

Abstract
Background
The pathogenesis of CKD-associated anemia is multifactorial and includes decreased production of erythropoietin, iron deficiency, inflammation and erythropoietin resistance. To better understand the trajectory of these parameters we described temporal trends in hemoglobin (Hb), ferritin, transferrin saturation, C-reactive protein (CRP) and darbepoetin dosing in the Trial to Reduce cardiovascular Events with Aranesp Therapy (TREAT).
Methods
We performed a post-hoc analysis of 4,038 participants in TREAT. Mixed effects linear regression models were used to determine the trajectory of parameters of interest prior to ESRD. Likelihood ratio tests were used to test for overall differences in biomarker values and differences in trajectories between those who did and did not develop ESRD.
Results
Hb declined precipitously in the year prior to the development of ESRD (irrespective of treatment assignment), and was on average 1.15 g/dL (95%CI −1.26 to −1.04) lower in those that developed ESRD vs. those that did not, at the time of ESRD/end of follow-up. Simultaneously, the mean darbepoetin dose and CRP concentration increased, while serum ferritin and transferrin saturations were greater than 140 mcg/L and 20% respectively.
Conclusions
Our analyses provide descriptive insights regarding the temporal changes of Hb, darbepoetin dose and related parameters as ESRD approaches in participants of TREAT. Hb declined as much as 1–2 years prior to the development of ESRD, without biochemical evidence of iron deficiency. The most precipitous decline occurred in the months immediately prior to ESRD, despite administration of escalating doses of darbepoetin and in parallel with an increase in CRP.
Keywords: Anemia, Hemoglobin, CRP, ESRD, Type 2 Diabetes Mellitus, Chronic Kidney Disease
Introduction
Chronic kidney disease (CKD) affects approximately 11.5% of the general population[1] and 40% of those with self-reported diabetes mellitus in the United States.[2] Anemia (defined as hemoglobin <12 g/dL in men and <11 g/dL in women) is a common comorbid condition in diabetic patients with CKD and is estimated to affect 22% of patients with CKD Stage 3 and 52% with CKD Stage 4.[3] The presence of anemia in CKD is associated with an increased risk of cardiovascular events, all-cause mortality,[4] and progression to end-stage renal disease (ESRD).[5]
Progressive loss of renal function is associated with inadequate erythropoiesis and lowering of the blood hemoglobin concentration (Hb). Other factors that may contribute to the development of anemia as CKD progresses include iron deficiency and inflammation.[6] The introduction of recombinant erythropoietin (EPO) heralded a new era in nephrology, as the requirement for repeated blood transfusions was negated, and Hb could be maintained at near normal levels. [7,8] However, several studies reported that targeting ‘normal’ Hb levels was not associated with benefit, but rather with potential harm,[9–11] while a lack of responsiveness to exogenous EPO is associated with greater risk of CV events and death.[12] A contemporary approach that examines the sequential changes of Hb decline relative to markers of iron deficiency, inflammation and darbepoetin dosing may provide important clinical insights, building on what is already known in this area.
The repeated scheduled ascertainment of Hb, and other biomarkers, in the Trial to Reduce cardiovascular Events with Aranesp Therapy (TREAT)[11] provided an opportunity to describe the temporal trends of Hb prior to the occurrence of ESRD. Furthermore, comparisons to a group of participants who did not develop ESRD (both in the presence and absence of randomized darbepoetin vs. placebo therapy) was possible.
Methods
Study design and population
In these post-hoc and descriptive analyses, we evaluated longitudinal changes in biomarkers of interest (hemoglobin, ferritin, transferrin saturation, C-reactive protein [CRP], creatinine, eGFR, urine protein/creatinine ratio [PCR] and average monthly darbepoetin dose) in participants of the TREAT study. The design and original results of TREAT (NCT00093015) have been published.[11,13] Briefly, TREAT was a prospective, double-blind, randomized controlled trial of darbepoetin alfa versus placebo for the treatment of anemia in 4,038 participants with type 2 diabetes mellitus (T2DM), eGFR of 20–60 mL/min/1.73m2 according to the 4-variable MDRD (Modification of Diet in Renal Disease) Study equation, Hb <11.0 g/dL, and transferrin saturation >15%. The initial dose of darbepoetin in the active treatment arm was 0.75 mcg/kg, with doses adjusted monthly according to a computer-based algorithm to target Hb at 13 g/dL. In the placebo arm, if Hb fell below 9g/dL, ‘rescue’ therapy with a single dose of 0.45 mcg/kg darbepoetin was given (Supplementary Figure 1). Per protocol, all participants were regularly assessed (and treated as needed) for serum markers of iron sufficiency (ferritin and transferrin saturation; Supplementary Figure 2). All participants gave written informed consent in the primary trial and the serum samples used in this analysis (Partners IRB 2005P000170).
