

Abstract
We hypothesized that PRR contributes to renal inflammation in the 2-kidney, 1-clip (2K1C) renal ischemia model. Male Sprague-Dawley rats were fed normal sodium diet. BP was obtained on day 0 and 28 after left renal artery clipping that reduced renal blood flow by 40%. Renal expression of TNF-α, COX-2, NF-κB, IL-1β, MCP-1 and collagen type I were assessed in sham and 2K1C rats with or without left renal administration of scramble or PRR shRNA. At baseline, there were no differences in BP. Compared to sham, MAP significantly increased in clipped animals (sham: 102± 1.9 vs 2K1C: 131.8 ± 3.09 mmHg, P<0.05) and was not influenced by scramble or PRR shRNA treatment. Compared to sham and contra lateral (non-clipped) kidney, there was upregulation in mRNA and protein expression of PRR (99% and 45%, P<0.01), TNF-α (72% and 50%, P<0.05), COX-2 (72% and 39%, P<0.05), p-NF-κB (92%, P<0.05), MCP-1 (87%, P<0.05) and immunostaining of collagen type I in the clipped kidney. These increases were not influenced by scramble shRNA. Compared to 2K1C and scramble shRNA, PRR shRNA treatment in the clipped kidney significantly reduced the expression of PRR (62% and 57%, P<0.01), TNF-α (51% and 50%, P<0.05), COX-2 (50% and 56%, P<0.05), p-NF-κB by 68% (P<0.05), MCP-1 by 73% (P<0.05) and collagen type I respectively. Ang II was increased in both kidneys and did not change in response to scramble or PRR shRNA treatments. We conclude that PRR mediates renal inflammation in renal ischemia independent of blood pressure and Ang II.
Keywords: Fibrosis, inflammation, (pro)renin receptor
Introduction
The kidneys play a pivotal role in physiological functions including blood pressure regulation, and salt and water balance. Renal inflammation and fibrosis are progressive processes that are associated with renal ischemia, leading to the development of end stage renal disease (2, 3). Previous studies demonstrated a key role for the renin-angiotensin system (RAS) in development of hypertension and kidney damage in renovascular hypertension (1). However, the role of (Pro)renin receptor (PRR) in renal ischemia is not well elucidated.
PRR is a 350 amino-acid protein with a single transmembrane domain that binds to both renin and prorenin (2). PRR is expressed in heart, kidney, brain, placenta, liver, lung, smooth muscle, pancreas, adipose tissue, and the gastrointestinal tract (1). In the kidney, it is mainly expressed in podocytes, mesangial cells, vascular smooth muscle cells, and the proximal and distal renal tubules (3). The coordinated presence of renin and the PRR in the kidney may provide a pathway for generation of intrarenal Angiotensin II (Ang II) (4). Ang II is the most potent effector component of the RAS and contributes to the regulation of blood pressure, inflammation and renal function. However, independent of RAS, PRR activation also elicits intracellular signaling via the mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) signaling pathways, leading to cellular proliferation and activation of inflammatory and pro-fibrotic molecules including transforming growth factor β (TGF-β), plasminogen activator inhibitor-1 (PAI-1), IL-1β, nuclear factor-κa (NF-κB) and COX-2 (5). Cellular activation of the cytokine TGF-β mediates production and deposition of collagen, extracellular matrix, and eventually fibrosis (6). NFκB plays a pivotal role in this process and reduction of its activity attenuates renal injury through the reduction of inflammatory cytokines (7). Recent reports demonstrated the presence of PRR in the collecting ducts of normal rat kidneys and in both clipped (CK) and non-clipped (NCK) kidneys of the hypertensive 2K1C Goldblatt rat model (1). However, little is known about the contribution of PRR to renal inflammation and fibrosis in the setting of renal ischemia present in the 2K1C rat model. In the present study, we demonstrated increased expression of PRR in the CK of 2K1C rat model and its contribution to renal inflammation and fibrosis, independent of Ang II.
