

Abstract
Tumor complexity and intratumor heterogeneity contribute to subclonal diversity. Despite advances in next-generation sequencing (NGS) and bioinformatics, detecting rare mutations in primary tumors and metastases contributing to subclonal diversity is a challenge for precision genomics. Here, in order to identify rare mutations, we adapted a recently described epithelial reprograming assay for short-term propagation of epithelial cells from primary and metastatic tumors. Using this approach, we expanded minor clones and obtained epithelial cell-specific DNA/RNA for quantitative NGS analysis. Comparative Ampliseq Comprehensive Cancer Panel sequence analyses were performed on DNA from unprocessed breast tumor and tumor cells propagated from the same tumor. We identified previously uncharacterized mutations present only in the cultured tumor cells, a subset of which has been reported in brain metastatic but not primary breast tumors. In addition, whole-genome sequencing identified mutations enriched in liver metastases of various cancers, including Notch pathway mutations/chromosomal inversions in 5/5 liver metastases, irrespective of cancer types. Mutations/rearrangements in FHIT, involved in purine metabolism, were detected in 4/5 liver metastases, and the same four liver metastases shared mutations in 32 genes, including mutations of different HLA-DR family members affecting OX40 signaling pathway, which could impact the immune response to metastatic cells. Pathway analyses of all mutated genes in liver metastases showed aberrant tumor necrosis factor and transforming growth factor signaling in metastatic cells. Epigenetic regulators including KMT2C/MLL3 and ARID1B, which are mutated in >50% of hepatocellular carcinomas, were also mutated in liver metastases. Thus, irrespective of cancer types, organ-specific metastases may share common genomic aberrations. Since recent studies show independent evolution of primary tumors and metastases and in most cases mutation burden is higher in metastases than primary tumors, the method described here may allow early detection of subclonal somatic alterations associated with metastatic progression and potentially identify therapeutically actionable, metastasis-specific genomic aberrations.
Keywords: mutation, sequencing, metastasis, reprogramming, and breast cancer
Introduction
Recent advances in genomic technologies and bioinformatics analyses have significantly reduced the cost of sequencing tumor samples and, in few cases, have altered treatment strategies 51. Precision therapeutics programs in many institutions rely on sequencing of genes frequently mutated/amplified/deleted in cancer and believed to be associated with cancer progression 17, 55. Recent studies, however, have revealed several limitations of this widely used approach. For example, sequencing performed on single biopsies from only one tumor site will not reveal tumor heterogeneity, clones evolving independently or minor clones with distinct mutations 25, 43, 64. In order to identify “low frequency” mutations sequencing depth is critical, yet most studies fail to identify mutations present in less than 15% of tumor cells due to lack of deep sequencing, and stromal cell contamination may also impact mutation detection 37. Therefore, improved ability to detect genomic aberrations in minor tumor clones is clearly needed.
To begin to address such challenges of precision genomics, we investigated whether short-term culture to allow outgrowth of minor tumor clones followed by subsequent purification of epithelial cells increased detection of novel mutations. Furthermore and highly relevant to recent studies reporting parallel evolution of primary and metastatic tumors 4, 30, 31, 42, we investigated whether “rare” mutations, previously described to be present only in metastasis, could be detected in cultured primary tumor cells.
To achieve our objectives, we utilized a new approach to search for mutations found in cultured tumor cells but not in unprocessed tumors of the same patient 40. By using recently described conditionally reprogrammed epithelial cell growth assay to propagate primary breast cancer, we detected non-synonymous mutations in PI3KR1 (L7R) and DNMT3A (L4V) in only breast tumor cells, which have previously been shown to reside only in breast cancer brain metastasis 4. After initial proof-of-concept studies in six primary breast cancer samples, we extended the study to metastatic samples. By propagating and sequencing liver metastasis of breast, colon, melanoma and spindle cell carcinoma of the abdomen, we determined that mutations common in liver metastasis of these four types of cancers were an integral part of a signaling network involving migration inhibitory factor (MIF)-mediated glucocorticoid regulation and/or Notch signaling, previously shown to be essential for establishing liver metastasis 5. Furthermore, we detected rare mutations in brain metastases, suggesting that our approach can be applied to detect both common and rare mutations in metastases of various cancers.
Results
Expanding primary tumors under reprogramming conditions permits detection of novel actionable mutations
Assay design and clinical characteristics of tumors are shown in Fig. 1A and Supplementary Table S1, respectively. Briefly, freshly obtained or cryopreserved (described previously 45) tumors were minced and divided into two parts. One part was used for DNA isolation and the other was placed in culture. Once epithelial cells were sufficiently expanded, typically ~one million cells within five passages, epithelial cells were sorted by flow cytometry using Jam-A/EpCAM antibodies 45, an antibody combination demonstrated to separate epithelial cells from stromal and feeder layer fibroblasts 45. Sorted cells were used to prepare DNA and RNA. In addition, cells generated from normal tissue further away from the tumor of two patients were propagated and sequenced, serving as a control for the unlikely chance of culture-introduced mutations. No mutations in unprocessed normal tissue or cells from normal tissue were detected. Additional controls included sequencing germline DNA from blood (two cases). Analyses were restricted to non-synonymous exonic mutations.
Fig. 1.

Enhanced mutation detection in cancer using tumor cells propagated under reprogramming conditions. A) Experimental scheme for DNA sequencing. DNA from blood was used as a source for germline DNA sequencing. Staining with EpCAM/Jam-A antibodies allowed separation of epithelial cells from stromal cells. See Table S1 for additional details of metastatic samples. B) Venn diagram showing mutations shared between unprocessed tumor and tumor-derived cells as well as mutations found exclusively in cultured cells. Data from two representative samples are shown. Details are in Table S2. C) Validation of FOXO1 mutation in the LCIS sample using droplet digital PCR. One-dimensional plots for both wild type (VIC) and mutant (FAM) -specific probes are shown from different tumor samples, water, and positive plasmid controls (separated by yellow vertical lines). The sample named C01 is the LCIS sample, which showed amplification with mutant-specific primers.
Sequencing using the Ampliseq Platform detected several common mutations between unprocessed tumor DNA and cultured tumor cells (Table 1 for summary and Table S2 for detailed sample-specific data). In one case, where the same patient was diagnosed with invasive ductal carcinoma (IDC) and ductal carcinoma in situ (DCIS) in one breast and lobular carcinoma in situ (LCIS) in the other breast, cancer-specific mutations were identified in both unprocessed tumor and cultured tumor cells (Fig. 1B, Table S2). For example, IDC/DCIS contained mutations in four different exons of 5-methyltetrahydrofolate-homocysteine methyltransferase (MTR) including R802Q mutation. Mutation of R802 resulting in a stop codon has been reported in gastric carcinoma 9, and MTR gene is amplified in ~13% breast cancers of the TCGA dataset and mutated in multiple cancers based on cBioportal analyses 11, 24. MTR encodes an enzyme involved final step of methionine biosynthesis pathway, and defects in this pathway increase breast cancer risk in BRCA1 and BRCA2 mutant carriers 48. Unlike MTR mutants, which could be detected in both unprocessed tumor and cultured cells, mutant PIK3R1 was detected in only cultured cells. Interestingly, PIK3R1 mutations are found in tumors that have progressed to brain metastasis 4.