Statistical Analyses
A contemporary descriptive analytic approach was employed, whereby temporal trends in biomarkers were plotted by working retrospectively from the time of an event of interest (i.e. ESRD). In these analyses, the exposure variable for those that developed ESRD was the date of the confirmed ESRD event subtracted from the date of the biomarker measurement (i.e. time prior to ESRD). ESRD (n=668) was defined as initiation of renal replacement therapy (RRT; sustained for at least 30 days), initiation of RRT with death within 30 days, a physician recommendation to initiate RRT with documented participant refusal, or receipt of a kidney transplant. The outcome variable was the measured biomarker of interest. Owing to right-skewed distributions, log-transformation was performed for ferritin, CRP, urine PCR and creatinine, and corresponding estimates were produced using geometric means. The CRP assay had a lower limit of detection of 3.0mg/L; for those with concentrations below this limit, a value of 1.5mg/L was imputed for these analyses.
In order to provide meaningful comparisons, similar analyses were performed in a comparator group of TREAT participants who did not experience ESRD during follow-up (n=3,370). For these individuals, time prior to end of follow-up was similarly calculated as date of the end of follow-up (EOF) subtracted from the date of the biomarker measurement. To ensure the follow-up of this group was comparable to those that developed ESRD (i.e. participants entered analyses at randomization, were followed for the same average duration, and EOF was not induced by death), a modified EOF (mEOF) was created by shortening the exposure duration by 310 days. Therefore, the average duration between randomization and end of follow-up was identical (563 days) for both subgroups. In exploratory analyses, a propensity-score algorithm (based on baseline age, gender, race, eGFR and urine PCR) was used to select non-ESRD comparators that were similar with respect to those that went on to develop ESRD.
Baseline variables were summarized such that continuous variables were examined graphically and recorded as means (± standard deviations) for normally distributed data, or medians (with interquartile ranges) for non-normally distributed data. Categorical variables were examined by frequency distribution and recorded as proportions. Tests for difference according to ESRD were conducted using t-tests, Wilcoxon Rank Sum tests and the Chi-square test for trend for continuous normal, continuous non-normal, and categorical data, respectively.
Average values of biomarker measurements prior to ESRD/mEOF were estimated through the use of a mixed effects linear regression model, using participant ID’s as random effects. To allow for potentially non-linear changes over time, days prior to ESRD/mEOF was modeled using restricted cubic spline terms with four knots. The relationship between time and the average biomarker values were modeled using interaction terms between the group indicator (ESRD vs. not) and the cubic spline terms representing days prior to ESRD/mEOF. From this model, estimates of the average difference between those that developed ESRD vs. those that did not were produced at 0, 1, 2, and 3 years prior to the ESRD/mEOF. Likelihood ratio tests were performed comparing the full model (with group variables and interaction terms) to: 1) models without any group variable or interaction terms to perform global tests for differences in biomarker values between groups; 2) models without any interaction terms to perform global tests for differences in biomarker trajectories over time. Estimated time of divergence of biomarkers of interest (ESRD vs. not) was defined as the latest time at which the difference in point estimates became significant at α=0.01. A pre-specified sub-group analysis was performed according to randomized treatment arm and baseline use of angiotensin converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB).
Recognizing that the trajectories of biomarkers changed most markedly during the last six months prior to ESRD/mEOF, the following exploratory analyses were performed to explore potential associations with clinical variables according to development of ESRD or not: use of a more stringent biochemical definition of iron sufficiency (transferrin saturation >30% and ferritin >200 mcg/L); the proportion of participants that developed a GI bleed. For all other analyses, nominal 2-sided P values of <0.05 were considered statistically significant. All analyses were performed using STATA 14 (College Station, Tex., USA).
Results
Baseline Characteristics
The primary cohort consisted of 4,038 individuals (57% women) with a median age of 68 years.[11] At baseline, those who ultimately developed ESRD were more likely to be younger, male and black, and to have a longer duration of T2DM and higher HbA1c compared with those that did not. Those that developed ESRD were more likely to have lower baseline Hb, serum albumin, eGFR, and to have higher ferritin, transferrin saturation, CRP, creatinine, urine protein/creatinine ratio and more likely to have a history of acute kidney injury (Table 1).
Table 1.