Results
Renal blood flow and Blood Pressure
Renal ischemia was induced by clipping of the renal artery and confirmed by measurement of renal cortical blood flow (RCBF). Compared to sham or contra lateral NCK, RCBF decreased significantly in the CK (Sham 39.4 ± 0.7, NCK 38.3 ± 0.57, CK 23.4± 0.7 ml/min/100g, respectively, P<0.01). Similarly, compared to sham there was a significant increase in mean arterial BP (MAP) in 2K1C rats (day 0: Sham 92.4 ± 6.1 vs 2K1C 100.2 ± 4.1mmHg; and day 28: Sham 102 ± 1.9 vs 2K1C 131.8 ± 3.09 mmHg, P<0.05). Neither scramble shRNA nor PRR shRNA treatment changed mean arterial BP (MAP) (day 0: 2K1C+ Scramble shRNA 94 ± 2.17 vs 2K1C+ PRR shRNA 95.6 ± 2.2 mmHg; and day 28: 2K1C+ Scramble shRNA 130.4± 4.0 vs 2K1C+ PRR shRNA 127.4± 6.3 mmHg). There were no significant differences in bodyweight between different treatment groups at baseline or at the end of the study.
Expression of PRR
Compared to sham (mRNA: 1.00 ± 0.11, WB: 1.00 ± 0.12 AU) and contra lateral NCK (mRNA: 1.07 ± 0.096, WB: 0.84 ± 0.13 AU), total renal mRNA and protein expression of PRR was significantly increased in the CK (mRNA: 1.99 ± 0.15, WB: 1.45 ± 0.13 AU) of 2K1C rats by 99% and 45% respectively (P<0.01). There were no significant changes in expression of PRR mRNA and protein in response to left renal administration of scramble shRNA. PRR shRNA treatment significantly reduced total renal expression of PRR mRNA and protein (mRNA: 0.75 ± 0.09, WB: 0.63 ± 0.15 AU) by 62% and 57%, respectively (P<0.01) (Fig. 1A–B).
Figure 1.

Renal mRNA (A) and protein (B) expressions of (Pro)renin receptor (PRR), mRNA (C) and protein (D) expressions of Tumor necrosis factor- α (TNF-α), mRNA (E) and protein (F) expressions of COX-2, phosphorylation of p-NFκB (G) and total expression of NFκB (H) in Non-clip kidney (NCK, RK) vs clipped kidney (CK, LK) in sham and 2K1C rats following scramble shRNA and PRR shRNA administration in CK. mRNA normalized to β-actin, representative blots (top), quantitative results normalized to β-actin. Data presented as mean ± SEM, *p<0.05 vs sham and #p<0.05 vs 2K1C+ scramble shRNA.
Effect of PRR shRNA on TNF-α, COX-2, NF-κB p65 and MCP-1 Expression
There were significant increases in CK mRNA (sham 1.0 ± 0.05 vs 2K1C 1.72 ± 0.13 AU) and protein expression (sham 1.0 ± 0.02 vs 2K1C 1.5 ± 0.15 AU) of TNF-α by 72% and 50%, respectively (P<0.05, Fig. 1C–D) and COX-2 (mRNA: sham 1.0 ± 0.17 vs 2K1C 1.72 ± 0.15, WB: sham 1.0 ± 0.08 vs 2K1C 1.39 ± 0.14 AU) by 72% and 39%, respectively (P<0.05, Fig. 1E–F) and increased phosphorylation of NF-κB p65 (sham 1.0 ± 0.04 vs 2K1C 1.92 ± 0.12 AU) by 92% (P<0.05, Fig. 1G) and MCP-1 (sham 1.0 ± 0.04 vs 2K1C 1.87 ± 0.08 AU) by 87% (P<0.05, Fig. 2D). Scramble shRNA administration did not change TNF-α, COX-2, NF-κB p65 and MCP-1 expressions. PRR shRNA treatment significantly attenuated the total renal mRNA and protein expression of TNF-α (mRNA: 2K1C+ Scramble shRNA 1.84 ± 0.18 vs 2K1C+ PRR shRNA 0.84 ± 0.12, WB: 2K1C+ Scramble shRNA 1.53 ± 0.14 vs 2K1C+ PRR shRNA 0.74 ± 0.05 AU) by 54% and 51% respectively (P<0.05, 1C-D) and COX-2 (mRNA: 2K1C+ Scramble shRNA 1.74 ± 0.07 vs 2K1C+ PRR shRNA 0.85 ± 0.14, WB: 2 K1C+ Scramble shRNA 1.32 ± 0.13 vs 2K1C+ PRR shRNA 0.61 ± 0.15 AU) by 51% and 53% respectively (P<0.05, Fig. 1E–F). Compared to control (sham) and treatment with 5% dextrose in water (D5W) or scramble shRNA, PRR shRNA treatment significantly reduced renal p-NF-κB p65 (2K1C+ Scramble shRNA 1.7 ± 0.13 vs 2K1C+ PRR shRNA 0.60 ± 0.21 AU) by 64% (P<0.05, Fig. 1G). There were no changes in total NF-κB p65 in response to any treatment (Fig. 1H). MCP-1 expression (2K1C+ Scramble shRNA 2.1 ± 0.21 vs 2K1C+ PRR shRNA 0.5 ± 0.12 AU) was reduced by 76% (P<0.05, Fig. 2D) in response to PRR shRNA treatment.
