levels are still greater than 250 mg/L or there is stone recurrence, cystine-binding thiol drugs constitute the next line of therapy (Pearle et al., 2014). The failure rate of conservative therapy (therapeutic lifestyle changes and urinary alkalinization) may be as high as 55% (Barbey et al., 2000).
Cystine-Binding Thiol Drugs
Cystine-binding thiol drugs include alpha-mercaptopropionylglycine, also known as tiopronin (Thiola®) and D-penicillamine. Tiopronin should be considered first if conservative therapy fails (Pearle et al., 2014) because tiopronin has been shown to be approximately 1.5 times as effective as D-penicillamine both in reducing urinary excretion of free cystine and increasing the amounts of soluble mixed disulfide in the urine (Harbar, Cusworth, Lawes, & Wrong, 1986). Dosing of tiopronin should be based on the amount required to reduce urinary cystine concentration to below its solubility limit (generally < 250 mg/L), which in turn is determined by urinary cystine level.
Thiol derivatives, such as tiopronin and D-penicillamine, cleave cystine into two cysteine moieties and combine with cysteine to form a highly soluble disulfide compound, decreasing the excretion of poorly soluble-free cystine and increasing its solubility. Although captopril has sometimes been promoted for the management of cystinuria, it is not approved by the United States Food and Drug Administration for the treatment of cystinuria, and it does not appear in the urine in sufficient amounts to be useful. The effect of these thiol drugs depends on urine pH, with less cystine dissolved at lower pH. Therefore, thiol drugs should be used in conjunction with alkalinization therapy (Asplin & Asplin 2013).
In a study by Barbey et al. (2000), addition of thiol drugs to standard hyperdiuresis/alkalinization therapy was shown to decrease cystine excretion by 32% versus baseline. A treatment regimen of hyperdiuresis, alkalinization, and thiol drugs with frequent follow up and monitoring prevents or markedly decreases cystine stone formation, and precludes the need for urologic procedures in the majority of patients (Barbey et al., 2000; Chow & Streem, 1996; Denneberg, Jeppsson, & Stenberg, 1983; Hautmann, 1983; Koide, Kinoshita, Takemoto, Yachiku, & Sonoda, 1982; Lindell, Denneberg, Hellgren, & Jeppsson, 1995; Strologo, Laurenzi, Legato, & Pastore, 2007).
Importance of Treatment Adherence
The importance of treatment adherence for patients with cystinuria cannot be overemphasized. In a retrospective study of 20 patients treated at a single urology center, compliance with a treatment regimen that included fluid intake, urinary alkalinization, and cystine-binding drugs was associated with a lower rate of stone formation and surgical interventions (Pareek, Steele, & Nakada, 2005). In this study, 73% of the 11 compliant patients were stone-free versus 33% of the 9 noncompliant patients. The compliant patients underwent significantly fewer surgical procedures per year versus the noncompliant patients (1.0/patient versus 4.0/patient, respectively; p < 0.05) (Pareek et al., 2005).
Primary Hyperoxaluria
Primary hyperoxaluria (PH) is a very rare but serious autosomal recessive metabolic disease with an estimated prevalence of about 1 to 3 per million population (Ferrandino, 2012). Three types of PH have been described, and all are due to impairment of different enzymes in the oxalate metabolic pathways (Edvardsson et al., 2013). In Type 1 PH, which accounts for 80% of PH cases, the hepatic conversion of glyoxalate to glycine is impaired due to deficiency or dysfunction of alanine: glyoxylate aminotransferase (AGT) (see Figure 2) (Edvardsson et al., 2013). This defect leads to increased conversion to oxalate and subsequent calcium oxalate stone formation (van der Hoeven, van Woerden, & Groothoff, 2012). The formation of these insoluble crystals leads to interstitial scarring, fibrosis, and renal insufficiency. Renal insufficiency, in turn, prevents excretion of oxalate, resulting in systemic deposition of oxalate in tissues, including bone, skin, retina, myocardium, vascular walls, and the central nervous system (van der Hoeven et al., 2012). Some cases of Type 1 PH are not due to deficiency of AGT, but rather, to mistargeting of AGT to mitochondria (Edvardsson et al., 2013). Type 2 PH and Type 3 PH are due to impairment of glyoxylate reductase/hydroxyl pyruvate reductase (GRHPR) and 4-hydroxy-2-oxaloglutarate aldolase (HOGA1), respectively (Edvardsson et al., 2013).
Figure 2. Impairment of AGT or GRHPR Leads to Primary Hyperoxaluria Type 1 and Type 2, Respectively.

Notes: AGT = alanine:glyoxylate aminotransferase, GRHPR = glyoxylate reductase/hydroxyl pyruvate reductase, LDH = lactate dehydrogenase, PH = primary hyperoxaluria.