Table 1.
Mutations found in unprocessed tumor and cultured tumor cells
| Mutations in unprocessed tumor and cultured tumor cells | Mutations found only in cultured cells |
|---|---|
| PML S456R | RET T3270G, Y1090X |
| MTR T868A | PIK3CD R88H |
| PIK3R1- T629C, L210P, T908C, L303P, T818C, L273P* | FZR1 N315S, N404S |
| PKHD1, D3475V* | PIK3R1 T20G, L100R, L70R |
| ROS1 K1026M | TRRAP P825L |
| DAXX C850T, R284W, R359W* | MUC1 P141R, P168R |
| PIK3CA H1047R* | SUFU T396A |
| FOXO1 R554C | |
| DNMT3A L4V | |
| NLRP1 F504S | |
| PTPRD A364V* | |
| NFKB1 S852N |
Mutations present in cBioportal
Bold, mutations in these genes were found in breast cancer brain metastasis
The mutations found in the LCIS sample from the same patient were markedly different compared to the IDC/DCIS mutations. The most common PIK3CA H1047R and PIK3R1 mutations were detected in both unprocessed LCIS tumor and cultured LCIS cells (Fig. 1B). By contrast, mutations in FOXO1 and DNMT3A were detected only in cultured cells. As shown in Table S2, two other cancers also contained mutations detectable in tumor cells but not in unprocessed tumors, supporting an enhanced ability of our approach to detect rare mutations in primary tumor. The unique FOXO1 mutation in LCIS was confirmed by droplet digital PCR using primers that detect either wild type or mutant FOXO1 in genomic DNA (Fig. 1C), which detected this mutation in ~10% of cells (similar to sequencing results).
Reprogramming conditions allow detection of unique mutations in metastasis
Development of innovative in vitro and in vivo model system was one of the recent recommendations of metastatic breast cancer alliance to overcome obstacles associated with metastasis research 22. Encouraged by results of primary tumor studies, we next determined whether cancer cells in metastasis can be expanded using the same reprogramming conditions described in Fig. 1A followed by DNA/RNA isolation and NGS. Whole genome sequencing was done on a HiSeq X instrument, whereas RNA-seq was done on a HiSeq 2500 instrument, both manufactured by Illumina. In all cases, RNA was sequenced from reprogrammed cells, whereas whole genome sequencing was performed on cells from two samples. For the remaining samples, due to limited cell availability, unprocessed metastasis DNA was sequenced. Because of limited tissue availability, unlike in primary tumors, comparison between unprocessed tissue and cultured cells was not feasible. Germline DNA was sequenced from three cases. Samples were derived from liver metastases of one breast cancer, two ocular melanomas, a spindle cell carcinoma of the abdomen and a colorectal cancer (Table S1). Detailed results of breast cancer liver metastasis are provided below and in Table S3. Table S3 also shows mutations found in DNA, which have been confirmed at mRNA levels (column labeled RNA_supporting_Reads(Ref/Alt). These in-depth analyses allowed us to identify actionable as well as high/moderate impact mutations in each of these metastases.
A phase contrast image of cells from an inflammatory triple negative breast cancer (TNBC) liver metastasis is shown in Fig. 2A. Flow cytometry of CD49f and EpCAM antibody stained cells (Fig. 2B) confirmed that the majority of cells are of luminal progenitor phenotype (CD49f+/EpCAM+) 62. CD44/CD24 staining showed two distinct populations: CD44+/CD24- cancer stem-like cells 1 (typically enriched in TNBCs 54) and CD44+/CD24+ cell population. Expectedly, the majority of cells displayed basal-like features based on CD271/EpCAM staining pattern 34. Thus, cultured metastatic cells maintained many features expected of TNBC. Jam-A/EpCAM staining was used to separate epithelial cells from feeder layer fibroblasts. Somewhat surprisingly, minimal large-scale genomic aberrations were observed in this metastasis (Fig. 2C), despite the fact that the primary tumor was inflammatory TNBC, a subtype expected to have high degree of genomic aberrations 8. A high-confidence chromosomal inversion in chromosome 1 involving NOTCH2 was noted in this metastasis (Fig. 2D). Among 37 non-synonymous mutations present in this metastasis and also found in Cancer Gene Census Genes and Cosmic database, mutations in PRDM1 and ARID1B, both involved in epigenetics and chromatin remodeling, were considered to have moderate impact (Table 2). PRDM1 is a cancer driver gene and cancer-specific alternative splicing has been reported previously 53. ARID1B is frequently mutated in multiple cancers including breast cancer leading to its inactivation and consequently Wnt pathway activation 58. Mutation of ARID1B in 50% of hepatocellular carcinomas has been reported and may be essential for cancer cells to home and epigenetically adapt to liver microenvironment 23.
Fig. 2.

Characterization of liver metastasis of a TNBC. A) Phase contract image of metastatic cells grown under reprogramming assay condition. B) Flow cytometry analyses show the presence of cancer stem cell-like (CD44+/CD24-) and basal-like (CD49f+/EpCAM- and CD271+) cancer cells. C) Circos plot showing chromosomal aberrations in liver metastatic cells. Chromosome positions and the region with NOTCH2 aberration are indicated. D) Details of chromosomal aberrations involving NOTCH2. Nucleotide numbers with disruptions are indicated on left and genes affected by these disruptions are indicated on right. Table S3 describes additional mutations.
Table 2. List of high/moderate confidence mutations in liver and brain metastasis.
These mutations were identified by whole genome sequencing.