Figure 2.

Renal interstitial fluid (RIF) (A) expression of interleukine-1β (IL-1β) and (B) angiotensin II in kidneys of sham and 2K1C rats. (C) Urinary albumin to creatinine ratio (UACR) in sham and 2K1C rats following scramble shRNA and PRR shRNA administration in CK. (D) Renal expressions of Monocyte chemotactic protein 1 (MCP-1), in non-clip kidney (NCK, RK) vs clipped kidney (CK, LK) in sham and 2K1C rats following scramble shRNA and PRR shRNA administration in CK. Representative blots (top), quantitative results normalized to β-actin. Data presented as mean ± SEM, *p<0.05 vs sham and #p<0.05 vs 2K1C+ scramble shRNA.
Renal interstitial fluid (RIF)-interleukin-1β (IL-1β) and angiotensin II (Ang II)
Compared to sham and contra lateral NCK, RIF IL-1β (sham 0.128 ± 0.005 vs 2K1C 0.16 ± 0.003 pg/min) and Ang II (sham 0.012 ± 0.003 vs 2K1C 0.041 ± .001 pg/min) significantly increased in CK of 2K1C+D5W control rats (P<0.01). Left renal administration of scramble shRNA did not cause significant changes in RIF- IL-1β or Ang II. PRR shRNA treatment significantly reduced RIF IL-1β (2K1C+ Scramble shRNA 0.159 ± 0.008 vs 2K1C+ PRR shRNA 0.138 ± 0.006 pg/min) expression but not Ang II (2K1C+ Scramble shRNA 0.038 ± 0.001 vs 2K1C+ PRR shRNA 0.042 ± 0.001 pg/min) in CK (Fig. 2A–B).
Urine albumin creatinine ratio (UACR)
Compared to sham, UACR significantly increased in the 2K1C+ D5W group (31.5± 2.5 vs 62.3± 5.2 μg/mg, P<0.01). Treatment with scramble shRNA did not change UACR. Compared to scramble shRNA treatment, PRR shRNA significantly reduced the UACR (67.2± 4.2 vs 48.8± 2.1 μg/mg respectively, P<0.05, Fig. 2C).
Renal Immunostaining for PRR, Collagen I
Compared to sham, the CK of 2K1C+ D5W control and 2K1C+ scramble shRNA groups demonstrated a significant increase in PRR and collagen I immunostaining in the renal cortex (Fig.3A–C, I–K) and medulla (Fig. 3E–G, M–O). Treatment with PRR shRNA significantly decreased the renal PRR, collagen I immunostaining in the renal cortex (Fig. 3D, L) and medulla (Fig. 3H, P).
Figure 3.
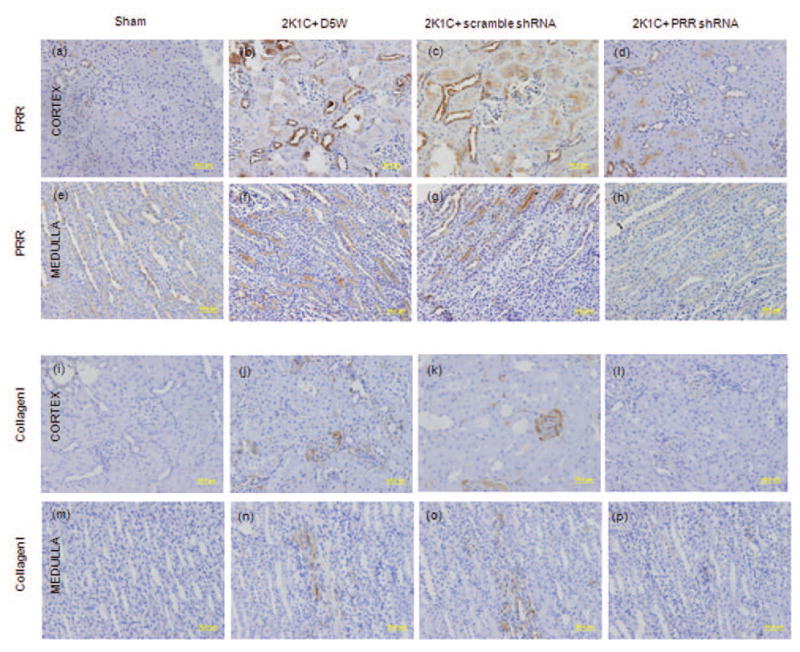
Representative images of PRR and Collagen Type I immunostaining in renal cortex and medulla of Clipped Kidney. Cortical (A–D), medullary (E–H) expression of PRR, and Cortical (I–L), medullary (M–P) expression of Collagen Type I in sham, 2K1C+ D5W, 2K1C+ scramble shRNA and 2K1C+ PRR shRNA.