Clinical Presentation In Patients with Primary Hyperoxaluria
The majority of patients with Type 1 PH present with recurrent kidney stones and/or nephrocalcinosis, although onset of symptoms related to stone formation may occur at any age (Cochat et al., 2012; van der Hoeven et al., 2012). Presentation manifests as occasional stones in adulthood, or in severe cases, irreversible renal failure as early as the first year of life (van der Hoeven et al., 2012). In the classic case, onset of symptoms is before age 20 years, with a median age at onset of 5 to 6 years (Edvardsson et al., 2013; Lieske et al., 2005; van der Hoeven et al., 2012). Over time, progressive kidney damage leads to reduced kidney function.
In one study, more than one-third of patients had ESRD at the time of diagnosis, underscoring the need for early recognition of PH symptoms (van der Hoeven et al., 2012). Patients who are diagnosed only in adulthood (> 18 years) have a higher rate of ESRD and mortality compared with those diagnosed in childhood due to prolonged exposure of the kidney to elevated urinary oxalate levels (van der Hoeven et al., 2012), again emphasizing the importance of preventing diagnostic delays. In some cases, diagnosis has been made only after the patient develops ESRD or undergoes transplant (Lieske et al., 2005).
Diagnosis of Primary Hyperoxaluria
Diagnosis of PH should be suspected in any child with a first kidney stone, in adults with recurrent stone disease, any subject with nephrocalcinosis especially when associated with decreased GFR, and any subject with oxalate crystals in any biological fluid or tissue (Cochat et al., 2012). Stones are typically calcium oxalate monohydrate (COM), although they may be mixed COM and calcium oxalate dihydrate (Edvardsson et al., 2013).
Measurement of urinary oxalate excretion, glycolate, glycerate, and creatinine is essential to the metabolic workup (timed 12-hour or 24-hour urine collection) (Cochat et al., 2012; Edvardsson et al., 2013). Type 1 PH is characterized by markedly elevated urine oxalate excretion of greater than 0.5 mmol/1.73 m2 per day (Cochat et al., 2012). Oxalate excretion must be normalized to body surface area (BSA) for accurate interpretation in children (Edvardsson et al., 2013). If GFR is less than 60 mL/min/1.73m2, plasma oxalate should also be measured. Although plasma oxalate increases with renal failure due to any cause, levels greater than 100 μmol/L are more likely to be due to PH (Cochat et al., 2012). Urinary levels of glycolate are abnormal in Type 2 PH; in Type 3 PH, urinary levels of glycerate are abnormal.
Liver, kidney, or bone marrow biopsy is used to identify enzymatic deficiencies or tissue deposition of oxalate (i.e., oxalate crystals in any biological fluid or tissue). Measurement of AGT catalytic activity and immunoreactivity in a liver biopsy was the gold standard diagnostic test for Type 1 PH (Cochat et al., 2012). Currently, however, genotyping an individual for the relevant genes has supplanted measurement of tissue enzyme activity. The diagnosis is confirmed by DNA testing to identify specific mutations in AGTX in the case of Type 1 PH, or in GRHPR or HOGA1 in the case of Types 2 and 3 PH, respectively (Cochat et al., 2012; Edvardsson et al., 2013). The presence of oxalate crystals on renal biopsy supports the diagnosis of PH (Edvardsson et al., 2013).
Treatment of Primary Hyperoxaluria
The goal of treatment in PH is to minimize stone formation and preserve kidney function. As with cystinuria, conservative therapy is initiated as soon as a diagnosis is suggested (Cochat et al., 2012). Fluid intake is increased to at least 3 L/m2/day. In infants, achieving this level of hydration requires a nasogastric tube or a gastrostomy feeding tube (Cochat et al., 2012). Oral therapy with potassium citrate or phosphate is administered to help inhibit calcium oxalate crystallization (in this case, increasing urinary citrate is the goal, not alkalinization as in cystinuria). The recommended dose of potassium citrate is 0.10 to 0.15 g/kg body weight per day (0.3 to 0.5 mmol/kg), as long as GFR is preserved (Cochat et al., 2012). In patients with a confirmed diagnosis of Type 1 PH, vitamin B6 (pyridoxine therapy) is administered, with the goal of decreasing urine oxalate excretion by greater than 30%. The starting dose is 5 mg/kg per day; doses are increased stepwise by 5 mg/kg per day up to a maximum of 20 mg/kg per day. In some cases, pyridoxine responsiveness can be predicted by AGTX genotyping. Dietary limitation of oxalate intake is of limited use in the management of PH because the main source of oxalate is endogenous; however, it is prescribed nonetheless. Patients with PH should avoid excessive intake of vitamin C and D (Cochat et al., 2012).