| Gene | SNPEFF_EFFECT | SNPEFF_CDS | SNPEFF_AA | VAF(%) | Impact |
|---|---|---|---|---|---|
| Breast cancer liver metastasis | |||||
| PRDM1 | Non-synonymous | C.1145C>T | p.S382F | 32.6 | Moderate |
| ARID1B | Non-Synonymous | C.832G>A | p.G278S | 10.6 | Moderate |
| Spindle cell carcinoma liver metastasis | |||||
| CBFA2T3 | Splice site donor+intron | C.304+2T>A | NA | 35.3 | High |
| CEP89 | Splice site donor+intron | C.492+2T>C | NA | 5.6 | High |
| Ocular melanoma liver metastasis | |||||
| GNAQ | Variant | 9:80409488 | p.Q209L | 32.9 | Actionable |
| BAP1 | Stop gained | C.1360>T | p.E454* | 69.2 | High |
| KLF4 | Frame shift+splice site region | C.689_*41del GCAGCC.. | p.G230fs | 43.8 | High |
| CEP89 | Splice site donor+intron | C.492+2T>C | NA | 16.0 | High |
| Choroidial melanoma (left ocular) liver metastasis | |||||
| GNA11 | Variant | 19:3118942 | p.Q209L | 35.1 | Actionable |
| BAP1 | Frame shift | C.1910-1911insCT | pK637fs | 48.8 | High |
| Colorectal cancer liver metastasis | |||||
| STAG2 | Splice site donor+intron | NA | p.453+2T>G | 26.7 | High |
| APC | Frame shift | C.1657delT | p.W553fs | 22.2 | High |
| PDE4DIP | Stop gained | C.2090G>A | p.W697* | 21.3 | High |
| MYH9 | Frame shift | C.3606_3610delGGAG.. | p.E1203fs | 19.2 | High |
| CEP89 | Splice site donor+intron | C.492+2T>C | NA | 19.0 | High |
| APC | Stop gained | C.4012C>T | p.Q1338* | 14.3 | High |
| MYH11 | Splice site donor+intron | C.654+1G>A | NA | 6.3 | High |
| Lung cancer brain metastasis-1 | |||||
| IKZF1 | Stop gained | C.318C>G | p.Y106* | 50% | High |
| ERBB2 | Frame shift+splice site region | C.2726_2730delGTGT… | p.G909fs | 41.9 | High |
| CDKN2A | Frame shift | C.57dupC | p.A20fs | 36.6 | High |
| BLM | Stop gained | C.2785G>T | p.E929* | 20.0 | High |
| GATA3 | Stop gained | C.274G>T | p.G92* | 18.1 | High |
| CDKN2A | Frame shift | C.63_64delCC | p.R22fs | 13.8 | High |
| Lung cancer brain metastasis-2 | |||||
| TP53 | Splice site donor+intron | C.782+1G>T | NA | 69.6 | High |
| KIAA1549 | Stop gained | C.1462A>T | p.R488* | 48.0 | High |
Sequence analyses of spindle cell carcinoma that had metastasized to liver further validated our method. Similar to the above sample, DNA/RNA from cells was used in this case. Sequencing revealed the presence of NAB2-STAT6 fusion, an aberration commonly found in solitary fibrous tumors of mesenchymal origin, confirming that in vitro growth conditions allowed expansion of cancer cells containing cancer-initiating mutations 16, 52. Unlike the breast cancer liver metastasis described above, increased genomic aberrations were detected in this liver metastasis (Fig. 3A), including “high impact” CBFA2T3 and CEP89 mutations (Table 2). CBFA2T3, a member of myeloid translocation gene family, is frequently translocated in pediatric acute megakaryoblastic leukemia 27 and considered to be a tumor suppressor in breast cancer 38. CEP89 is a centrosomal protein involved in mitochondrial metabolism and required for neuronal function 60. Thus, reprogrammed metastatic cell sequencing allowed for robust detection of gene aberrations previously described to occur in different cancer types.
Fig. 3.

Chromosomal aberrations and structural variants in liver metastases. A) Genomic aberrations in liver metastasis of a spindle cell carcinoma. Location of STAT6-NAB2 fusion, a characteristic of these tumors, is indicated. B) Genomic aberrations in liver metastasis of a melanoma. As in Figure 2, structural variants generated due to chromosome disruptions are shown along with involved chromosomes and position numbers (right panel). Few of these aberrations are inter-chromosomal. Tables S4–S6 contain additional details of mutations and structural variants.
Similar to primary melanomas, extensive genomic aberrations were observed in liver metastases of two melanomas (Fig. 3B and Supplementary Figures, Fig. S1). Both melanoma samples contained actionable mutations in the same pathway: one sample contained a mutation in GNAQ and the other in GNA11 (Table 2). GNAQ and GNA11 are guanine nucleotide binding proteins, and activating mutations in these two genes are considered driver mutations in uveal melanoma and transmit mitogenic signaling through Rho- and Rac-regulated signaling pathways 61. Note that one of the melanomas showed mutation in CEP89 at the same location as in the spindle cell carcinoma (Table 2).
A colon cancer liver metastasis showed expected mutations in APC (a frame shift and a stop codon; Table 2), and this metastasis also harbored KRAS A146T mutation, a KRAS mutation previously linked to MEK/ERK dependence and resistance to EGFR targeted therapies 12, 47. This metastasis had seven “high” and 33 “moderate” impact mutations as well as numerous structural variants including deletion in Polycomb group family member EZH2 gene (Supplementary Figures, Fig. S1). Detection of expected mutations in this metastasis and identification of authentic additional mutations serves as further validation of the assay system. This metastasis also harbored a mutation in the BRD4 gene and inhibitors of BRD4 are currently being developed as cancer therapies 46. Furthermore, mutation in CEP89 was noted, similar to liver metastases of spindle cell carcinoma and melanoma (Table 2).
Pathways commonly altered in liver metastasis irrespective of cancer types
To investigate whether liver metastases share common mutations irrespective of cancer type, four different types of analyses were performed. In the first analysis, pathways based on high impact and moderate impact Cancer Census and COSMIC mutations seen in five liver metastases were constructed. A signaling network involving TGFβ1 and TNFα was identified (Fig. 4A), and at least one mutation from each of the five metastases was represented in this network (example ARID1B, CEP89, and BAP1). In addition, all liver metastases showed mutations in MUC family genes (MUC21 in breast cancer liver metastasis and MUC20 in the remaining samples) (Supplementary Tables S3–S6). MUC family members play a major role in cell adhesion to specific extracellular matrix proteins and metastasis 3.
Fig. 4.

Pathway affected commonly in liver metastases irrespective of cancer types. A) Ingenuity pathway analyses integrate at least one high impact and/or moderate impact Cancer Census and COSMIC mutation from each of the liver metastasis into TNF and TGFβ signaling networks. Mutated genes are shown in grey. PRDM1 and ARID1B are mutated in breast cancer liver metastasis, whereas CEP89 is mutated in liver metastasis of spindle cell carcinoma, melanoma, and colon cancer. B) Four out of five liver metastases showed deregulated MIF-Glucocorticoid pathway. The commonly mutated cPLA2 gene is indicated in pink.
