

Abstract
Chlamydia trachomatis (Ct) is a Gram-negative obligate intracellular pathogen of humans that causes significant morbidity from sexually transmitted and ocular diseases globally. Ct acquires host fatty acids (FA) to meet the metabolic and growth requirements of the organism. Lipid droplets (LDs) are storehouses of FAs in host cells and have been proposed to be a source of FAs for the parasitophorous vacuole, termed inclusion, in which Ct replicates. Previously, cells devoid of LDs were shown to produce reduced infectious progeny at 24 hours post-infection (hpi). Here, while we also found reduced progeny at 24 hpi, there were significantly more progeny at 48 hpi in the absence of LDs compared to the control wild-type (WT) cells. These findings were confirmed using transmission electron microscopy where cells without LDs were shown to have significantly more metabolically active reticulate bodies at 24 hpi and significantly more infectious but metabolically inert elementary bodies at 48 hpi than WT cells. Furthermore, by measuring basal oxygen consumption rates (OCR) using extracellular flux analysis, Ct infected cells without LDs had higher OCRs at 24 hpi than cells with LDs, confirming ongoing metabolic activity in the absence of LDs. While the FA oleic acid (OA) is a major source of phospholipids for Ct and stimulates LD synthesis, treatment with OA, but not other FAs, enhanced growth and led to an increase in basal OCR in both LD depleted and WT cells, indicating that FA transport to the inclusion is not affected by the loss of LDs. Our results show that Ct regulates inclusion metabolic activity and growth in response to host FA availability in the absence of LDs.
Keywords: Chlamydia trachomatis, metabolism, oxygen consumption rate, lipid droplets, fatty acids, sexually transmitted diseases, ocular diseases
1. Introduction
Chlamydia trachomatis (Ct) is a Gram-negative pathogen of humans that is the leading cause of bacterial sexually transmitted diseases and preventable blindness worldwide (CDC, 2016; Dean, 2013). The obligate intracellular nature of the organism has limited our ability to completely understand the complex host-pathogen interactions that allow it to survive and replicate within a diversity of host cells.
The elementary body (EB) is the infectious but metabolically inactive form of the bacterium. Once inside the host cell, the EB differentiates into a metabolically active reticulate body (RB), which is surrounded by a membrane composed of host and bacterial proteins (AbdelRahman and Belland, 2005) termed an inclusion. The RB replicates by binary fission, producing progeny that condense into EBs before being released to infect neighboring cells (Hackstadt et al., 1997; Bastidas et al., 2013).
Ct has undergone genome reduction during its evolution (Zomorodipour and Andersson, 1999) similar to other intracellular pathogens such as Buchnera aphidicola (Wassenaar et al., 2009). One consequence of this is that the bacterium is dependent on the host cell for survival. Ct must therefore intercept trafficking pathways in the host cell to incorporate essential metabolites and enzymes for inclusion formation, replication, and membrane maintenance (Elwell & Engel, 2012). Ct actively modulates its lipid composition within hours of entry into the host cell and during replication. The organism recruits into the inclusion different pools of host-derived lipids such as sphingomyelin (Hackstadt et al., 1995), cholesterol (Carabeo et al., 2003), cardiolipin (Wylie et al., 1997), and phosphatidylcholine (Wylie et al., 1997). However, one study suggests that Ct is able to synthetize the lipids required for its membrane systems without the need for host phospholipids (Yao et al., 2015). The bacteria also recruit host enzymes into the inclusion that are involved in lipid biosynthesis, such as CERT and high density lipoproteins (Derré et al., 2011; Elwell et al., 2011; Cox et al., 2012).
One of the proposed mechanisms by which Ct acquires lipids and enzymes is via lipid droplets (LDs), organelles involved in lipid homeostasis, storage and protein trafficking present in all eukaryotic cells and some bacteria. LDs were purported to be recruited into the Ct inclusion and modified in response to Ct infection (Kumar et al., 2006; Cocchiaro et al., 2008; Saka et al., 2015). Other viral and bacterial pathogens also target LDs during infection, either for nutritional purposes or as part of an anti-immunity strategy (Herker and Ott, 2012).