Key point
Patients with PH present with calcium oxalate stones, which could lead to failure to recognize the underlying cause of stone presentation. Therefore, it is critical to consider that a higher volume of calcium oxalate stone disease in a pediatric patient may have a genetic cause, and the diagnosis of PH should not be missed.
Adenine Phosphoribosyltransferase Deficiency
Adenine phosphoribosyltransferase (APRT) deficiency is a rare autosomal recessive disorder of adenine metabolism. APRT catalyzes the synthesis of 5-adenosine monophosphate from adenine and 5-phosphoribosyl-1-pyrophosphate, the only metabolic pathway for adenine in humans. Absence or impairment of APRT, therefore, results in conversion of the 8-hydroxyadenine intermediate to the highly insoluble 2, 8-dihydroxyadenine (DHA) byproduct, which in turn results in stone formation and crystalline nephropathy. This conversion to DHA is catalyzed by xanthine dehydrogenase (XDH). The DHA crystals can precipitate in renal tubules and interstitium, leading to severe renal impairment. APRT deficiency often leads to irreversible renal failure, and the resulting nephropathy can recur even after transplant (Zaidan et al., 2014). About one-third of patients have decreased renal function, and about 10% have ESRD at the time of diagnosis (Bollée et al., 2010; Edvardsson, Palsson, Olafsson, Hjaltadottir, & Laxdal, 2001; Kamatani, Terai, Kuroshima, Nishioka, & Mikanagi, 1987).
Epidemiology of Adenine Phosphoribosyltransferase Deficiency
APRT deficiency is reported in all ethnic groups, but the majority of studies are from Japan, Iceland, and France. The estimated prevalence of APRT deficiency is 0.5 to 1 per 100,000 in the white population and 0.25 to 0.5 per 100,000 in the Japanese population. In Iceland, the estimated point prevalence is 8.9/100,000. This indicates there are at least 70,000 to 80,000 cases of APRT deficiency worldwide, suggesting that the condition may be seriously under-recognized and under-diagnosed (Bollée et al., 2012; Edvardsson et al. 2013).
Two types of APRT deficiency are recognized. In Type I disease, which affects primarily whites, APRT activity in red cell lysates is completely absent. In Type II APRT deficiency, which accounts for the majority of cases in Japan, enzyme activity is about 25% of normal values (Bollée et al., 2012).
Clinical Presentation Of Adenine Phosphoribosyltransferase Deficiency
APRT deficiency can present at any age (Bollée et al., 2012). The most common manifestations of APRT deficiency are radiolucent kidney stones and crystalline nephropathy (Bollée et al., 2012). In children, the initial presentation is often acute kidney injury due to bilateral DHA calculi and urinary tract obstruction. Other clinical features include recurrent urinary tract infection, hematuria, and reddish-brown diaper stains (Edvardsson et al., 2013). In children, APRT deficiency is often characterized by progressive CKD with reduced GFR, whereas in adults, ESRD secondary to crystalline nephropathy is more common (Runolfsdottir, Palsson, Agustsdottir, Indridason, & Edvardsson, 2016). First stone presentation can be at any age. In studies from France and Japan, mean age at first onset of stone presentation ranged between 18 to 22 years, but some patients may be asymptomatic even in late adulthood (Bollée et al., 2010; Kamatani et al., 1987).
Diagnosis of Adenine Phosphoribosyltransferase Deficiency
A diagnosis of APRT deficiency should be considered in all children presenting with renal colic, recurrent radiolucent kidney stones, or acute kidney injury, and in infants presenting with reddish-brown diaper stains. Uric acid stones are radiolucent but usually associated with a low urine pH; therefore, radiolucent stones seen in conjunction with a high urine pH also support the diagnosis of APRT deficiency (Bollée et al., 2012; Edvardsson et al., 2013).
Microscopic examination of urine can yield diagnostic clues. Urine microscopy will usually show characteristic small and round, brown DHA crystals, which are pathognomonic of APRT deficiency. In addition, crystals will show a central Maltese cross-pattern on polarized light microscopy (see Figure 3) (Bollée et al., 2012; Edvardsson et al., 2013). Stone analysis using infrared spectroscopy or X-ray crystallography can distinguish DHA from uric acid or xanthine (Bollée et al., 2012).
Figure 3. Characteristic Crystals of APRT Deficiency.

Notes: (E) DHA crystals are round and brown and (F) show a central Maltese cross pattern on polarized light microscopy.
Source: Reprinted with permission from Bollée et al., 2012.