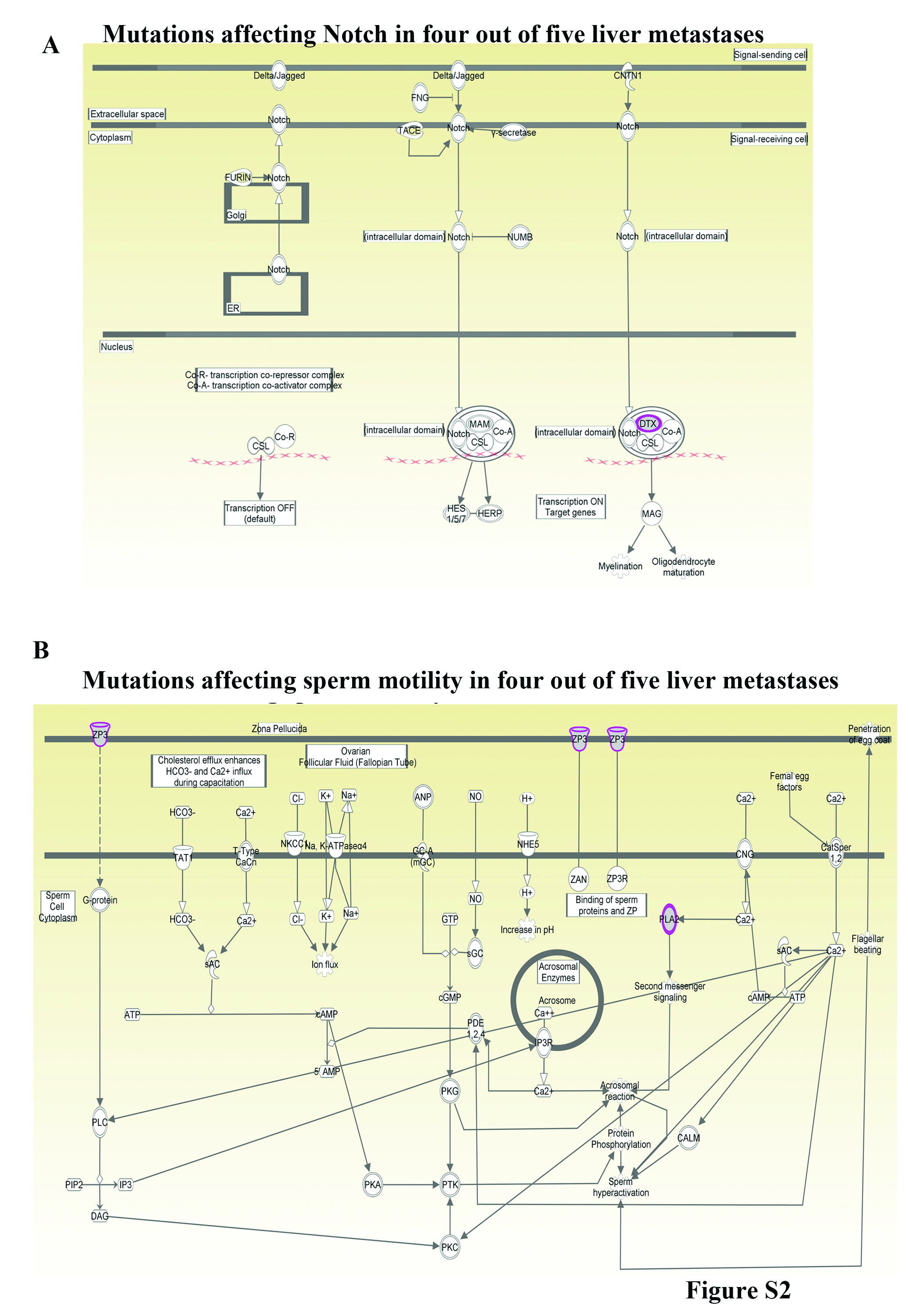
In the second analysis, all five liver metastases samples were examined for common non-synonymous mutations and pathways unique to liver metastasis. We identified 32 genes commonly mutated in 4/5 liver metastasis samples (present in all except breast cancer liver metastasis) (Supplementary Tables S3–S6). These genes are part of sperm motility, Notch and MIF-glucocorticoid receptor signaling networks (Fig. 4B and Supplementary Figures, Fig. S2). Phospholipase A2 group IVE (PLA2G4E; mutated in liver metastases) mediates the activity of migration inhibitory factor (MIF), a major factor required for establishing liver metastasis 5, 44. ZP3 is expressed mostly in oocytes and sperm and relevance of its mutation in liver metastasis is unknown 32, 50. Mutated DTX2 is a negative regulator of Notch signaling and lower DTX2 expression in colon cancer patients is associated with unfavorable outcome 19, 26. Although non-synonymous mutations involving Notch pathway were not seen with breast cancer liver metastasis, inversion of NOTCH2 gene was observed (Fig 2D).
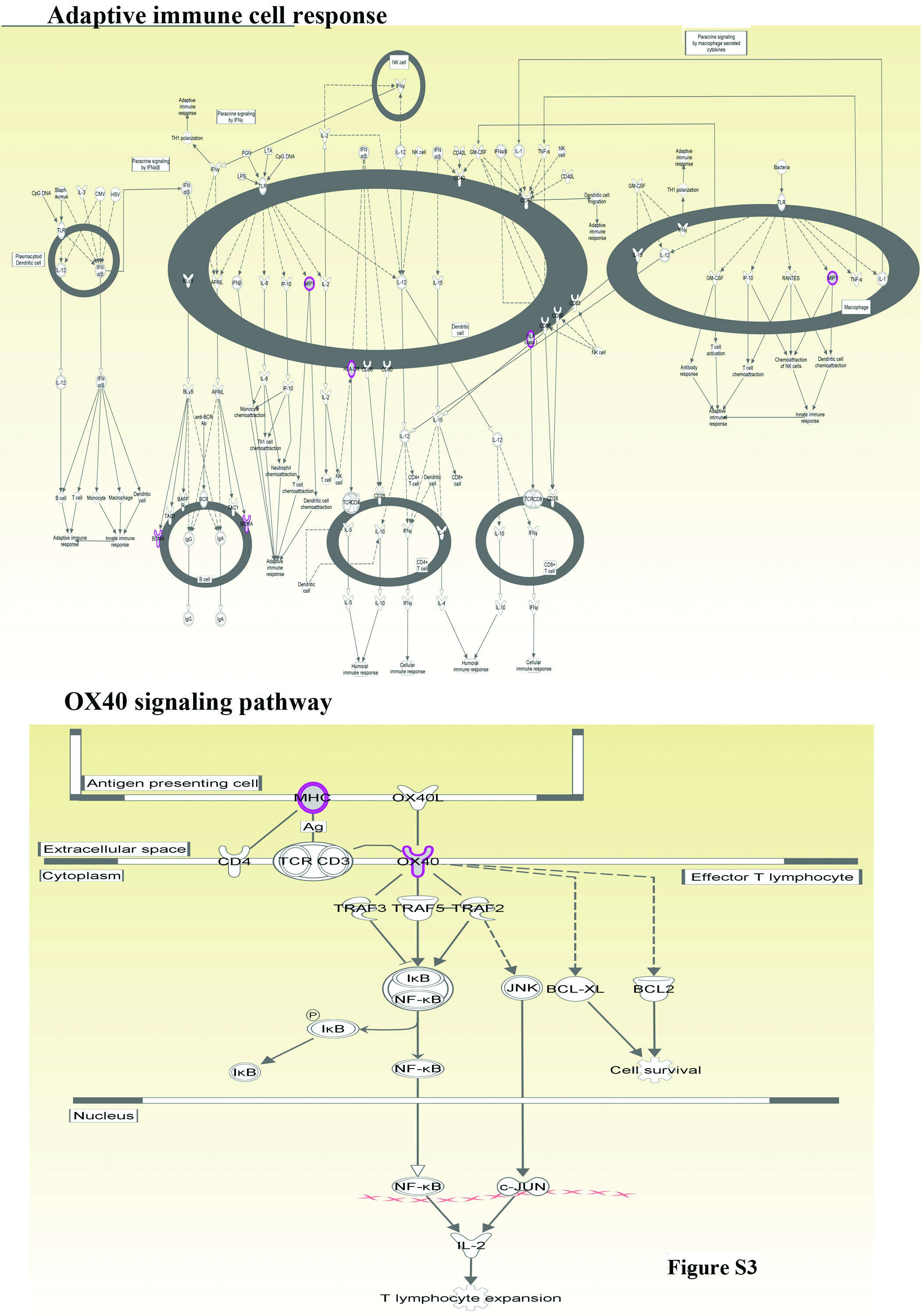
The third analyses involved combined analyses of all non-synonymous mutations found in liver metastases. Significant pathways affected in metastases are listed in Table S7. Various members of HLA-DR family were mutated in 4/5 liver metastases (Tables S3–S6, Supplementary Figures, Fig. S3).