Baseline Characteristics of the Cohort According to Development of End-Stage Renal Disease
| ESRD (N=668) |
Non-ESRD (N=3,370) |
Pa | |
|---|---|---|---|
| Age (years) | 63.6 ±10.6 | 68.1 ±10.5 | <0.001 |
| Male (%) | 54.8 | 40.4 | <0.001 |
| Race (%) | <0.001 | ||
| Black | 27.7 | 18.7 | |
| Hispanic | 15.7 | 12.8 | |
| Other | 2.7 | 2.9 | |
| White | 53.9 | 65.6 | |
| Hemoglobin (g/dL) | 10.2 ±1.0 | 10.4 ±1.0 | <0.001 |
| Ferritin (mcg/L) | 175 [95–315] | 126 [63–248] | <0.001 |
| Transferrin Saturation (%) | 25.5 ±9.9 | 24.0 ±9.3 | <0.001 |
| Iron therapy (%) | 46.0 | 43.7 | 0.29 |
| Treatment (Darbepoetin vs. Placebo, %) | 50.6 | 49.7 | 0.66 |
| Creatinine (mg/dL) | 2.5 [2.1–3.0] | 1.8 [1.5–2.2] | <0.001 |
| eGFR (mL/min/1.73m2) | 23 [19–29] | 33 [26–42] | <0.001 |
| Urine Pr/Cr ratio | 2.7 [0.9–5.7] | 0.3 [0.1–1.1] | <0.001 |
| CRP (mg/L)b | 3.3 [Ub–7.3] | Ub [Ub–6.5] | 0.01 |
| Albumin (g/dL) | 3.7 ±0.5 | 4.0 ±0.4 | <0.001 |
| History of AKI (%) | 13 | 9 | <0.001 |
| Duration of T2DM (years) | 16 [10–22] | 15 [8–22] | <0.001 |
| HbA1c (%) | 7.2 [6.4–8.2] | 6.9 [6.2–7.9] | <0.001 |
| ACEi/ARB therapy (%) | 78.7 | 80.0 | 0.45 |
P value for difference; significance testing was by t test or Wilcoxon rank sum test for continuous variables or chi-squared test for categorical variables. Continuous variables are presented as means ±standard deviation if normally distributed or medians [25th to 75th percentiles] if non-normally distributed. Abbreviations: eGFR, estimated glomerular filtration rate; Pr/Cr, protein to creatinine ratio; CRP, C-reactive protein; U, undetectable; AKI, acute kidney injury; T2DM, type 2 diabetes mellitus; HbA1c, glycated hemoglobin; ACEi/ARB, angiotensin converting enzyme inhibitor/angiotensin receptor blocker
The lower limit of detection for baseline CRP was 3 mg/L
Trajectory of Hemoglobin
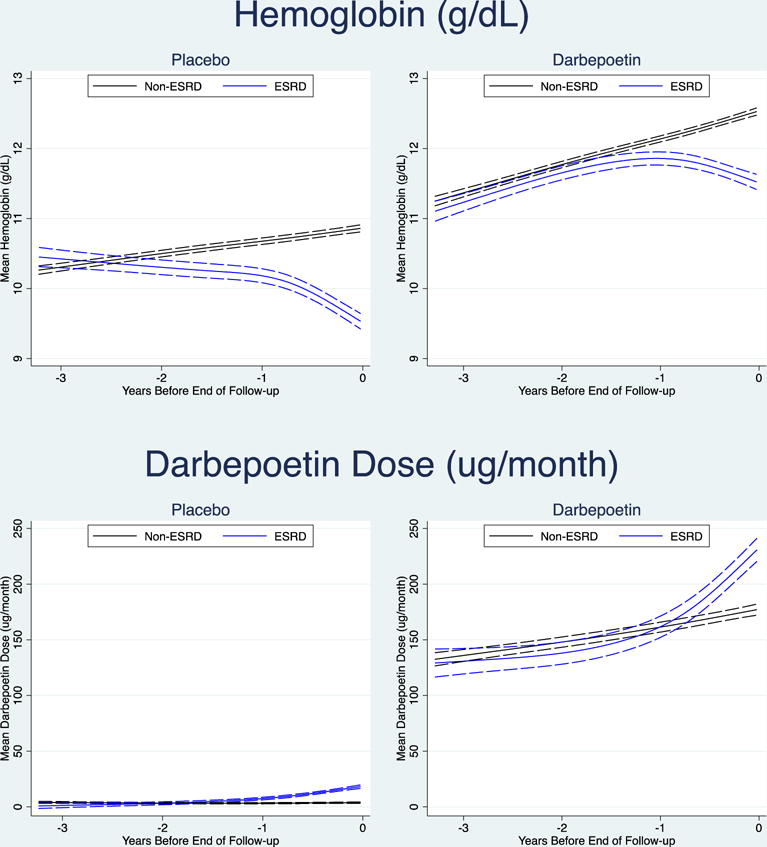
In the placebo arm, three years prior to the development of ESRD/mEOF, Hb was similar in those that ultimately developed ESRD compared with those who did not (10.4 g/dL vs. 10.3g/dL; p=0.10; Table 2). However, from the outset, the overall trajectory of Hb in those who went on to develop ESRD was downwards, with a more precipitous decline evident in the year prior to the ESRD event. For those who did not develop ESRD, Hb modestly increased over time.
Table 2.