Enzyme activity measurements help confirm the diagnosis; however, these assays may not be widely available. Patients with APRT deficiency Type I will have no APRT activity in red cell lysates and functionally significant mutations in both copies of the APRT gene. In Type II APRT deficiency, APRT activity will be 15% to 30% of normal (Bollée et al., 2012). The ability to assay urine for DHA is progressing in Iceland and should soon be a useful clinical test. Genotyping for an abnormal APRT gene is also available through the RKSC.
Treatment of Adenine Phosphoribosyltransferase Deficiency
Early recognition of APRT deficiency and prompt initiation of pharmacologic therapy can help stabilize renal function and prevent further renal damage. Allopurinol, an XDH inhibitor, is an effective and well-tolerated treatment for APRT deficiency. The recommended dose is 5 to 10 mg/kg/day in children (maximum suggested daily dose is 600 to 800 mg) taken as a single dose or divided into two doses. In adults, a dose of 200 to 300 mg/day prevents the formation of DHA crystals in most patients. Dosing is reduced when kidney function is impaired (Bollée et al., 2012). All patients with APRT deficiency, even if asymptomatic, receive allopurinol therapy because it can prevent stone formation, renal crystal deposition, and the development of kidney failure (Bollée et al., 2012). In patients who do not tolerate allopurinol, febuxostat, another XDH inhibitor, may be an option (Arnadóttir, 2014; Goldfarb, 2011).
Low purine diet and ample fluid intake provide adjunctive benefits to pharmacologic therapy (Bollée et al., 2012). Fluid intake should be increased to at least 2.5 L per day in adults, and patients should avoid purine-rich foods. Urine alkalinization therapy is not necessary because DHA remains insoluble at pH less than 8.5 (Bollée et al., 2012).
Dent Disease
Dent disease is a very rare X-linked disorder of proximal tubule dysfunction due to mutations in the CLCN5 chloride/protein exchanger gene (Dent disease 1, accounting for 60% of cases) or in the OCRL1 gene (Dent disease 2, accounting for 15% of cases) (Devuyst & Thakker, 2010; Edvardsson et al., 2013; Lieske et al., 2014). These mutations result in defective receptor-mediated endocytosis, causing generalized dysfunction of proximal tubule cells (Devuyst, 2010; Devuyst & Thakker, 2010). Exactly how this dysfunction produces the phenotype of hypercalciuria remains uncertain.
Dent disease is characterized by low-molecular-weight (LMW) proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and progressive renal failure (Devuyst & Thakker, 2010; Lieske et al., 2014). To date, approximately 250 families with Dent disease have been identified (Devuyst & Thakker, 2010; Lieske et al., 2014).
Clinical Presentation Of Dent Disease
Dent disease generally presents in childhood or early adulthood, with males more severely affected than females. A universal feature is LMW proteinuria (pathognomonic feature). Other features include hypercalciuria, stone disease, rickets, nephrocalcinosis, and renal failure, with possible foamy urine and bone pain. Dent disease may also be accompanied by short stature and growth retardation. In males younger than 10 years, the only manifestation of the disease may be asymptomatic LMW proteinuria and/or hypercalciuria. Up to 80% of affected males may develop ESRD between the third and fifth decades of life (Devuyst & Thakker, 2010; Lieske et al., 2014). Patients with Dent disease 2, caused by OCRL mutations, have Lowe syndrome, an “oculocerebral” syndrome. Their phenotype includes reduced cognitive function, bone disease, cataracts, and growth retardation.
Diagnosis of Dent Disease
Clinical diagnosis of Dent disease is based on the presence of all three of the following criteria: a) LMW proteinuria (elevation of urinary excretion of 2-microglobulin or retinol binding protein (RBP) by at least 5-fold above the upper limit of normality); b) hypercalciuria (> 4 mg/kg in a 24 h-hour collection or > 0.25 mg Ca2+ per mg creatinine on a spot sample); and c) at least one of the following: nephrocalcinosis, kidney stones, hematuria, hypophosphatemia, renal insufficiency, or a family history consistent with X-linked inheritance (Devuyst & Thakker, 2010; Lieske et al., 2014). Genetic screening for either of the two implicated genes (CLCN5 and OCRL1) can be used to confirm the diagnosis (Edvardsson et al., 2013; Lieske et al., 2014).
A typical urinary total protein screen can detect LMW proteinuria. However, these LMW proteins are not detected by dipstick. Stone analysis will reveal calcium oxalate and/or calcium phosphate stones (Edvardsson et al., 2013). Some patients with Dent disease not only have LMW proteinuria, but albuminuria as well; occasionally, they may present with the nephrotic syndrome. Kidney biopsy in such patients may demonstrate focal segmental glomerulosclerosis (Copelovitch, Nash, & Kaplan, 2007). Some patients have unnecessarily been subjected to treatment with glucocorticoids or cytotoxic therapy, treatments that will not be effective for Dent disease.