In the fourth analyses, high confidence structural variants disrupting cancer gene census were compared. Four out of five liver metastases showed structural defects in FHIT gene. FHIT is located on a fragile site on chromosome 3 and a member of the histidine triad gene family encoding a diadenosine 5′,5‴-P1,P3-triphosphate hydrolase involved in purine metabolism. Silent/intronic mutations or structural alterations in KMT2C (MLL3), a histone methyl transferase involved in transcriptional co-activation and co-repression, were seen in liver metastasis of breast, melanoma, and colon cancer but not in spindle cell carcinoma (Fig. 3B, Tables S3–S6).
Genomic aberrations detected in brain metastasis
Although systemic treatments targeting visceral metastasis have improved dramatically, the same treatments are ineffective against brain metastasis and consequently the number of cancer patients suffering from brain metastasis has increased significantly over the past decade 57. Our previous work in animal models demonstrated brain metastatic cells undergo unique gene expression changes that allow them to adapt to brain microenvironment 6. However, to our knowledge, attempts to cultivate human brain metastatic tumors cells in vitro and identify mutations have been limited. We used the reprograming assay to grow brain metastasis of two lung cancer patients. Sequencing of DNA from metastasis and blood (for germline) and RNA from metastatic cells showed abundant genomic aberrations in the metastases including several high and moderate impact mutations (Fig. 5, Table 2; details of mutations are provided in Supplementary Table S8). Except for mutation in TP53, although affecting different regions, no other mutations were common between two brain metastases. In addition, other than PDE4DIP (mutated in one brain and four liver metastases), no overlapping mutations between liver and brain metastases were observed.
Fig. 5.

Chromosomal aberrations and structural variants in brain metastases of two lung cancers. Circos plots show extensive chromosomal rearrangements. However, two metastases did not show common structural variants (right panel). Table S8 describes additional mutations and structural variants.
Discussion
Recent advances in DNA sequencing technology have enabled sequencing of large numbers of tumor samples at an affordable cost. However, to increase clinical utility, it is essential to reduce sequencing errors and detect low frequency mutations that may be present in a minor population of tumor cells. Recent studies have shown that DNA damage causes erroneous identification of variants in cancer samples and these errors may have been introduced to widely used resources such as 1000 Genome Project and The Cancer Genome Atlas 13. Comparative analyses of unprocessed and processed/cultured tumor from the same patient as well as sequencing of both DNA and RNA from two sources of materials from the same tumor may help to reduce these errors, and the assay presented in this study may also increase detection of rare mutations.
In the current study, we detected several significant and potentially actionable mutations only in cultured cells, including FOXO1, DNMT3A and PIK3R1. FOXO1 plays a major role in apoptosis and mutations/translocations involving this gene are found in rhabdomyosarcoma 18. The new FOXO1 mutation R554C we detected could be a deleterious mutation, similar to other FOXO1 mutations reported previously 49. It is possible that FOXO1 mutation rates in other cancers including breast cancers are underreported in databases such as TCGA due to deficiencies in detection techniques. DNMT3A mutations are more frequent in hematologic malignancies and DNMT3A L4V mutation could alter DNA methylation and influence the epigenetic landscape 59. Although DNMT3A mutations in primary breast cancers are rare according to the TCGA dataset, tumor clones with DNMT3A mutations are selected in patient-derived xenografts 20. Thus, low frequency DNMT3A mutations in primary breast cancer are more common than recognized, partly due to mutation detection inefficiency. PIK3R1 L370 frame shift mutation we detected in cultured cells has previously been shown to increase MAPK pathway in ovarian cancer 15.
We extended the reprogramming technique to identify mutations in metastasis for several reasons. First, detection of mutations in metastasis is difficult because of limited availability of tissue as well as stromal cell contamination. With recent data demonstrating parallel evolution of primary tumor and metastasis 4, 41, improved methods for detecting mutations unique to metastasis are urgently needed. Consistent with this possibility, recent studies have shown that luminal type A breast cancers undergo substantial (~55%) subtype conversion upon metastasis and frequency of this subtype conversion is higher in liver metastasis compared to lung metastasis 10. Second, since metastases account for the majority of cancer deaths, there is a critical need to develop assays to detect mutations unique to sites of metastasis as well as to have live cells for follow up analyses including functional studies. Here we document feasibility of both aspects. Third, this study supports our previous publication demonstrating organ-site specific adaptive signaling pathway activation in metastatic cells 6. Despite differences in the cancer type of origin, liver metastases showed few consistent mutation patterns and/or pathway aberrations, suggesting that organ site of metastasis influences mutation spectrum in metastases. For example, four out of five liver metastases showed mutation in DTX2, a negative regulator of NOTCH signaling 35, whereas the fifth liver metastasis had genomic aberrations involving NOTCH2. Therefore, aberration in NOTCH signaling could be common across liver metastasis. Furthermore, mutations in HLA-DR family members were observed in most of the liver metastases, clearly indicating deregulated immune response to metastases. The spectrum of HLA-DR mutations observed in metastatic cells would likely disrupt antigen presentation and immune-tumor cell interactions. Deregulation in OX40 signaling network could impact therapeutic immunization strategies for metastasis 39.
FHIT, mutated in four out of five liver metastases, is believed to be a tumor suppressor involved in purine metabolism and lower FHIT expression/promoter methylation is associated with poor outcome in non-small cell lung and breast cancer and metastasis in colon cancer 2, 56, 63. Furthermore, purinergic mechanisms play an important role in intravasation, extravasation and angiogenesis and mutations in FHIT could impact this process 7.