Comparison of Biomarkers During Follow-Up According to Development of End-Stage Renal Disease
| Comparison of Biomarkers in those that develop ESRD (n=668) vs. Not (n=3,370) | |||||||
|---|---|---|---|---|---|---|---|
| Time Prior to ESRD/Modified End of Follow-up | P-values | ||||||
| Biomarker | 3 years | 2 years | 1 year | Time of ESRD/mEnd of Follow-up | Estimated Time of Divergencea | Equality | Interaction |
| Placebo Group | |||||||
| ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ||||
| Hemoglobin (g/dL) | 10.4 vs 10.3; p=0.10 | 10.3 vs 10.5; p=0.001 | 10.2 vs 10.7; p<0.001 | 9.5 vs 10.9; p<0.001 | 2.1 years | <0.001 | <0.001 |
| Darbepoetin (Mean Dose in mcg/month) | 1.3 vs 3.9; p=0.019 | 3.2 vs 3.4; p=0.82 | 7.4 vs 3.3; p<0.001 | 18.5 vs 3.8; p<0.001 | 1.4 years | <0.001 | <0.001 |
| Darbepoetin Dose Difference ([ESRD – non-ESRD] in mcg/month) | −2.6 | −0.2 | +4.1 | +14.7 | 1.4 years | <0.001 | <0.001 |
| Ferritin (mcg/L)b | 147 vs 105; p<0.001 | 148 vs 114; p<0.001 | 172 vs 132; p<0.001 | 204 vs 153; p<0.001 | >3 years | <0.001 | 0.36 |
| Transferrin Saturation (%) | 24.9 vs 22.6; p=0.007 | 24.5 vs 24.0; p=0.3 | 26.1 vs 25.2; p=0.07 | 24.3 vs 26.0; p=0.014 | N/A | <0.001 | <0.001 |
| CRP (mg/L)b, c | 3.7 vs 3.0; p=0.05 | 3.9 vs 3.2; p=0.007 | 3.9 vs 3.5; p=0.06 | 5.8 vs 3.7; p<0.001 | 0.6 years | <0.001 | 0.006 |
| Darbepoetin Group | |||||||
| ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ESRD vs. not (P-difference) | ||||
| Hemoglobin (g/dL) | 11.2 vs 11.4; p=0.05 | 11.7 vs 11.8; p=0.036 | 11.9 vs 12.1; p<0.001 | 11.5 vs 12.5; p<0.001 | 1.6 years | <0.001 | <0.001 |
| Darbepoetin (Mean Dose in mcg/month) | 131 vs 136; p=0.42 | 138 vs 148; p=0.08 | 162 vs 161; p=0.95 | 232 vs 177; p=<0.001 | 0.6 years | <0.001 | <0.001 |
| Darbepoetin Dose Difference ([ESRD – non-ESRD] in mcg/month) | −5 | −10 | +1 | +55 | 0.6 years | <0.001 | <0.001 |
| Ferritin (mcg/L)b | 92 vs 78; p<0.001 | 119 vs 98; p<0.001 | 151 vs 122; p<0.001 | 179 vs 150; p=0.005 | >3 years | 0.002 | 0.73 |
| Transferrin Saturation (%) | 22.0 vs 20.9; p=0.3 | 25.6 vs 25.7; p=0.94 | 29.8 vs 29.8; p=0.94 | 29.5 vs 32.2; p=0.002 | 0.2 years | 0.008 | 0.005 |
| CRP (mg/L)b, c | 2.9 vs 3.2; p=0.35 | 3.5 vs 3.1; p=0.14 | 3.6 vs 3.4; p=0.41 | 5.3 vs 3.7; p<0.001 | 0.4 years | <0.001 | <0.001 |
P values refer to the difference in average biomarker concentrations between those that did and do not develop ESRD at specific time points (time of event or end of follow-up; 1 year prior; 2 years prior; or 3 years prior). Equality tests the null hypothesis that the curves are superimposable (i.e. differences in biomarkers between groups); P for Interaction tests the null hypothesis that the curves are parallel (i.e. differences in biomarker trajectories over time).
Estimated time of divergence of biomarkers of interest (developed ESRD vs. not) was defined as the latest time at which the difference in point estimates became significant at α=0.01.
Groups are summarized and compared using geometric means
Individuals with undetectable CRP (≤3 mg/L) were assumed to have a CRP=1.5 mg/L
In the treatment arm, Hb initially increased for both those who did and did not go on to develop ESRD. However, approximately 1.6 years prior to the ESRD/mEOF, the trajectories were significantly different, such that Hb continued to increase in those who did not develop ESRD, but declined in those who did. This decline was more marked in the year prior to ESRD, such that at the time of ESRD/mEOF the Hb was lower (1.0 g/dL; 95%CI −1.1 to −0.9 g/dL) in those with ESRD (Figure 1 and Table 2). There was no evidence of difference in patterns according to baseline use of ACEi/ARB medications (Supplementary Figure 3).
Figure 1.