Management of Dent Disease
Goals of management are to decrease hypercalciuria, prevent kidney stones and nephrocalcinosis, and delay the progression of CKD. However, no specific treatments have been evaluated in randomized clinical trials (Lieske et al., 2014).
Thiazide diuretics, chlorthalidone, hydrochlorothiazide, and indapamide have been used to reduce urinary calcium excretion. At doses greater than 0.4 mg/kg/day, thiazide diuretics are shown to reduce urinary calcium excretion by more than 40% (Blanchard et al., 2008; Raja et al., 2002). However, these agents should be dosed with caution, and patients need to be monitored for side effects, such as hypokalemia, hyponatremia, hyperuricemia, and cramping (Blanchard et al., 2008). Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers have been used to stabilize kidney function in children with proteinuria (Lieske et al., 2014).
Nursing Implications
Urology nurses can play an important role in the identification and management of patients with rare stone disorders. Kidney stones, especially those that occur in younger individuals, recur in adults, or are accompanied by renal insufficiency, should elicit suspicion and appropriate diagnostic workup for rare inherited stone disorders. Once the diagnosis is confirmed, urology nurses can help provide appropriate disease information to patients and their families. Because the rare kidney disorders are inherited, family members of patients may also need to be evaluated even if they are asymptomatic. After appropriate treatment is initiated, urology nurses can counsel patients on the importance of compliance with therapy, including fluid intake, urinary alkalinization, and specific therapy (e.g., cystine-binding thiol drugs, vitamin B6, or allopurinol), and help facilitate treatment adherence. Compliance with treatment, in the case of cystinuria, has been shown to improve outcomes, decreasing stone formation and the need for surgical procedures (Pareek et al., 2005). Finally, urology nurses can help monitor patients for side effects that may warrant a dose adjustment or change in therapy. For example, the dose of cysteine-binding thiol drugs may often need to be adjusted to optimize therapy. Further, patients with cystinuria who are intolerant of D-penicillamine may need to be switched to tiopronin (Pearle et al., 2014), and patients with APRT deficiency who cannot tolerate allopurinol may benefit from febuxostat therapy. By maintaining a high index of suspicion, providing timely disease information, facilitating compliance, and monitoring patients for side effects, urology nurses can help improve outcomes for patients with rare kidney stone disorders.
Summary
Rare inherited metabolic disorders should always be included in the differential diagnosis of kidney or urinary stones that present in childhood or adolescence, and never forgotten in adults with unclear causes of recurrent stone disease. Once identified, certain rare stone disorders can be appropriately managed to help decrease stone burden and get patients stone-free. A high index of suspicion, along with early diagnosis and appropriate treatment, can help prevent kidney injury and perhaps the serious long-term complications of rare stone disorders.
Acknowledgments
This work was supported in part by the Rare Kidney Stone Consortium (U54KD083908), which is a part of the NIH Rare Diseases Clinical Research Network, supported through collaboration between the NIH Office of Rare Diseases Research at the National Center for Advancing Translational Sciences and National Institute of Diabetes and Digestive and Kidney Disease.
The authors thank Viji Anantharaman of Scientific Communications Group LLC for providing medical writing and editorial assistance, which is funded by Retrophin.
Footnotes
Statements of Disclosure: Ross Goldstein disclosed that he is a paid employee of Retrophin. David S. Goldfarb disclosed that he is a paid consultant for AstraZeneca, Cymabay, Pfizer, Retrophin, and Revive; is the owner of the Ravine Group; and receives funding from NIDDK and NCATS.
Contributor Information
Ross Goldstein, Director, Medical Affairs, Retrophin, Inc., San Diego, CA.
David S. Goldfarb, Clinical Chief, Nephrology Division, NYU Langone Medical Center, and Professor of Medicine and Physiology, New York University School of Medicine, New York, NY.
References
- Al-Marhoon MS, Bayoumi R, Al-Farsi Y, Al-Hinai A, Al-Maskary S, Venkiteswaran K, … Al-Hashmi I. Urinary stone composition in Oman: With high incidence of cystinuria. Urolithiasis. 2015;43(3):207–211. doi: 10.1007/s00240-015-0763-7. [DOI] [PubMed] [Google Scholar]
- American Urological Association (AUA) The state of the urology workforce and practice in the United States 2015. 2015 Retrieved from https://www.auanet.org/common/pdf/research/census/AUA-Census-2015-State-of-the-Urology-Workforce-and-Practice-in-the-United-States.pdf.