Previously our group showed that Ct recruits long-chain acyl-CoA synthetases (ACSL) into the inclusion, converting FAs into acyl-CoA for use in various metabolic pathways (Recuero-Checa et al., 2016). We also discovered that ACSLs, although present on the surface of LDs, were translocated into the inclusion independent of the presence or absence of LDs. Additionally, inhibition of ACSL activity, and not the subsequent loss of LDs, blocked Ct growth. Our results suggested that Ct dependence on LDs is more nuanced than what has been reported previously. Here, we have focused on the role of FAs and LDs during Ct infection, growth and development.
2 Results
2.1 Absence of lipid droplets (LD) does not affect C. trachomatis (Ct) inclusion formation
Previous research suggested that depletion of LDs was the reason for the observed decrease in Ct EB formation (Kumar et al., 2006). To determine whether LDs are essential for Ct development, we used two mouse embryonic fibroblast (MEF) cell lines that either lack LD synthesis or lack synthesis after treatment with the chemical inhibitor T863 (Cao et al., 2011). In the last step of LD synthesis, diacylglycerol (DAG) is converted to triacylglycerol (TAG) by two enzymes, DGAT1 and DGAT2. When both are inactivated, there is no TAG synthesis and the cells cannot synthetize LDs. However, with one functional DGAT, LDs can be synthetized (Cao et al., 2011). Wild Type (WT), DGAT2-/- (SKO) treated with DGAT1 inhibitor T863, and DGAT1&2-/-(DKO) MEF cells were infected with Ct strain L2 at a multiplicity of infection (MOI) of 1 for 24 h. Inclusions were imaged by confocal microscopy. Similar to our previous findings (Recuero-Checa et al., 2016), Figure S1a shows that LDs are visible in WT and SKO cells. While we did not observe LDs in SKO cells treated with T863, we did notice that there were some LDs in DKO cells. It is likely that this is due to the instability of the KO in these cells at higher passage numbers (personal communication, Dr. Robert Farese). Consequently, subsequent experiments were performed with SKO cells with and without T863. This provided a robust system as the same SKO cells containing DGAT1 could be used to show the effect of infection with and without LDs (when treated with T863), essentially obviating the need for complementation to add back DGAT2. However, we did attempt to transfect the SKO cells with DGAT2 but were unsuccessful due to enhanced cytotoxicity (data not shown), similar to what was found by Saka et al. (2015). Inclusion formation was not affected by the absence of LDs. Figure S1b shows that Ct Heat Shock protein 60 (CtHSP60) was produced in the absence of LDs.
2.2 C. trachomatis (Ct) inclusions are significantly larger in cells depleted of lipid droplets (LD) at the end of development
Because the lack of LDs may affect Ct growth and development in the inclusion, a time course of infection was performed. Cells were infected with Ct at an MOI of 1 and fixed at 16, 24, 36 and 48 hpi and stained for Ct. At the later time points, the inclusions were larger in the absence of LDs compared to WT cells (Figure 1a). For quantitative analysis, the inclusion area was measured for at least 500 infected cells for each cell type using an automated high throughput microscopy system. The area was similar for all cells up to 24 hpi with a significantly larger area for T863 treated SKO cells compared to WT cells and to SKO cells at 48 hpi (Figure 1b). Although the microscopy images for the 48 hpi time point appear to have similar sized inclusions for DKO, SKO and T863 treated SKO cells, the difference in inclusion area reflect the actual quantitation of a large number of cells for each replicate experiment.
Figure 1. C. trachomatis (Ct) inclusions are significantly larger in cells depleted of lipid droplets (LD) at the end of development.