Discussion
This study demonstrated that PRR expression is upregulated in the ischemic clipped kidney of the 2K1C Goldblatt rat model and promotes renal injury by upregulating renal inflammation and fibrosis in this model.
Renal ischemia is associated with increased renin activity and elevated blood pressure (8). In the 2K1C Goldblatt model, development of inflammation is prominent in the ischemic kidney and is followed by progressive kidney fibrosis (6, 9). Previous studies implicated the role of PRR in the pathogenesis of kidney disease (10–11). However, the pathologic role of PRR in renal ischemia is unknown. In the Goldblatt model, renal PRR expression was demonstrated to be increased (1). Recently, our laboratory demonstrated regulation of PRR expression by the cGMP-PKG signaling pathway and by enhanced binding of CREB-1, NF-κB p65, and c-Jun to the PRR promoter (12). In addition, we demonstrated PRR involvement in renal production of inflammatory cytokines (12). In this study, we aimed to evaluate the role of PRR in promoting renal inflammation in renal ischemia of the 2K1C rat model. We found that PRR expression is upregulated only in the clipped kidney but not in the contralateral non-clipped kidney. This finding is in agreement with the previous observation that renal PRR expression is upregulated in the clipped kidney of Goldblatt hypertensive rats (13). It is possible that increased renal production of Ang II in the ischemic kidney could have contributed to increased expression of PRR (14–15). Upregulation of PRR activates the MAPK/ERK1/2 signaling pathway which in turn increases inflammatory cytokines and promotes inflammation (10, 16). Our study clearly demonstrated that upregulation of PRR in 2K1C contributed to increased expression of COX-2, NF-κB and production of the inflammatory factors IL-1β, TNF-α and MCP-1. Intrarenal knockdown of PRR by PRR shRNA reversed the process of inflammation and attenuated the expression of IL-1β, TNF-α, COX-2 and NF-κB indicating the direct role of this receptor in regulating inflammatory cytokines in kidney. These results clearly demonstrate the intricate and direct interaction between PRR and renal inflammation in renal ischemia of the 2K1C model.
Our study demonstrated that the effects of PRR downregulation were independent of changes in blood pressure. This observation could be the result of activation of the intra-renal renin-angiotensin system (RAS) as demonstrated by increased RIF Ang II level in the CK. Activation of Ang II is associated with increased salt and water retention which in turn increases the blood pressure (17–18). These results again confirm the importance of the RAS in the 2K1C model of Goldblatt hypertension.
Previous studies demonstrated that intrarenal upregulation of inflammatory factors including TNF-α and NF-κB is associated with increased renal Ang II production (19–20) and contributed to tissue fibrosis and loss of kidney function (21). In this study, we observed that the intrarenal Ang II level was upregulated in both CK and NCK. This observation is in agreement with a previous study showing increased (renal?) Ang II levels in the 2K1C model (22). Our results did not show changes in renal Ang II with reduction in renal PRR expression, indicating an Ang II independent role of PRR in renal inflammation. It is noteworthy to state that the current study differs from previous 2K1C studies in that it utilized a less severe degree of renal artery stenosis that reduced RBF by only about 40% to induce renal ischemia. Although the achieved level of renal ischemia was less severe than that observed with the classic 2K1C Goldblatt rat model, we still observed increases in biomarkers of renal fibrosis. These biomarkers were attenuated by PRR downregulation in the clipped kidney and without changes in renal Ang II levels or BP, confirming the RAS independent direct pathologic role of PRR in this animal model. Similarly, we confirmed the significance of the renal injury in this model by demonstrating an increase in the UACR in the 2K1C rat groups. The attenuation of the UACR with reduction in renal PRR expression confirms direct involvement of PRR in renal injury.