- Arnadóttir M. Febuxostat in adenosine phosphoribosyltransferase deficiency. American Journal of Kidney Diseases. 2014;64(2):316. doi: 10.1053/j.ajkd.2014.04.026. [DOI] [PubMed] [Google Scholar]
- Aronson JK. Rare diseases and orphan drugs. British Journal of Clinical Pharmacology. 2006;61(3):243–245. doi: 10.1111/j.1365-2125.2006.02617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asplin DM, Asplin JR. The interaction of thiol drugs and urine pH in the treatment of cystinuria. Journal of Urology. 2013;189(6):2147–2151. doi: 10.1016/j.juro.2012.12.031. [DOI] [PubMed] [Google Scholar]
- Assimos DG, Leslie SW, Ng C, Streem SB, Hart LJ. The impact of cystinuria on renal function. Journal of Urology. 2002;168(1):27–30. [PubMed] [Google Scholar]
- Barbey F, Joly D, Rieu P, Méjean A, Daudon M, Jungers P. Medical treatment of cystinuria: Critical reappraisal of long-term results. Journal of Urology. 2000;163(5):1419–1423. doi: 10.1016/s0022-5347(05)67633-1. [DOI] [PubMed] [Google Scholar]
- Biyani CS, Cartledge JJ. Cystinuria – Diagnosis and management. European Association of Urology-European Board of Urology Update Series. 2006;4:175–183. Retrieved from http://eu-acme.org/europeanurology/uploadarticles/Cystinuria.pdf. [Google Scholar]
- Blanchard A, Vargas-Poussou R, Peyrard S, Mogenet A, Baudouin V, Boudailliez B, … Azizi M. Effect of hydrochlorothiazide on urinary calcium excretion in Dent disease: An uncontrolled trial. American Journal of Kidney Diseases. 2008;52(6):1084–1095. doi: 10.1053/j.ajkd.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Bollée G, Dollinger C, Boutaud L, Guillemot D, Bensman A, Harambat J, … Ceballos-Picot I. Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. Journal of the American Society of Nephrology. 2010;21:679–688. doi: 10.1681/ASN.2009080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollée G, Harambat J, Bensman A, Knebelmann B, Daudon M, Ceballos-Picot I. Adenine phosphoribosyltransferase deficiency. Clinical Journal of the American Society of Nephrology. 2012;7(9):1521–1527. doi: 10.2215/CJN.02320312. [DOI] [PubMed] [Google Scholar]
- Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, … Hildebrandt F. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clinical Journal of the American Society of Nephrology. 2016;11(4):664–672. doi: 10.2215/CJN.07540715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celiksoy MH, Yilmaz A, Aydogan G, Kiyak A, Topal E, Sander S. Metabolic disorders in Turkish children with urolithiasis. Urology. 2015;85(4):909–913. doi: 10.1016/j.urology.2014.12.032. [DOI] [PubMed] [Google Scholar]
- Chow GK, Streem SB. Medical treatment of cystinuria: Results of contemporary clinical practice. Journal of Urology. 1996;156:1576–1578. doi: 10.1016/s0022-5347(01)65451-x. [DOI] [PubMed] [Google Scholar]
- Claes DJ, Jackson EJ. Cystinuria: Mechanisms and management. Pediatric Nephrology. 2012;27:2031–2038. doi: 10.1007/s00467-011-2092-6. [DOI] [PubMed] [Google Scholar]
- Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, … van Woerden CS for OxalEurope. Primary hyperoxaluria type 1: Indications for screening and guidance for diagnosis and treatment. Nephrology, Dialysis, and Transplantation. 2012;27(5):1729–1736. doi: 10.1093/ndt/gfs078. [DOI] [PubMed] [Google Scholar]
- Copelovitch L, Nash MA, Kaplan BS. Hypothesis: Dent disease is an underrecognized cause of focal glomerulosclerosis. Clinical Journal of the American Society of Nephrology. 2007;2(5):914–918. doi: 10.2215/CJN.00900207. [DOI] [PubMed] [Google Scholar]
- Denneberg T, Jeppsson JO, Stenberg P. Alternative treatment of cystinuria with alpha-merkaptopropionylglycine, Thiola®. Proceedings of the European Dialysis and Transplant Association – European Renal Association. 1983;20:427–433. [PubMed] [Google Scholar]
- Devuyst O. Dent’s disease: Chloride-proton exchange controls proximal tubule endocytosis. Nephrology Dialysis Transplantation. 2010;25(12):3832–3835. doi: 10.1093/ndt/gfq556. [DOI] [PubMed] [Google Scholar]
- Devuyst O, Thakker RV. Dent’s disease. Orphanet Journal of Rare Diseases. 2010;5:28. doi: 10.1186/1750-1172-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardsson VO, Goldfarb DS, Lieske JC, Beara-Lasic L, Anglani F, Milliner DS, Palsson R. Hereditary causes of kidney stones and chronic kidney disease. Pediatric Nephrology. 2013;28(10):1923–1942. doi: 10.1007/s00467-012-2329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T. Clinical features and genotype of adenine phosphoribosyltransferase deficiency in Iceland. American Journal of Kidney Diseases. 2001;38:473–480. doi: 10.1053/ajkd.2001.26826. [DOI] [PubMed] [Google Scholar]
- EURORDIS. About rare diseases. 2016 Retrieved from http://www.eurordis.org/about-rare-diseases.