MLL3 (KMT2C; mutated in three liver metastases) is required for the luminal epithelial gene expression program. MLL3 functions with FOXA1 transcription factor and mutational inactivation leads to increased mammary stem cell activity and accelerated tumor progression in PI3K-driven mammary tumor models 33, 65. Furthermore, MLL3 is required for conditional repression of inflammatory response genes and thus MLL3 mutational inactivation could provide inflammatory microenvironment required to establish metastasis 14. As per cBioportal, MLL3 is frequently mutated in multiple cancer types including breast cancer (~12%), melanoma (~45%), colon cancer (~14%) and hepatocellular carcinomas (50%) 23. Furthermore, chromothripsis leading to rearrangement of MLL3 has been reported in colon cancer 36. Mutation of MLL3 may enhance homing and acclimatizing capacity of cancer cells in the liver. In addition, it is possible that these common MLL3 mutations/rearrangements function not only as drivers of primary tumors but also drivers of liver metastasis.
In summary, the reprogramming assay used in this study demonstrates enhanced ability to detect mutations in primary and metastatic cancer cells. We recognize that in order to determine whether mutations are unique to metastasis, an ideal study would utilize cells from matched primary tumor and metastasis. However, it is highly impractical to obtain tissues from primary and metastasis from the same patient at the same time. We have found that tissues from both primary tumors and metastasis can be cryopreserved, making it possible to establish cultures from primary tumor and metastasis from the same patient for future mutation detection and evaluating drugs for efficacy against metastasis. Our data also raise the possibility that not all previously reported mutations found exclusively in metastatic samples are acquired during metastatic progression; rather, failure to detect these mutations in primary tumor samples because of low frequency may be a major contributing factor. For example, few of the previously reported breast cancer brain metastasis-enriched mutations 4 were detected in cultured primary tumor cells but not in unprocessed tumors (Supplementary Table S2). Further extension of the method described here to additional primary tumor samples may enable clear separation of metastasis-specific mutations from primary tumor-specific mutations.
Materials and Methods
Tumor tissues and reprogramming growth conditions
All tumor tissues were obtained with informed consent and Indiana University Institutional Review Board (IRB) considered the study non-human subjects in all cases except in case of liver biopsies. Collection of liver biopsy was approved by the IRB (protocol number IUCRO-0514). Primary tumor cells were propagated using epithelial reprogramming conditions as described previously 40. Characterization of these cells by flow cytometry and antibodies used for flow cytometry has been described previously 45. Antibodies used are CD44-APC (Cat#559942, BD Pharmingen), CD24-PE (555428, BD Pharmingen), CD49f-APC (FAB13501A, R&D Systems), EpCAM-PE (130-091-253, Miltenyi Biotech), EpCAM-APC (130-091-253, Miltenyi Biotech), Jam-A-PE (552556, BD Pharmingen) and CD271-APC (345108, Biolegand).
Differentially mutated genes were subjected to intensive investigation such as gene interaction networks and functions. Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Mountain View, CA, USA) was performed to create pathways affected by gene mutations.
Sequencing data analysis from freshly obtained or cryopreserved tumors with and without culturing on irradiated mouse embryonic fibroblasts
To characterize the degree of contamination from mouse cells, we first applied in silico PCR to tabulate the percentage of amplicon primer pairs that can also specifically pull down sequences from mouse genome. The in silico PCR tool from the UCSC genome browser was used to search over ~16,000 amplicon primer pairs from the Ion AmpliSeq Comprehensive Cancer panel. The default parameters for in silico PCR tool in UCSC genome browser were used, which required 15bp perfect match for both 5′ and 3′ primers and also allowing a maximum of 4000-bp amplified region. We found that 235 primer pairs can also pull down mouse genome sequences, which is 1.47% of all amplicons. Despite the low percentage of amplicon primer pairs that may introduce mouse DNA contamination, it is still necessary to consider the possibility that the primer pairs may have some level of random pairing which may potentially pull down mouse genome sequences. To avoid the potential bias due to the mouse genome contamination, the sequencing reads derived for cultured reprogrammed cells were mapped both to the human genome (genome build hg19) and the mouse genome (genome build mm10) using TMAP from Torrent Suite software. To distinguish reads from mouse rather than human, we explored 3 strategies to filter the sequencing data, (1) ‘no mouse’ which removes reads that can be aligned to the mouse genome with mapping quality greater than 20, (2) ‘MAPQ’ which removes reads that have a larger mapping qualities when mapped to the mouse genome than the human genome, (3) ‘longer match’ which removes reads that have a larger total number of aligned bases to the mouse genome than the human genome.
Detecting Somatic SNVs with RareVar
SNV detection was conducted using with RareVar, a tool we developed earlier, to effectively deal with diluted SNV signals from low-prevalence tumor subpopulations 28, 29. For each patient, all types of samples independently went through the Bayes factor based candidate SNV identification and machine-learning based recalibration in RareVar framework to derive SNVs, then a series of filters and statistical tests were applied to determine somatic SNVs.
Step 1: we filtered candidate somatic SNVs by only including SNVs (1) not in potentially mouse contaminated amplicons, (2) RareVar detected those SNVs in either tumor tissue or tumor cells and the allele frequencies are larger than those in the germline sample, (3) the depths on SNV loci in tumor tissue and tumor cells are greater than 100, and (4) maximum of allele frequencies from tumor tissue and tumor cells are at least 2-fold of the allele frequencies from germline sample.
Step 2: for SNVs detected in both tumor tissue and tumor cells, a binomial test (p value threshold 0.01, single sided test) was first used to check if the allele frequencies are significantly larger than those in the germline samples. Then only the ones showing larger frequencies were kept and a second binomial test was conducted to see if the allele frequencies in tumor tissue are different from those in tumor cells. If allele frequencies are significantly (p value threshold 0.01, single sided test) greater in tumor cells, then those SNVs are potentially from enriched tumor subpopulations in tumor cells. If the allele frequencies in tumor cells are similar or even lower, then the prevalence of the variants in the host tumor subpopulations did not show changes.
Step 3: for SNVs only detected in tumor tissue by RareVar, we first used binomial test to make sure the allele frequencies were greater than those in the germline sample, then checked whether there are also reads supporting those SNVs in tumor cells. If there are, it is an indicator of the host subpopulation shrinkage (the percentage in tumor cells is smaller than in tumor tissue) and also increases our confidence that those are true somatic SNVs rather than sequencing artifacts.
Step 4: for SNVs only detected in tumor cells by RareVar, we first used binomial test to make sure the allele frequencies were greater than those in the germline samples, then checked whether there are also reads supporting those SNVs in tumor tissue. If there are, it is an indicator of the host subpopulation enrichment and also increased our confidence that those are true new somatic SNVs rather than sequencing artifacts.