In summary, renal ischemia is associated with increased expression of PRR, NF-κB, the inflammatory cytokine TNF-α, and UACR. Downregulation of PRR expression in this model reduced inflammation, fibrosis and albuminuria. Therefore, we conclude that PRR plays a significant role in kidney injury in renal ischemia, independent of Ang II or blood pressure. These findings may help delineate new mechanisms underlying the regulation of renal inflammation and fibrosis in renal ischemia while identifying PRR as a potential therapeutic target in the treatment of renal disorders.
Methods
Experimental Animals Preparation, Urine collections, and BP Measurements
All experimental protocols were approved by the University of Virginia Animal Care and Use Committee. Male Sprague-Dawley rats (150 to 175 g; Charles River Laboratories, Wilmington, MA, n= 24) were cage housed and maintained in a temperature-controlled room with a 12-hour light/dark cycle. Rats were provided tap water ad libitum and fed normal sodium diet (0.4% NaCl) (Harlan-Teklad, Madison, WI) throughout the duration of the study. Following a training period for blood pressure measurement, blood pressure was monitored by tail-cuff plethysmography (IITC Life Sciences, Woodland Hills, CA). For 24hr urine collections, rats were placed in individual metabolic cages. The total urine volume was determined and urine aliquots were stored at −80°C until assayed.
Surgical Procedures
Rats were anesthetized with a combination of ketamine (80 mg/kg; I.P.) and xylazine (8 mg/kg; I.P) and placed on a heating pad throughout the surgery period. Utilizing a sterile technique, a midline laparotomy was performed and the left renal artery was clipped using u-shaped silver clip. A catheter (PE-10) was inserted into the left kidney interstitium (cortex and medulla) and D5W (200μl), scramble shRNA (200 μl, Viral vector core, Iowa), or PRR shRNA (200μl, Viral vector core, Iowa), containing a lentiviral construct was slowly administered (5μl/min). The contra lateral kidney remained untouched. The abdominal wall was then sutured and the rats were allowed to recover. Rats were allocated into four treatment groups; 2K1C+ D5W (n=6), 2K1C+ scramble shRNA (n=6), 2K1C+ PRR shRNA (n=6), and sham (n=6) (operation was performed on a control group). Rats were maintained for 28 days after surgery.
Renal blood flow
Changes in renal cortical (RCBF) blood flow were determined by laser flowmeter (Advance Laser Flowmeter ALF 21D, Tokyo, Japan) as described earlier (23).
In vivo Renal Interstitial Fluid (RIF) Collections and Assays for IL-1β and Ang II
In vivo RIF for IL-1β and Ang II measurements was collected as previously described (11). IL-1β levels were measured using an enzyme immunoassay kit (R&D Systems, Minneapolis, MN, n= 4). Ang II concentrations were determined using commercially available ELISA kit (Cayman Chemical, Ann Arbor, MI, USA, n=4) (24–25).
Determination of mRNA expressions
Quantitative real-time reverse transcriptase- polymerase chain reaction (RT-PCR, n=6) was used to determine changes in renal mRNA expressions of PRR, TNF-α, and COX-2. The RNA was extracted using Trizol (Invitrogen, Carlsbad, CA). Reverse transcription of the RNA was performed by the first strand cDNA synthesis kit (Bio-Rad, Hercules, CA). The PCR was analyzed using SYBR Green Super mix (Bio-Rad). Primer sequences were as follows: PRR, forward sequence 5′-TGGCCTATACCAGGAGATCG-3′; reverse sequence 5′- AATAGGTTGCCCACAGCAAG-3′; TNFα forward, 5′-ACTCCCAGAAAAGCAAGCAA-3′, reverse, 5′-CGAGCAGGAATGAGAAGAGG-3′; COX-2 forward, 5′-GTG TGA GTG GTA GCC AGC AA-3′, reverse, CCC ACA GGA GGA TCT GAA AA and β-actin, forward sequence 5′-AGCCATGTACGTAGCCATCC-3′, reverse sequence 5′-ACCCTCATAGATGGGCACAG -3′. RT-PCR was performed using iCycler (Bio-Rad) and threshold cycle number was determined using iCycler software version 3.0 (Bio-Rad). Reactions were performed in triplicate and threshold cycle numbers were averaged. The mRNA results for specific target genes were calculated with normalization to β-actin mRNA.