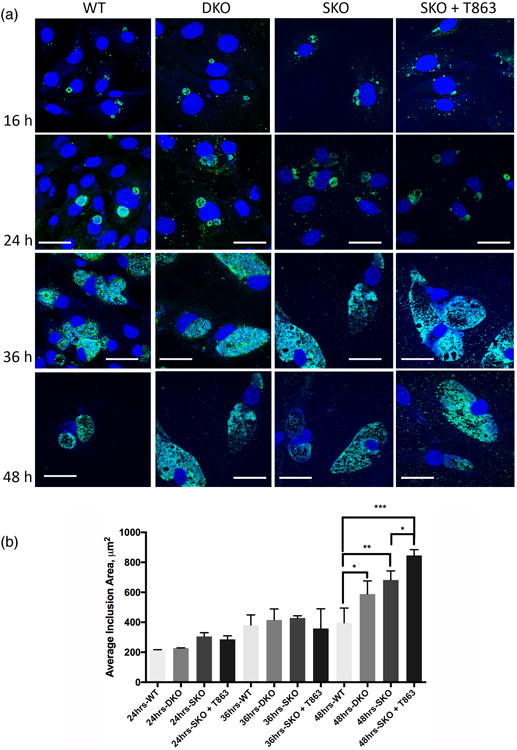
(a) WT, DKO, SKO, and T863 treated SKO MEF cells were infected with Ct L2 at an MOI of 1 for 16, 24, 36 and 48 h, fixed and labeled with Ct– specific MOMP antibody (green), and with Hoechst dye for nuclear and bacterial DNA (blue). Scale bar, 10 μm. (b) WT, SKO, T863 treated SKO and DKO MEF cells were infected with Ct L2 at an MOI of 1 for 24, 36 and 48 h and then fixed and stained as above. The inclusions areas were measured and plotted (mean ± standard error for three independent experiments). Asterisks denote statistically significant differences at *p<0.05, **p < 0.01, ***p<0.001 by one-way ANOVA test.
2.3 Generation of C. trachomatis (Ct) infectious progeny varies throughout development depending on the presence or absence of lipid droplets (LDs)
The significantly increased inclusions at 48 hpi suggested that Ct growth and differentiation of RBs into EBs may be affected. We infected WT, DKO, and SKO with and without T863 cells with Ct at an MOI of 1 for 24, 36 and 48 h. Ct obtained from each infected cell type at each time point was used to infect a fresh, uninfected monolayer of HeLa 229 cells. Similar to previous reports, we observed a decrease in the infectivity of Ct grown in the absence of LDs at 24 hpi (Figure 2a). However, at 48 hpi, there was a significant increase in the infectious progeny production for DKO and T863 treated SKO cells (Figure 2a). Figure S2 shows the percentage of infection in the initial cells that were then used for the infectivity assay. For subsequent experiments, we used T863 treated SKO and not DKO cells because, as mentioned above, the former consistently showed ablation of LDs.
Figure 2. Generation of C. trachomatis (Ct) infectious progeny increases significantly throughout development in cells devoid of lipid droplets (LDs).

(a) WT, SKO, T863 treated SKO, and DKO MEF cells were infected with Ct L2 at an MOI of 1 for 24, 36 or 48 h. The cultures were used for re-infecting new HeLa cell monolayers and analyzed for infectious progeny production. Values (mean ± standard error for three independent experiments) are shown as inclusion forming units (IFU)/mL. Asterisks denote statistically significant differences at *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001 by one-way ANOVA test. (b) Cells were infected as above for 24 and 48 h, fixed and prepared for TEM. Scale Bar 1 μm.
Since the apparent decrease in infectivity in cells devoid of LDs could be due to increased replication or an increased rate of RB-to-EB differentiation, we monitored DNA genomic copy numbers using qPCR for the Ct gene ompA in SKO cells with and without T863 treatment. The time points were 24, 36 and 48 h. Although the copy numbers were similar at 24 hpi, the difference between the DNA copy numbers in the absence of LDs was significantly higher than control SKO cells at 48 hpi (Figure S3a). This suggests that the apparent decrease in infectivity at 24 h in the absence of LDs could be due to increased replication and, therefore, a decreased rate of RB-to-EB differentiation.
We performed Transmission Electron Microscopy (TEM) of WT and T863 treated SKO cells to assess the effect of LD depletion on the generation of infectious progeny at 24 and 48 hpi. Figure 2b shows that, similar to the results from the infectivity assay, WT cells produced in general more EBs compared to T863 treated SKO cells at 24 hpi. However, at 48 hpi, SKO cells treated with T863 exhibited higher numbers of EBs as compared to WT cells.