Whole genome sequencing
DNA and RNA prepared from samples using Qiagen RNA and DNA preparation kits were sent to New York Genome Center (info@nygenome.org) for whole genome and RNA sequencing. The center then provided processed data for SNVs and Indel detection using Mutect, Lofreq, and Strelka software tools for variant calling. Summary of results are provided in the manuscript and additional details can be provided upon request.
Droplet Digital droplet PCR (ddPCR)
ddPCR to verify mutation was done as described previously 21. Samples were analyzed using a dual fluorescent probe-based multiplex assay. Primers; Forward, 5′-GCACTTGTACAGGTGTCTTCACTT-3′; Reverse, 5′-CCCACACGGTAAGCACCAT-3′; Reporter 1, tcaggc[G]gttcatacc (Dye-VIC); Reporter 2, tcaggc[A]gttcatacc (Dye-FAM). Seventy-five base pair long oligonucleotides spanning either wild type or mutant FOXO1 region were synthesized, cloned into a plasmid vector and were used as positive controls.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank tissue collection team at the IU Simon Cancer Center, Clinical Research Office and Neurooncology Center at IU School of Medicine for collection of fresh tissues for the study. We also thank the flow cytometry core at the IU Simon Cancer Center. Excellent support from New York Genome Center, particularly Mr. Benjamin Hubert, is highly appreciated. IUPUI Signature Center for the Cure of Glioblastoma supported brain metastases tissue collection. Susan G. Komen for the Cure (SAC110025 to HN), Indiana CTSI Project development pilot grant (to HN, LL and KNP) and IU Simon Cancer Center Breast Cancer Program Pilot grant (to YL and HN) supported this study. This study utilized core services by National Institutes of Health Grant P30 DK097512 to the Indiana University School of Medicine.
Footnotes
Conflicts of interest: Authors have no conflict to declare
Author’s contribution: MA, primary cell culturing, DNA and RNA extraction and flow cytometry; YH, RareVar development, bioinformatics and analyses of genomic data; ERS, bioinformatics and analyses of genomic data; PN, primary cell collection and DNA/RNA extraction; JN, digital droplet PCR; SAT, digital droplet PCR, RGM, digital droplet PCR assay design and implementation; AAC, patient accrual, collection and processing of brain metastases; MRS, clinical protocol development and brain metastases collection; LL, bioinformatics and analyses of genomic data; FF, pathway analyses of genomic data; KPN, pathway analyses of genomic data and manuscript writing; KDM, clinical protocol development and implementation of liver metastasis sample collection; YL, assay design, supervision of bioinformatics efforts and manuscript writing; HN, experimental design, primary cell culturing, data interpretation, manuscript writing and overall supervision of the project. All authors read and approve the manuscript.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)”
References
- 1.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arun B, Kilic G, Yen C, Foster B, Yardley DA, Gaynor R, et al. Loss of FHIT expression in breast cancer is correlated with poor prognostic markers. Cancer Epidemiol Biomarkers Prev. 2005;14:1681–1685. doi: 10.1158/1055-9965.EPI-04-0278. [DOI] [PubMed] [Google Scholar]
- 3.Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene. 2010;29:2893–2904. doi: 10.1038/onc.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer discovery. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodt P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin Cancer Res. 2016;22:5971–5982. doi: 10.1158/1078-0432.CCR-16-0460. [DOI] [PubMed] [Google Scholar]
- 6.Burnett RM, Craven KE, Krishnamurthy P, Goswami CP, Badve S, Crooks P, et al. Organ-specific adaptive signaling pathway activation in metastatic breast cancer cells. Oncotarget. 2015;6:12682–12696. doi: 10.18632/oncotarget.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buxton IL, Yokdang N, Matz RM. Purinergic mechanisms in breast cancer support intravasation, extravasation and angiogenesis. Cancer Lett. 2010;291:131–141. doi: 10.1016/j.canlet.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cejalvo JM, Martinez de Duenas E, Galvan P, Garcia-Recio S, Burgues Gasion O, Pare L, et al. Intrinsic Subtypes and Gene Expression Profiles in Primary and Metastatic Breast Cancer. Cancer Res. 2017;77:2213–2221. doi: 10.1158/0008-5472.CAN-16-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen D, Huang X, Cai J, Guo S, Qian W, Wery JP, et al. A set of defined oncogenic mutation alleles seems to better predict the response to cetuximab in CRC patient-derived xenograft than KRAS 12/13 mutations. Oncotarget. 2015;6:40815–40821. doi: 10.18632/oncotarget.5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Liu P, Evans TC, Jr, Ettwiller LM. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 2017;355:752–756. doi: 10.1126/science.aai8690. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Blum R, Bowman C, Hu D, Shilatifard A, Shen S, et al. A role for H3K4 monomethylation in gene repression and partitioning of chromatin readers. Mol Cell. 2014;53:979–992. doi: 10.1016/j.molcel.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung LW, Yu S, Zhang D, Li J, Ng PK, Panupinthu N, et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell. 2014;26:479–494. doi: 10.1016/j.ccell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chmielecki J, Crago AM, Rosenberg M, O’Connor R, Walker SR, Ambrogio L, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet. 2013;45:131–132. doi: 10.1038/ng.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins DC, Sundar R, Lim JS, Yap TA. Towards Precision Medicine in the Clinic: From Biomarker Discovery to Novel Therapeutics. Trends Pharmacol Sci. 2017;38:25–40. doi: 10.1016/j.tips.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Coomans de Brachene A, Demoulin JB. FOXO transcription factors in cancer development and therapy. Cellular and molecular life sciences: CMLS. 2016;73:1159–1172. doi: 10.1007/s00018-015-2112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cormier S, Vandormael-Pournin S, Babinet C, Cohen-Tannoudji M. Developmental expression of the Notch signaling pathway genes during mouse preimplantation development. Gene Expr Patterns. 2004;4:713–717. doi: 10.1016/j.modgep.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, et al. Elevations in Circulating Methylated and Unmethylated Preproinsulin DNA in New-Onset Type 1 Diabetes. Diabetes. 2015;64:3867–3872. doi: 10.2337/db15-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flowers M, Birkey Reffey S, Mertz SA Marc Hurlbert for the Metastatic Breast Cancer A. Obstacles, Opportunities and Priorities for Advancing Metastatic Breast Cancer Research. Cancer Res. 2017;77:3386–3390. doi: 10.1158/0008-5472.CAN-17-0232. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 24.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giampieri R, Scartozzi M, Loretelli C, Piva F, Mandolesi A, Lezoche G, et al. Cancer stem cell gene profile as predictor of relapse in high risk stage II and stage III, radically resected colon cancer patients. PloS one. 2013;8:e72843. doi: 10.1371/journal.pone.0072843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruber TA, Downing JR. The biology of pediatric acute megakaryoblastic leukemia. Blood. 