Western blot analysis
Antibodies to PRR (anti-ATP6IP2/ab40790, Abcam, Cambridge, MA, USA), TNF-α (Santa Cruz biotechnology, Inc., Santa Cruz, CA, USA), COX-2 (Novus bilogicals, Colorado, USA), NF-κB p65 (Abcam, Cambridge, MA, USA) and MCP-1 (Cell Signaling, USA) were used in the Western blot (n=4) as previously described (12). Protein expression was normalized to β-actin protein.
Renal PRR and Collagen I Immunostaining
Left and right kidney tissue blocks were prepared and deparaffinized as described previously (12). Sections (n=4) were incubated overnight at 4°C with primary antibody directed against PRR monoclonal antibody (1:100 dilution; anti-ATP6IP2/ab40790; Abcam, Cambridge, MA, USA), collagen I (1:100 dilution; Abcam, Cambridge, MA, USA). On the following day, sections were incubated for 1hr with secondary antibody at room temperature. A negative control (IgG) was included by omitting the primary antibody. The immunostaining images were captured by light microscopy using a Q-imaging Micropublisher 5.0 RTV camera coupled to a Zeiss Axiophot microscopy (Carl Zeiss, Jena, Germany).
Urinary Albumin Creatinine Ratio (UACR)
Urinary albumin was determined by using a sensitive rat albumin enzyme immunoassay (EIA) kit (Cayman Chemical, Ann Harbor, MI, USA (n=4). Urine creatinine was determined by creatinine assay kit (Cayman). UACR was calculated and presented as albumin in micrograms divided by creatinine in milligrams.
Statistical Analysis
Comparisons among different treatment groups were assessed by ANOVA followed by a Tukey test for post hoc comparisons. Data is expressed as mean ± SE. P<0.05 is considered statistically significant.
Acknowledgments
Grants
This study was supported by National Institutes of Health grants DK078757, DK114875 and HL091535 to H.M. Siragy.
Footnotes
Disclosure
None.
References
- 1.Prieto MC, Botros FT, Kavanagh K, Navar LG. Prorenin receptor in distal nephron segments of 2-kidney, 1-clip goldblatt hypertensive rats. Ochsner J. 2013 Spring;13(1):26–32. [PMC free article] [PubMed] [Google Scholar]
- 2.Quadri S, Siragy HM. Regulation of (pro)renin receptor expression in mIMCD via the GSK-3beta-NFAT5-SIRT-1 signaling pathway. Am J Physiol Renal Physiol. 2014 Sep 1;307(5):F593–600. doi: 10.1152/ajprenal.00245.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, et al. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Renal Physiol. 2015 Jul 1;309(1):F48–56. doi: 10.1152/ajprenal.00126.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002 Jun;109(11):1417–27. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Siragy HM. Glucose promotes the production of interleukine-1beta and cyclooxygenase-2 in mesangial cells via enhanced (Pro)renin receptor expression. Endocrinology. 2009 Dec;150(12):5557–65. doi: 10.1210/en.2009-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lerman LO, Textor SC, Grande JP. Mechanisms of tissue injury in renal artery stenosis: ischemia and beyond. Prog Cardiovasc Dis. 2009 Nov-Dec;52(3):196–203. doi: 10.1016/j.pcad.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chade AR, Herrmann J, Zhu X, Krier JD, Lerman A, Lerman LO. Effects of proteasome inhibition on the kidney in experimental hypercholesterolemia. J Am Soc Nephrol. 2005 Apr;16(4):1005–12. doi: 10.1681/ASN.2004080674. [DOI] [PubMed] [Google Scholar]
- 8.Navar LG, Zou L, Von Thun A, Tarng Wang C, Imig JD, Mitchell KD. Unraveling the Mystery of Goldblatt Hypertension. News Physiol Sci. 1998 Aug;13:170–6. doi: 10.1152/physiologyonline.1998.13.4.170. [DOI] [PubMed] [Google Scholar]