2.4 C. trachomatis (Ct) growth and infectious progeny generation increases after treatment of host cells with the fatty acid (FA) oleic acid (OA), but not other FAs, independent of the presence of lipid droplets (LDs)
OA treatment is known to induce LD synthesis in cells. However, SKO cells treated with T863 cannot synthesize LDs and, therefore, treatment with OA provides a strong distinction between WT and T863 treated SKO. We wanted to determine whether Ct growth and development would be affected under this condition. WT and T863 treated SKO cells were treated with medium containing 100 μM OA for 16 h. The medium was then changed with normal growth medium, and the cells were infected with Ct at an MOI of 1. At 24, 36 and 48 hpi, the cells were fixed and analyzed for Ct growth (Figure 3a), and the inclusion area was quantified as described above (Figure 3b). OA treatment led to a significant increase in the inclusion area at all time points but not the number of inclusions, irrespective of the presence or absence of LDs. To illustrate this point, Figure S4 shows time-lapse real time imaging of GFP-L2 infected WT cells with and without pretreatment with OA. In agreement with these results, OA treatment lead to an increase in Ct DNA copy number at 24 h (Figure S3b). We also found that there was a significant increase in infectious progeny in both WT and T863 treated SKO cells pretreated with OA starting at 36 hpi (Figure 3c).
Figure 3. C. trachomatis (Ct) growth and infectious progeny generation increases after treatment of host cells with the fatty acid (FA) oleic acid (OA).


(a) WT and T863 treated SKO cells were grown in medium containing 100 μM OA for 16 h. The cells were washed and infected with Ct L2 at an MOI of 1 for 24, 36 and 48 h. Cells were fixed at the indicated time points (at 24 hpi for uninfected cells) and labeled with Ct-specific MOMP antibody (red), Bodipy (green) and Hoechst dye for nuclear and chlamydial DNA (blue). Scale Bar 20 μM. (b) The inclusion areas were measured for at least 500 infected cells for each cell type for three independent experiments using a Nikon Eclipse Ti-E inverted microscope system with the High Content Analysis system NIS Elements. Asterisks denote statistically significant differences at *p < 0.05, **p < 0.01, ***p<0.0001 by one-way ANOVA test. (c) Infectious progeny generation was calculated by passaging onto fresh monolayers of HeLa cells as described in EXPERIMENTAL PROCEDURES.
We checked if different FAs had a similar effect on Ct growth. In similar experiments, cells were treated with increasing concentrations of other FAs including linoleic acid, myristic acid, palmitic acid, and arachidonic acid. No increase in inclusion size (data not shown) or CtHSP60 protein synthesis was observed (Figure 4a). OA appeared to have a dose dependent effect on Ct growth at concentrations starting at 0.5 μM. The effect of OA was independent of the presence of LDs (Figure 4b).
Figure 4. Oleic acid (OA) mediated increase in C. trachomatis (Ct) growth and infectious progeny is specific to the time point of treatment.

(a) WT MEFs were treated with different fatty acids for 16 h, washed and then infected with Ct L2 at an MOI of 1. At 24 hpi, samples were collected for western blotting and probed with antibodies specific for GAPDH and CtHSP60. (b) WT and T863 treated SKO cells were treated with increasing concentrations of OA and palmitic acid for 16 h. Cells were washed, infected, and samples were collected for Western blotting as above and probed with antibodies specific for β-Actin and CtHSP60. (c) WT and T863 treated SKO cells were treated with 100 μM OA at different time points relative to the infection (-16 refers to the pre-treatment for 16 h before infection). At each time point prior to and post infection, samples were collected and analyzed by Western blotting and probed with antibodies specific for β-Actin and CtHSP60.
It has previously been shown that Ct growth is affected by Triacsin C treatment, which also depletes LDs inside host cells (Kumar et al., 2006). We checked if OA treatment could rescue inclusion growth in Triacsin C pre-treated WT and T863 treated SKO cells with and without pretreatment with OA. We found that OA was able to reverse the effect of Triacsin C to some extent (Figure S6). The effect was seen even in WT cells where LDs were absent due to Triacsin C treatment.
2.5 Oleic acid (OA) mediated increase in C. trachomatis (Ct) growth is specific to the time point of treatment
We next checked the effect of OA treatment on the cells at different time points relative to the time of infection. WT and T863 treated SKO cells were infected with Ct at an MOI of 1, and OA was added to the cells at different times pi. For comparison, cells were pretreated with 100 μM OA for 16 h, washed and infected at the same time. At 24 hpi, the samples were collected, and CtHSP60 levels were assessed by Western blot (Figure 4c). The increase in CtHSP60 production correlated with the duration of OA treatment. Treatment at early time points of infection up to 5 hpi lead to enhanced growth of Ct compared to cells not treated with OA (Figure 4c).