- Ferrandino M. Evaluation and medical management of urinary lithiasis. In: McDougal A, Scott W, editors. Campbell-Walsh urology: 10th edition review. Philadelphia, PA: Elsevier/Saunders; 2012. pp. 1287–1335. [Google Scholar]
- Fjellstedt E, Denneberg T, Jeppsson JO, Tiselius HG. A comparison of the effects of potassium citrate and sodium bicarbonate in the alkalinization of urine in homozygous cystinuria. Urology Research. 2001;29(5):295–302. doi: 10.1007/s002400100200. [DOI] [PubMed] [Google Scholar]
- Goldfarb DS. Potential pharmacologic treatments for cystinuria and for calcium stones associated with hyperuricosuria. Clinical Journal of the American Society of Nephrology. 2011;6(8):2093–2097. doi: 10.2215/CJN.00320111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbar JA, Cusworth DC, Lawes LC, Wrong OM. Comparison of 2-mercaptopropionylglycine and D-penicillamine in the treatment of cystinuria. Journal of Urology. 1986;136(1):146–149. doi: 10.1016/s0022-5347(17)44760-4. [DOI] [PubMed] [Google Scholar]
- Hilton L. Non-physician providers: allied or disparate? Urology Times. 2013 Jul 30; Retrieved from http://urologytimes.modernmedicine.com/urology-times/content/tags/aua/non-physician-providers-allied-or-disparate.
- Hautmann R. Cystine-stone therapy with alpha-mercaptopropionylglycine: Experience with 99 patients. World Journal of Urology. 1983;1:186–191. [Google Scholar]
- Kamatani N, Terai C, Kuroshima S, Nishioka K, Mikanagi K. Genetic and clinical studies on 19 families with adenine phosphoribosyltransferase deficiencies. Human Genetics. 1987;75:163–168. doi: 10.1007/BF00591080. [DOI] [PubMed] [Google Scholar]
- Knoll T, Zollner A, Wendth-Nordahl G, Michel MS, Alken P. Cystinuria in childhood and adolescence: Recommendations for diagnosis, treatment, and follow-up. Pediatric Nephrology. 2005;20:19–24. doi: 10.1007/s00467-004-1663-1. [DOI] [PubMed] [Google Scholar]
- Koide T, Kinoshita K, Takemoto M, Yachiku S, Sonoda T. Conservative treatment of cystine calculi: Effect of oral alpha-mercaptopropionylglycine on cystine stone dissolution and on prevention of stone recurrence. Journal of Urology. 1982;128(3):513–516. doi: 10.1016/s0022-5347(17)53023-2. [DOI] [PubMed] [Google Scholar]
- Lieske JC, Milliner DS, Beara-Lasic L, Harris P, Hopp K, Cogal A, Mattison K. Dent disease. Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, … Stephens K, editors. GeneReviews®. 2014 Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK99494/?report=reader.
- Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, … Milliner DS. International registry for primary hyperoxaluria. American Journal of Nephrology. 2005;25(3):290–296. doi: 10.1159/000086360. [DOI] [PubMed] [Google Scholar]
- Lindell A, Denneberg T, Edholm E, Jeppsson JO. The effect of sodium intake on cystinuria with and without tiopronin treatment. Nephron. 1995;71(4):407–415. doi: 10.1159/000188760. [DOI] [PubMed] [Google Scholar]
- Lindell A, Denneberg T, Hellgren E, Jeppsson JO, Tiselius HG. Clinical course and cystine stone formation during tiopronin treatment. Urology Research. 1995;23(2):111–117. doi: 10.1007/BF00307941. [DOI] [PubMed] [Google Scholar]
- Mohammadjafari H, Barzin M, Salehifar E, Khademi Kord M, Aalaee A, Mohammadjafari R. Etiologic and epidemiologic pattern of urolithiasis in north Iran: Review of 10-year findings. Iranian Journal of Pediatrics. 2014;24(1):69–74. [PMC free article] [PubMed] [Google Scholar]
- Nalcacioglu H, Ozden E, Genc G, Yakupoglu YK, Sarikaya S, Ozkaya O. An uncommon cause of acute kidney injury in young children: Cystinuria. Journal of Pediatric Urology. 2013;9(1):e58–e63. doi: 10.1016/j.jpurol.2012.08.006. [DOI] [PubMed] [Google Scholar]
- National Institutes of Health (NIH) Genetics home reference. 2014 Retrieved from http://ghr.nlm.nih.gov/condition/cystinuria.