- 9.Chade AR, Rodriguez-Porcel M, Grande JP, Krier JD, Lerman A, Romero JC, et al. Distinct renal injury in early atherosclerosis and renovascular disease. Circulation. 2002 Aug 27;106(9):1165–71. doi: 10.1161/01.cir.0000027105.02327.48. [DOI] [PubMed] [Google Scholar]
- 10.Kaneshiro Y, Ichihara A, Takemitsu T, Sakoda M, Suzuki F, Nakagawa T, et al. Increased expression of cyclooxygenase-2 in the renal cortex of human prorenin receptor gene-transgenic rats. Kidney Int. 2006 Aug;70(4):641–6. doi: 10.1038/sj.ki.5001627. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen G. Increased cyclooxygenase-2, hyperfiltration, glomerulosclerosis, and diabetic nephropathy: put the blame on the (pro)renin receptor? Kidney Int. 2006 Aug;70(4):618–20. doi: 10.1038/sj.ki.5001723. [DOI] [PubMed] [Google Scholar]
- 12.Matavelli LC, Huang J, Siragy HM. In vivo regulation of renal expression of (pro)renin receptor by a low-sodium diet. Am J Physiol Renal Physiol. 2012 Dec 15;303(12):F1652–7. doi: 10.1152/ajprenal.00204.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krebs C, Hamming I, Sadaghiani S, Steinmetz OM, Meyer-Schwesinger C, Fehr S, et al. Antihypertensive therapy upregulates renin and (pro)renin receptor in the clipped kidney of Goldblatt hypertensive rats. Kidney Int. 2007 Sep;72(6):725–30. doi: 10.1038/sj.ki.5002408. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez AA, Luffman C, Bourgeois CR, Vio CP, Prieto MC. Angiotensin II-independent upregulation of cyclooxygenase-2 by activation of the (Pro)renin receptor in rat renal inner medullary cells. Hypertension. 2013 Feb;61(2):443–9. doi: 10.1161/HYPERTENSIONAHA.112.196303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension. 2011 Mar;57(3):594–9. doi: 10.1161/HYPERTENSIONAHA.110.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol. 2007 Jun;18(6):1789–95. doi: 10.1681/ASN.2006091062. [DOI] [PubMed] [Google Scholar]
- 17.Waugh WH. Angiotensin II: local renal effects of physiological increments in concentration. Can J Physiol Pharmacol. 1972 Jul;50(7):711–6. doi: 10.1139/y72-103. [DOI] [PubMed] [Google Scholar]
- 18.Otsuka Y, Carretero OA, Albertini R, Binia A. Angiotensin and sodium balance: their role in chronic two-kidney Goldblatt hypertension. Hypertension. 1979 Jul-Aug;1(4):389–96. doi: 10.1161/01.hyp.1.4.389. [DOI] [PubMed] [Google Scholar]
- 19.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol. 2007 Jan;292(1):F330–9. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol. 1998 Jan;274(1 Pt 2):F148–55. doi: 10.1152/ajprenal.1998.274.1.F148. [DOI] [PubMed] [Google Scholar]
- 21.Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest. 2006 Feb;116(2):288–96. doi: 10.1172/JCI27699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan S, Fox J, Mitchell KD, Navar LG. Angiotensin and angiotensin converting enzyme tissue levels in two-kidney, one clip hypertensive rats. Hypertension. 1992 Dec;20(6):763–7. doi: 10.1161/01.hyp.20.6.763. [DOI] [PubMed] [Google Scholar]
- 23.Siragy HM, Inagami T, Carey RM. NO and cGMP mediate angiotensin AT2 receptor-induced renal renin inhibition in young rats. Am J Physiol Regul Integr Comp Physiol. 2007 Oct;293(4):R1461–7. doi: 10.1152/ajpregu.00014.2007. [DOI] [PubMed] [Google Scholar]
- 24.Quadri S, Prathipati P, Jackson DW, Jackson KE. Haemodynamic consequences of recurrent insulin-induced hypoglycaemia. Clin Exp Pharmacol Physiol. 2014 Jan;41(1):81–8. doi: 10.1111/1440-1681.12183. [DOI] [PubMed] [Google Scholar]
- 25.Quadri S, Prathipati P, Jackson D, Jackson K. Regulation of heme oxygenase-1 induction during recurrent insulin induced hypoglycemia. 2014 Jun 23;2(2):6. [Google Scholar]