2.6 Fatty acid (FA) transport into the C. trachomatis (Ct) inclusion occurs in the absence of lipid droplets (LD)
Since LDs have been proposed to aid in the transport of FAs to the inclusion, we used a saturated FA analog that has a tail composed of 12 carbons and a Bodipy 493/503 fluorophore bound to the hydrophobic end to test whether this analog is present in the Ct inclusion even in the absence of LDs. WT and T863 treated SKO cells were treated with Bodipy FA for 16 h, washed and then infected with Ct (see Experimental Procedures). Cells were stained for Ct and imaged by confocal microscopy. Z-stack images were obtained to determine the location of Bodipy FA. Figure S5a and Figure S5b show that Bodipy FA was observed inside the inclusion in both cell types. To confirm that the signal in the green channel was not background staining, signal intensity plots were included, and infected WT and T863 treated SKO cells without Bodipy staining were used as a negative control.
2.7 Oleic acid (OA) treatment increases the basal oxygen consumption rate (OCR) in C.trachomatis (Ct) infected cells independent of the presence of lipid droplets (LD)
Because the metabolic rate appeared to be increased in LD depleted cells compared to WT, we evaluated oxygen consumption rate (OCR) in both cell types using a Seahorse XF96 extracellular flux analyzer according to the manufacturer's protocol. The assay is very sensitive to the cell density and, therefore, to ensure that the cell densities were exactly the same, we used SKO cells to create a single dilution to seed all the wells in a 96 well plate. As seen in Figure 5a, cells infected with Ct had a significantly increased basal OCR compared to uninfected cells. To confirm that the increased OCR upon infection was due to Ct activity, cells were treated with Azithromycin for 4 h before the OCR measurement. Azithromycin treatment had no effect on uninfected cells but significantly decreased the OCR of Ct infected cells. Interestingly, infected cells treated with T863 had a higher OCR compared to untreated cells although T863 treatment had no effect on OCR in the uninfected cells. (Figure 5b).
Figure 5. The oxygen consumption rate (OCR) is increased in cells infected with C. trachomatis (Ct) even in the absence of lipid droplets (LD).

SKO MEFs were infected with Ct L2 at an MOI of 1 for 24 h. Oxygen consumption rates (OCR) were measured using an XF96 Extracellular Flux Analyzer (Seahorse Bioscience). (a) OCR levels after Azithromycin (0.5ug/ml) treatment was used to block chlamydial metabolism. (b) OCR in the presence or absence of 20 μM T863. (c) OCR in the presence or absence of 100 μM OA. Asterisks denote statistically significant differences at *p < 0.001, **p < 0.01, ***p<0.0001, ****p<0.0001 by one-way ANOVA test.
We also checked for the effect of OA on OCR levels during infection. Uninfected cells had no change in OCR levels upon OA treatment. However, cells infected in the presence of OA showed a significant increase in OCR levels (Figure 5C). OA treatment of T863 treated SKO cells also showed a similar increase in OCR levels. In the uninfected SKO cells, depletion of LDs by T863 treatment had no effect on OCR levels (Figure 5c).
3 Discussion
Chlamydia depend on host cells for their nutritional demands including lipids and FAs. Various mechanisms of lipid acquisition have been reported including interaction with the host Golgi, recruitment of Rab GTPases, acquisition of multivesicular bodies (Elwell & Engel, 2012) and interaction of the inclusion with LDs (Kumar et al., 2006; Cocchiaro et al., 2008).
LDs act as storehouses of neutral lipids, which could make them an attractive target for Ct interactions. Indeed, there is a growing body of work describing the interaction between host LDs and the Ct inclusion as well as the importance of LDs for Ct differentiation and growth. One study reported that LDs accumulate at the periphery of the chlamydial inclusion and associate with three secreted chlamydial proteins, Lda1, Lda2 and Lda3 (Kumar et al., 2006). Saka et. al. (2011) found that the LD proteome was altered during infection although no association of Lda1 and Lda3 with the LDs could be detected. A more recent publication analyzed the host-cell derived proteome from isolated Ct inclusions using LC-MS/MS-based proteomics, which was sensitive enough to detect most of the previously reported host cell proteins associated with the inclusion, but none of the LD marker proteins were identified (Aeberhard et al., 2015). The authors explained that the process of inclusion extraction might have had a bias for only those inclusions that were not associated with LDs. However, these data suggested that at least a significantly large subset of Ct inclusions did not associate with LDs.