Trajectory of Hemoglobin and Darbepoetin Dose According to Development of ESRD
Trajectory of Darbepoetin Dosing
In the placebo arm (where, per protocol, only ‘rescue’ doses of darbepoetin were provided), in the year prior to ESRD/mEOF there was an increase in the average monthly dose of darbepoetin in those who went on to develop ESRD, compared with those who did not. The trajectory in those that did not develop ESRD remained relatively flat. The estimated time of divergence of the dosing trajectories was ~1.4 years prior to the time of ESRD/mEOF (Figure 1 and Table 2).
In the treatment arm, the average monthly dose of administered darbepoetin (determined by a predefined algorithm) increased steadily in all participants between three years and one year prior to the development of ESRD/mEOF. However, for those that ultimately progressed to ESRD, there was a notable increase in the darbepoetin dosing during the year prior to this event, with significant divergence of the dose trajectory occurring ~0.6 years prior to this event (Figure 1 and Table 2).
Trajectory of Ferritin
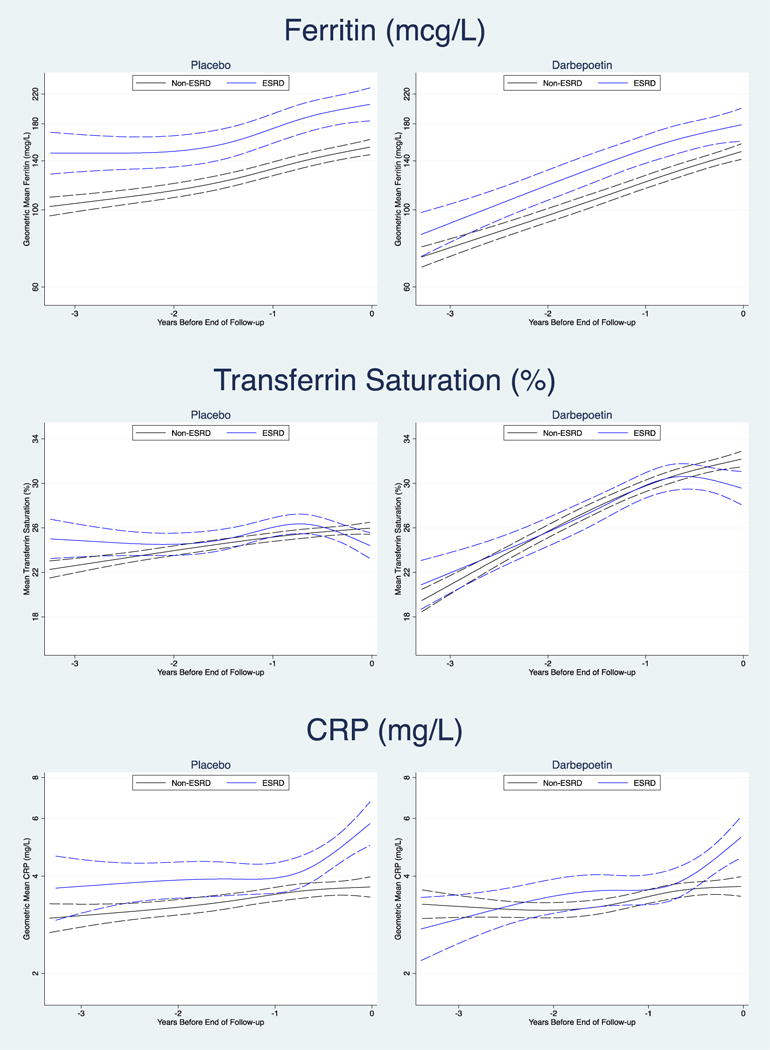
The trajectory of serum ferritin was similar for participants in both the placebo and treatment arms, such that ferritin increased over time for both those who did and did not develop ESRD, but was always significantly higher in the ESRD group. Overall, ferritin was relatively lower at each time point examined in the darbepoetin arm as compared with the placebo arm (Figure 2 and Table 2).
Figure 2.
Trajectory of Serum Ferritin, Transferrin Saturation and C-Reactive Protein According to Development of ESRD
Trajectory of Transferrin Saturation
In the placebo arm, transferrin saturations were marginally higher in those who developed ESRD at three years prior to ESRD/mEOF, but subsequently the trajectories were overlapping until the months immediately preceding ESRD/mEOF, when the transferrin saturations declined in those that developed ESRD. In the darbepoetin arm, the trajectories were overlapping and increased over time for both those who did and did not develop ESRD. However, in the months immediately prior to ESRD/mEOF, the transferrin saturation trajectory declined in those that went on to develop ESRD (Figure 2 and Table 2).
Trajectory of C-Reactive Protein
In the placebo arm CRP was marginally higher in those who developed ESRD during the period of three years to one year prior to ESRD/mEOF. After this point CRP began to markedly increase in those who went on to develop ESRD, with the trajectory significantly diverging ~0.6 years prior to the ESRD event. In the darbepoetin arm, three years prior to the development of ESRD/mEOF, CRP concentrations were similar in those who did and did not develop ESRD (p-difference=0.35). During the initial follow-up, CRP increased in both groups, but for those that developed ESRD a more marked rise was evident in the year prior to the ESRD event, with the trajectories significantly diverging ~0.4 years prior (Figure 2 and Table 2).