2015;126:943–949. doi: 10.1182/blood-2015-05-567859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao Y, Zhang P, Xuei X, Nakshatri H, Edenberg HJ, Li L, et al. Statistical modeling for sensitive detection of low-frequency single nucleotide variants. BMC Genomics. 2016;17(Suppl 7):514. doi: 10.1186/s12864-016-2905-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao YXX, Li L, Nakshatri H, Edenberg HJ, Liu Y. A framework for detecting low frequency single nucleotide variants. Journal of Computational Biology. 2017;24:637–646. doi: 10.1089/cmb.2017.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature. 2016 doi: 10.1038/nature20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016 doi: 10.1038/nature20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu X, Lu H, Cao S, Deng YL, Li QJ, Wan Q, et al. Stem cells derived from human first-trimester umbilical cord have the potential to differentiate into oocyte-like cells in vitro. Int J Mol Med. 2015;35:1219–1229. doi: 10.3892/ijmm.2015.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 Directs H3K4 Monomethylation at Enhancers via Recruitment of the Methyltransferase MLL3. Cell reports. 2016;17:2715–2723. doi: 10.1016/j.celrep.2016.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J, Villadsen R, Sorlie T, Fogh L, Gronlund SZ, Fridriksdottir AJ, et al. Tumor initiating but differentiated luminal-like breast cancer cells are highly invasive in the absence of basal-like activity. Proc Natl Acad Sci U S A. 2012;109:6124–6129. doi: 10.1073/pnas.1203203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishi N, Tang Z, Maeda Y, Hirai A, Mo R, Ito M, et al. Murine homologs of deltex define a novel gene family involved in vertebrate Notch signaling and neurogenesis. Int J Dev Neurosci. 2001;19:21–35. doi: 10.1016/s0736-5748(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 36.Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. 2011;12:R103. doi: 10.1186/gb-2011-12-10-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kostadinov R, Maley CC, Kuhner MK. Bulk Genotyping of Biopsies Can Create Spurious Evidence for Hetereogeneity in Mutation Content. PLoS Comput Biol. 2016;12:e1004413. doi: 10.1371/journal.pcbi.1004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar R, Manning J, Spendlove HE, Kremmidiotis G, McKirdy R, Lee J, et al. ZNF652, a novel zinc finger protein, interacts with the putative breast tumor suppressor CBFA2T3 to repress transcription. Mol Cancer Res. 2006;4:655–665. doi: 10.1158/1541-7786.MCR-05-0249. [DOI] [PubMed] [Google Scholar]
- 39.Linch SN, Kasiewicz MJ, McNamara MJ, Hilgart-Martiszus IF, Farhad M, Redmond WL. Combination OX40 agonism/CTLA-4 blockade with HER2 vaccination reverses T-cell anergy and promotes survival in tumor-bearing mice. Proc Natl Acad Sci U S A. 2016;113:E319–327. doi: 10.1073/pnas.1510518113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCreery MQ, Halliwill KD, Chin D, Delrosario R, Hirst G, Vuong P, et al. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat Med. 2015;21:1514–1520. doi: 10.1038/nm.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–376. doi: 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller CA, Gindin Y, Lu C, Griffith OL, Griffith M, Shen D, et al. Aromatase inhibition remodels the clonal architecture of estrogen-receptor-positive breast cancers. Nature communications. 2016;7:12498. doi: 10.1038/ncomms12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 45.Nakshatri H, Anjanappa M, Bhat-Nakshatri P. Ethnicity-Dependent and -Independent Heterogeneity in Healthy Normal Breast Hierarchy Impacts Tumor Characterization. Scientific reports. 2015;5:13526. doi: 10.1038/srep13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odore E, Lokiec F, Cvitkovic E, Bekradda M, Herait P, Bourdel F, et al. Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin Pharmacokinet. 2016;55:397–405. doi: 10.1007/s40262-015-0327-6. [DOI] [PubMed] [Google Scholar]
- 47.Park JT, Johnson N, Liu S, Levesque M, Wang YJ, Ho H, et al. Differential in vivo tumorigenicity of diverse KRAS mutations in vertebrate pancreas: A comprehensive survey. Oncogene. 2015;34:2801–2806. doi: 10.1038/onc.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pepe C, Guidugli L, Sensi E, Aretini P, D’Andrea E, Montagna M, et al. Methyl group metabolism gene polymorphisms as modifier of breast cancer risk in Italian BRCA1/2 carriers. Breast Cancer Res Treat. 2007;103:29–36. doi: 10.1007/s10549-006-9349-y. [DOI] [PubMed] [Google Scholar]
- 49.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petit FM, Serres C, Bourgeon F, Pineau C, Auer J. Identification of sperm head proteins involved in zona pellucida binding. Hum Reprod. 2013;28:852–865. doi: 10.1093/humrep/des452. [DOI] [PubMed] [Google Scholar]
- 51.Prasad V, Fojo T, Brada M. Precision oncology: origins, optimism, and potential. Lancet Oncol. 2016;17:e81–86. doi: 10.1016/S1470-2045(15)00620-8. [DOI] [PubMed] [Google Scholar]
- 52.Robinson DR, Wu YM, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung YS, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45:180–185. doi: 10.1038/ng.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sebestyen E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015;43:1345–1356. doi: 10.1093/nar/gku1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, et al. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8:R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062. doi: 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinha R, Hussain S, Mehrotra R, Kumar RS, Kumar K, Pande P, et al. Kras gene mutation and RASSF1A, FHIT and MGMT gene promoter hypermethylation: indicators of tumor staging and metastasis in adenocarcinomatous sporadic colorectal cancer in Indian population. PloS one. 2013;8:e60142. doi: 10.1371/journal.pone.0060142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steeg PS, Camphausen KA, Smith QR. Brain metastases as preventive and therapeutic targets. Nat Rev Cancer. 11:352–363. doi: 10.1038/nrc3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129:667–679. doi: 10.1182/blood-2016-10-695940. [DOI] [PubMed] [Google Scholar]
- 60.van Bon BW, Oortveld MA, Nijtmans LG, Fenckova M, Nijhof B, Besseling J, et al. CEP89 is required for mitochondrial metabolism and neuronal function in man and fly. Human molecular genetics. 2013;22:3138–3151. doi: 10.1093/hmg/ddt170. [DOI] [PubMed] [Google Scholar]
- 61.Vaque JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol Cell. 2013;49:94–108. doi: 10.1016/j.molcel.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan W, Xu N, Han X, Zhou XM, He B. The clinicopathological significance of FHIT hypermethylation in non-small cell lung cancer, a meta-analysis and literature review. Scientific reports. 2016;6:19303. doi: 10.1038/srep19303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Z, Christin JR, Wang C, Ge K, Oktay MH, Guo W. Mammary-Stem-Cell-Based Somatic Mouse Models Reveal Breast Cancer Drivers Causing Cell Fate Dysregulation. Cell reports. 2016;16:3146–3156. doi: 10.1016/j.celrep.2016.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.