While others have claimed that LDs are localized within the inclusion and treatment of Ct infected cells with Triacsin C, which pharmacologically inhibits LD formation, impairs Ct growth (Cocchiaro et al., 2008), we found that the effect was due to inhibition of the ACSL family of proteins by Triacsin C with no evidence of LDs localizing within the inclusion (Recuero-Checa et al., 2016). Interestingly, we found that treatment with OA resulted in an increase in Ct inclusion formation even in Triacsin C treated cells where LD formation remained inhibited. Furthermore, Ct is able to infect many different types of epithelial cells that, similar to HeLa cells, have very low levels of LDs (Kumar et al., 2006). These collective findings raise doubts about the physiological relevance of LD-Ct interactions. Consequently, we were prompted to investigate the requirement of LDs for the development of Ct inclusions and infectious progeny.
MEFs lacking DGAT1 and DGAT2 activity are unable to synthesize LDs and thus provide an elegant system for studying LD function (Harris et al., 2011). In our study, we first compared the levels of LDs in WT, SKO treated with the DGAT1 inhibitor T863, and DKO MEFs. The T863 treated SKO cells were consistently devoid of LDs unlike the DKOs where some cells were noted to have LDs. Therefore we used the SKO cells instead of DKO cells for subsequent experiments.
It is possible that during knock-out generation and immortalization, uncontrolled changes may occur which in turn may affect phenotypic features at multiple levels. Complementation is normally performed to exclude this possibility. The majority of our experiments were performed with DGAT2 knock-out MEFs (SKO cells) and WT MEFs that came from littermate control mice. Importantly, the exact same SKO cells were used with and without T863 in each experiment. Previously, Saka et al. (2015) was unable to do complementation because transfection with the DGAT2 gene proved toxic to the MEF cells. Similarly, we found that transfection with DGAT2 produced enhanced cytotoxicity, also preventing our ability to perform any complementation assays.
We found that Ct inclusion size was not adversely affected in the absence of LDs at 24 hpi nor was there any difference in CtHSP60 production. Interestingly, we found that while the inclusions in WT cells reached a maximum size around 36 hpi, the inclusions in cells devoid of LDs continued to grow until 48 hpi. Moreover, the infectious progeny generation was significantly higher in the absence of LDs at later time points of infection. These findings are consistent with the increase in genomic DNA copy number at later time points, indicating an increase in replication as opposed to an increase in RB to EB differentiation. Furthermore, using TEM, WT cells contained more EBs than T863 treated SKO cells at 24 hpi (where RBs predominated) while at 48 hpi the latter cells had significantly more EBs, again indicating increased replication.
The fact that the inclusion continued to grow along with the generation of infectious progeny up to 48 h indicates that the RBs were metabolically active for a longer period of time in the absence of LDs. Since Ct utilizes multiple strategies to acquire lipids from host cells, it is likely that when LDs are unavailable, FAs are redistributed in the cell in such a way that more are available for acquisition by the growing inclusion throughout development, resulting in a longer replicative phase, which yields a larger inclusion and higher number of infectious progeny. Indeed, host cell lipids such as sphingomyelin, cholesterol and glycerophospholipids that are acquired by Ct (Elwell & Engel, 2012) may be a source of FAs (likely generated by the catabolic activity of various lipases) that are then utilized by Ct for downstream needs such as metabolism and membrane synthesis.
It is possible that the FAs stored in LDs are initially available to the inclusion in WT cells but limited, which triggers the differentiation of RBs to EBs, resulting in the smaller inclusion size and decrease in progeny at the end of development. To test this hypothesis, we used OA to stimulate LD synthesis in the cells as has been performed in previous studies (Fujimoto et al., 2006). As expected, WT cells showed an increase in LDs in the cytoplasm whereas no LDs were observed in the T863 treated SKO cells. Inclusion growth and infectious progeny were enhanced in both cell types, confirming that it was not the LDs per se, but the availability of FAs in the host cells that was critical for Ct growth and development.