Trajectory of Serum Creatinine, eGFR and urine protein/creatinine ratio
The trajectories of serum creatinine and eGFR followed expected patterns for those who did and did not develop ESRD (Supplementary Table 1 and Supplementary figures 4 and 5). In contrast, there were no notable changes in the trajectory of urine protein/creatinine ratio in the last six months in either those who did or did not develop ESRD (Supplementary Table 1 and Figure 6).
Exploratory Analyses
Recognizing that there were baseline differences between the groups that did and not develop ESRD, additional analyses were performed utilizing a non-ESRD comparator group that was matched for baseline characteristics. Using this refined comparator group (n=670), the overall trajectories of hemoglobin, darbepoetin dose, transferrin saturation and ferritin were similar to those obtained from the non-ESRD comparator group in the original analyses (Supplementary Table 2).
In the year prior to ESRD/mEOF, 37% of those who developed ESRD and 32% of those that did not (p-difference=0.01) had simultaneously transferrin saturations >30% and ferritin >200 mcg/L on at least one occasion. This occurred despite a greater proportion of GI bleeds in the 6 months prior to ESRD/mEOF (19 of the 28 GI bleeding adverse events [68%] in those that developed ESRD vs. 19 of the 91 [21%] in those that did not develop ESRD occurred during this period).
Discussion
Our contemporary analytic approach (examining the temporal changes of Hb relative to biomarkers of iron stores, darbepoetin dosing and inflammation) provides incremental knowledge regarding the changes in Hb in individuals with CKD, anemia and T2DM. We illustrate that these parameters follow markedly different trajectories in participants who did and did not develop ESRD. Specifically, in those who developed ESRD, Hb became progressively lower, despite biochemical measures suggesting iron sufficiency and the administration of increasing darbepoetin doses, while CRP was noted to increase.
The primary regulator of erythrocyte production is EPO, a 34 kDa glycoprotein produced predominantly by renal interstitial fibroblasts, that functions primarily to stimulate erythropoiesis in the bone marrow in response to tissue hypoxia.[14] Prior reports indicate that the development of CKD-associated anemia is more likely related to insufficient increases in EPO production, rather than an absolute decline in circulating EPO levels.[15] This relationship provides challenges in the interpretation of EPO levels in CKD-associated anemia and limits their diagnostic utility; therefore, serum EPO levels were not measured as part of the TREAT study.
TREAT also provided an important opportunity to examine trends in Hb in the setting of exogenous administration of EPO derivatives. As expected, our analyses of the active therapy arm indicate that administration of darbepoetin resulted in an increase in Hb during the early phases of treatment. However, as CKD progressed over time, Hb started to decline in those who developed ESRD, and continued to decrease despite escalating doses of darbepoetin. A different pattern was noted in the placebo arm – in participants who developed ESRD the Hb trajectory was downward from the beginning, and again occurred despite administration of escalating ‘rescue’ doses of darbepoetin. In contrast, in those that did not develop ESRD, Hb was relatively stable over time. Prior data from our group examined participants in the placebo arm of TREAT, reporting that lower baseline Hb, lower eGFR and higher proteinuria were the major predictors for requiring ≥5 ‘rescue’ doses of darbepoetin. Furthermore, compared with participants that did not receive any ‘rescue’ doses, those that received ≥5 were more likely to progress to ESRD.[16] Together these findings highlight that, in many cases, administration of increasing doses of darbepoetin (often in excess of 200 mcg per month) as ESRD approaches appears to be insufficient to prevent a continued decline in Hb.
The most common reason for hypo-responsiveness to exogenous EPO in CKD patients is iron deficiency, suggested by a serum ferritin concentration <100 mcg/L and/or transferrin saturations <20%.[17] Results from NHANES suggest that up to two-thirds of patients with moderate CKD may have insufficient iron stores as defined by these criteria.[18] In our analyses we noted that serum ferritin increased over time in both those that did and did not develop ESRD, which may partially relate to protocol driven iron supplementation. However, it was also apparent that ferritin was consistently higher in those that developed ESRD compared with those that did not, which could suggest a greater degree of underlying inflammation. In this regard, it is notable that hepcidin (a major regulator of systemic iron homeostasis via its inhibitory effect on ferroportin), is also up regulated by inflammatory cytokines[19] and could contribute to anemia development. In TREAT all participants were assessed and treated for iron deficiency according to a predefined study algorithm. Interestingly, we noted a decline in transferrin saturation in the months prior to development of ESRD that corresponded to a greater observed frequency of GI bleeds in those that developed ESRD (vs. not). However, the overall event rate was relatively small, while overall a greater proportion of ESRD participants actually met a more stringent biochemical definition of iron sufficiency during this period. However, we recognize that biochemical measures of iron stores may under-diagnosis deficiency at the bone marrow level,[20] and therefore we cannot exclude the possibility of a greater prevalence of iron deficiency in these participants.