- National Institutes of Health (NIH) 2016 news release: New NIH research program targets the genomic basis for human disease. 2016 Retrieved from https://www.genome.gov/27563453/2016-news-release-new-nih-research-program-targets-the-genomic-basis-for-humandisease/
- Pareek G, Steele TH, Nakada SY. Urological intervention in patients with cystinuria is decreased with medical compliance. Journal of Urology. 2005;174(6):2250–2252. doi: 10.1097/01.ju.0000181817.89703.66. [DOI] [PubMed] [Google Scholar]
- Pearle MS, Goldfarb DS, Assimos DG, Curhan G, Denu-Ciocca CJ, Matlaga BR, … White JR for the American Urological Association (AUA) Medical management of kidney stones: AUA guideline. Journal of Urology. 2014;192(2):316–324. doi: 10.1016/j.juro.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Prot-Bertoye C, Lebbah S, Daudon M, Tostivint I, Bataille P, Bridoux F, … Courbebaisse M. CKD and its risk factors among patients with cystinuria. Clinical Journal of the American Society of Nephrology. 2015;10(5):842–885. doi: 10.2215/CJN.06680714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quallich SA. A survey evaluating the current role of the nurse practitioner in urology. Urologic Nursing. 2011;31:328–336. [PubMed] [Google Scholar]
- Raja KA, Schurman S, D’Mello RG, Blowey D, Goodyer P, Van Why S, … Scheinman SJ. Responsiveness of hypercalciuria to thiazide in Dent’s disease. Journal of the American Society of Nephrology. 2002;13:2938–2944. doi: 10.1097/01.asn.0000036869.82685.f6. [DOI] [PubMed] [Google Scholar]
- Rare Kidney Stone Consortium (RKSC) Welcome to the Rare Kidney Stone Consortium. 2016 Retrieved from rarekidneystones.org.
- Rodman JS, Blackburn P, Williams JJ, Brown A, Pospischil MA, Peterson CM. The effect of dietary protein on cystine excretion in patients with cystinuria. Clinical Nephrology. 1984;22(6):273–278. [PubMed] [Google Scholar]
- Rodriguez LM, Santos F, Malaga S, Martinez V. Effect of a low sodium diet on urinary elimination of cystine in cystinuric children. Nephron. 1995;71(4):416–418. doi: 10.1159/000188761. [DOI] [PubMed] [Google Scholar]
- Rule D, Krambeck AE, Lieske JC. Chronic kidney disease in kidney stone formers. Clinical Journal of the American Society of Nephrology. 2011;6(8):2069–2075. doi: 10.2215/CJN.10651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runolfsdottir HL, Palsson R, Agustsdottir IM, Indridason OS, Edvardsson VO. Kidney disease in adenine phosphoribosyltransferase deficiency. American Journal of Kidney Diseases. 2016;67(3):431–438. doi: 10.1053/j.ajkd.2015.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal S, Thier S. Cystinuria. In: Scriver CH, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. New York, NY: McGraw-Hill; 1995. pp. 3581–3601. [Google Scholar]
- Strologo LD, Laurenzi C, Legato A, Pastore A. Cystinuria in children and young adults: Success of monitoring free-cystine urine levels. Pediatric Nephrology. 2007;22:1869–1873. doi: 10.1007/s00467-007-0575-2. [DOI] [PubMed] [Google Scholar]
- Sumorok N, Goldfarb DS. Update on cystinuria. Current Opinion in Nephrology and Hypertension. 2013;22(4):427–431. doi: 10.1097/MNH.0b013e3283621c5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasian GE, Ross ME, Song L, Sas DJ, Keren R, Denburg MR, … Furth SL. Annual incidence of nephrolithiasis among children and adults in South Carolina from 1997 to 2012. Clinical Journal of the American Society of Nephrology. 2016;11(3):488–496. doi: 10.2215/CJN.07610715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoeven SM, van Woerden CS, Groothoff JW. Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: Results of the Dutch Cohort. Nephrology Dialysis Transplantation. 2012;27(10):3855–3862. doi: 10.1093/ndt/gfs320. [DOI] [PubMed] [Google Scholar]
- Zaidan M, Palsson R, Merieau E, Cornec-Le Gall E, Garstka A, Maggiore U, … Knebelmann B. Recurrent 2, 8-dihydroxyadenine nephropathy: A rare but preventable cause of renal allograft failure. American Journal of Transplantation. 2014;14(11):2623–2632. doi: 10.1111/ajt.12926. [DOI] [PMC free article] [PubMed] [Google Scholar]