The presence of inflammation is postulated to be a major risk factor for EPO hypo-responsiveness, with several studies reporting an association with the presence of elevated inflammatory biomarkers.[21,22] Indeed, prior analyses of TREAT by our group have reported that elevated levels of CRP (>3 mg/L) were present in 48% of participants at baseline,[23] and that CRP was marginally higher at baseline in those who had an impaired Hb response to darbepoetin.[12] Our current analyses extend these findings by examining all available CRP measurements during follow-up. We report that CRP concentrations increased markedly in participants who went on to develop ESRD in the months prior to this event, compared with those that did not. Although causality cannot be proved from our data, the temporal relationship with the decline in Hb in those that developed ESRD, in addition to increasing ferritin (also an inflammatory marker), provides additional support for the hypothesis of EPO hypo-responsiveness secondary to the presence of inflammation.
The strengths of this study include the utilization of repeated laboratory measures collected in the setting of a large, randomized controlled trial. Concerns related to potential prescriber bias are limited by the fact that a computer-based dosing algorithm was utilized for all participants. However, there are limitations to our report. Our analyses are primarily descriptive and do not allow for adjustment for potential variables that could potentially alter these measurements over the course of the study. For example, hospitalizations may be more likely to occur in those that developed ESRD, thereby contributing to lower Hb from inpatient stays, while also contributing to missing data from outpatient study visits. The follow up time for those who did not develop ESRD was truncated in order to be of similar duration to those that developed ESRD. While this may have reduced the overall number of available measurements, truncation ensured that the exposure time captured in both subgroups did not include any fatal events. The progression of secondary hyperparathyroidism or undetected/inadequately treated iron deficiency in those that develop ESRD may contribute to EPO hyporesponsiveness[24], but unfortunately parathyroid hormone and data related to the trajectory of intravenous iron therapy were not available. Similarly, aside from relative EPO insufficiency, other potential contributors to the development of anemia with progressive CKD may relate to accumulation of as yet unidentified uremic toxin(s). Finally, as this study was performed in participants with CKD, anemia and T2DM, it is unclear if the results are generalizable to patients without these comorbidities.
In conclusion, we report that Hb declined rapidly in the months prior to the development of ESRD, coincident with a rise in CRP. This pattern occurred in the absence of biochemical measures of iron deficiency and despite escalating doses of darbepoetin. This constellation of findings supports the contribution of an increasing inflammatory state to the pathogenesis of EPO hypo-responsiveness as CKD progresses to ESRD. Incorporation of this analytic approach, which provides additional insight to the temporal changes in biomarkers approaching an event of interest, may help to uncover further pathophysiologic changes that contribute to the progression of CKD.
Supplementary Material
Acknowledgments
Support: Dr. Mc Causland is supported by the National Institute of Diabetes and Digestive and Kidney Diseases grant K23DK102511. He has served as a consultant for GSK.
Footnotes
Conflicts of Interest
Financial Disclosure: The TREAT Study was funded by Amgen. This analysis was conducted independently by the authors and used the data set held at the Brigham & Women’s Hospital; the authors designed and conducted all analyses described herein and were solely responsible for the drafting and editing of this manuscript.
References
- 1.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009 May 5;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saran R, Li Y, Robinson B, Ayanian J, Balkrishnan R, Bragg-Gresham J, et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis. 2015 Jun;65:A7. doi: 10.1053/j.ajkd.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Achkar TM, Ohmit SE, McCullough PA, Crook ED, Brown WW, Grimm R, et al. Higher prevalence of anemia with diabetes mellitus in moderate kidney insufficiency: The Kidney Early Evaluation Program. Kidney Int. 2005 Apr;67:1483–1488. doi: 10.1111/j.1523-1755.2005.00226.x. [DOI] [PubMed] [Google Scholar]
- 4.Vlagopoulos PT, Tighiouart H, Weiner DE, Griffith J, Pettitt D, Salem DN, et al. Anemia as a risk factor for cardiovascular disease and all-cause mortality in diabetes: the impact of chronic kidney disease. JASN. 2005 Nov;16:3403–3410. doi: 10.1681/ASN.2005030226. [DOI] [PubMed] [Google Scholar]
- 5.Mohanram A, Zhang Z, Shahinfar S, Keane WF, Brenner BM, Toto RD. Anemia and end-stage renal disease in patients with type 2 diabetes and nephropathy. Kidney Int. 2004 Sep;66:1131–1138. doi: 10.1111/j.1523-1755.2004.00863.x. [DOI] [PubMed] [Google Scholar]
- 6.Johnson DW, Pollock CA, Macdougall IC. Erythropoiesis-stimulating agent hyporesponsiveness. Nephrology (Carlton) 2007 Aug;12:321–330. doi: 10.1111/j.1440-1797.2007.00810.x. [DOI] [PubMed] [Google Scholar]
- 7.Eschbach JW, Egrie JC, Downing MR, Browne JK, Adamson JW. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. Results of a combined phase I and II clinical trial. N Engl J Med. 1987 Jan 8;316:73–78. doi: 10.1056/NEJM198701083160203. [DOI] [PubMed] [Google Scholar]
- 8.Eschbach JW, Kelly MR, Haley NR, Abels RI, Adamson JW. Treatment of the anemia of progressive renal failure with recombinant human erythropoietin. N Engl J Med. 1989 Jul 20;321:158–163. doi: 10.1056/NEJM198907203210305. [DOI] [PubMed] [Google Scholar]
- 9.Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006 Nov 16;355:2085–2098. doi: 10.1056/NEJMoa065485. [DOI] [PubMed] [Google Scholar]
- 10.Drüeke TB, Locatelli F, Clyne N, Eckardt K-U, Macdougall IC, Tsakiris D, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006 Nov 16;355:2071–2084. doi: 10.1056/NEJMoa062276. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer MA, Burdmann EA, Chen C-Y, Cooper ME, de Zeeuw D, Eckardt K-U, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009 Nov 19;361:2019–2032. doi: 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- 12.Solomon SD, Uno H, Lewis EF, Eckardt K-U, Lin J, Burdmann EA, et al. Erythropoietic Response and Outcomes in Kidney Disease and Type 2 Diabetes. N Engl J Med. 2010 Sep 16;363:1146–1155. doi: 10.1056/NEJMoa1005109. [DOI] [PubMed] [Google Scholar]
- 13.Pfeffer MA, Burdmann EA, Chen C-Y, Cooper ME, de Zeeuw D, Eckardt K-U, et al. Baseline characteristics in the Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT) Am J Kidney Dis. 2009 Jul;54:59–69. doi: 10.1053/j.ajkd.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Bunn HF, Gu J, Huang LE, Park JW, Zhu H. Erythropoietin: a model system for studying oxygen-dependent gene regulation. J Exp Biol. 1998 Apr;201:1197–1201. doi: 10.1242/jeb.201.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGonigle RJS, Wallin JD, Shadduck RK, Fisher JW. Erythropoietin deficiency and inhibition of erythropoiesis in renal insufficiency. Kidney Int. 1984 Feb 1;25:437–444. doi: 10.1038/ki.1984.36. [DOI] [PubMed] [Google Scholar]
- 16.Skali H, Lin J, Pfeffer MA, Chen C-Y, Cooper ME, McMurray JJV, et al. Hemoglobin stability in patients with anemia, CKD, and type 2 diabetes: an analysis of the TREAT (Trial to Reduce Cardiovascular Events With Aranesp Therapy) placebo arm. Am J Kidney Dis. 2013 Feb;61:238–246. doi: 10.1053/j.ajkd.2012.08.043. [DOI] [PubMed] [Google Scholar]
- 17.KDIGO: Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl. 2012;2:279–335. [Google Scholar]
- 18.Hsu C-Y, McCulloch CE, Curhan GC. Iron status and hemoglobin level in chronic renal insufficiency. JASN. 2002 Nov;13:2783–2786. doi: 10.1097/01.asn.0000034200.82278.dc. [DOI] [PubMed] [Google Scholar]
- 19.Babitt JL, Lin HY. Molecular Mechanisms of Hepcidin Regulation: Implications for the Anemia of CKD. American Journal of Kidney Diseases. 2010 Apr;55:726–741. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bârsan L, Stanciu A, Stancu S, Căpuşă C, Brătescu L, Mandache E, et al. Bone marrow iron distribution, hepcidin, and ferroportin expression in renal anemia. Hematology. 2015 Oct;20:543–552. doi: 10.1179/1607845415Y.0000000004. [DOI] [PubMed] [Google Scholar]
- 21.Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. American Journal of Kidney Diseases. 1997 Apr;29:565–568. doi: 10.1016/s0272-6386(97)90339-5. [DOI] [PubMed] [Google Scholar]
- 22.Gunnell J, Yeun JY, Depner TA, Kaysen GA. Acute-phase response predicts erythropoietin resistance in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis. 1999 Jan;33:63–72. doi: 10.1016/s0272-6386(99)70259-3. [DOI] [PubMed] [Google Scholar]
- 23.Mc Causland FR, Claggett B, Burdmann EA, Eckardt K-U, Kewalramani R, Levey AS, et al. C-Reactive Protein and Risk of ESRD: Results From the Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT) Am J Kidney Dis. 2016 Sep 16; doi: 10.1053/j.ajkd.2016.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao DS, Shih M, Mohini R. Effect of serum parathyroid hormone and bone marrow fibrosis on the response to erythropoietin in uremia. N Engl J Med. 1993 doi: 10.1056/NEJM199301213280304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.