It has previously been reported that a major proportion of Ct modified lipids detected in infected cells are derived from OA, a FA that Ct cannot produce but must acquire from the host cell (Yao et al., 2015). Besides OA, palmitic, myristic and lauric acids have also been shown to be incorporated into Ct-derived phospholipids (Yao et al., 2015). However, we found that only treatment with OA lead to a significant increase in the inclusion size. The host cell maintains a pool of different FAs through de novo FA synthesis or through conversion from other lipid species within the cell (Schiller & Bensch, 1971; Beld et al., 2015). It is therefore likely that the other FAs were available to the inclusion in sufficient quantity and are not the rate-determining factor for growth in the absence of exogenous treatment.
OA has been widely used in the literature to stimulate LD production in cells that normally don't accumulate a large number of LDs, such as HeLa cells, to study the relationship between Ct and LDs (Kumar et al., 2006; Cocchiaro et al., 2008; Saka et al., 2015; Soupene et al., 2015). Cocchiaro et al. (2008) enhanced LD formation in HeLa cells by using 100 μM of OA. They found no adverse effect on Ct replication, which is not contradictory to what we found. Indeed, the results of Ouellette et al. (2010) were very similar to ours in that HeLa cells treated with OA showed a 5-fold increase in IFUs/μl compared to untreated cells.
The absence of LDs in our experiments did not appear to adversely affect Ct metabolic activity. To confirm this, we used extracellular flux analysis to measure OCR as an indicator of Ct metabolism. We found a significant increase in OCR in Ct infected cells compared to uninfected cells. While it was possible that host cell glycolysis during Ct infection may have contributed to the increased OCR, treating infected cells with azithromycin once the inclusions had formed lowered the OCR significantly, suggesting that Ct metabolic activity was contributing to the OCR. Furthermore, the OCR was significantly higher in infected cells without LDs than in those with LDs, which confirmed that at 24 hpi, there were more metabolically active RBs when no LDs were available. These data are also consistent with our TEM results, the observed increase in inclusion size, genomic DNA copy number and infectious progeny over the course of development for MEFs devoid of LDs. As expected, OA treatment significantly increased the OCR irrespective of the presence of LDs, confirming that Ct acquisition of FAs and increase in metabolic rate is not affected by the lack of LDs.
Taken together, our findings indicate that Ct is able to scavenge FAs from host cells primarily via an LD independent mechanism. LDs might enter the inclusion as claimed by one study where there was morphologic evidence of LDs that accumulated in HeLa cells in response to OA treatment. However, no direct LD markers were used. The authors mentioned that LDs were quickly dispersed once inside the Ct inclusion (Cocchiaro et al., 2008), which could be one reason why LDs are not frequently observed. Rank et al. (2011) similarly showed morphological evidence of LDs inside a single inclusion of the Chlamydia species Chlamydia muridarum, a pathogen restricted to mice. However, no study has replicated the findings of Cocchiaro et al. (2008) using Ct. Saka et al. (2015) showed EM images with LDs in close proximity to the inclusion but none were inside. Furthermore, no markers of LDs were found in purified Ct inclusions analyzed by a sensitive LC-MS/MS-based proteomics approach (Aeberhard et al., 2015). Aeberhard et al. (2015) argued that it was likely that only the inclusions that did not carry LDs were preferentially isolated by their protocol. LDs may be one source of FAs in the Ct infected cell but the fact that HeLa cells do not contain many LDs and Ct is still able to develop normally in those cells suggest that they could be dispensable. Moreover, we show that even in cells completely devoid of LDs, either by use of SKO cells treated with T863 or inhibition of LDs by Triacsin C, Ct is still able to grow and develop normally. We propose that it is not the LDs per se, but the availability of FAs in the host cells that contribute to Ct growth and development.
4 Experimental Procedures
4.1 Cell culture and C. trachomatis infection
WT, SKO, and DKO MEF cells were a gift from Dr. Robert V. Farese (Harvard Medical School, Boston, MA). HeLa 229 cells were grown at 37°C, 5% CO2, in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA) with 10% heat-inactivated fetal bovine serum (Hyclone, GE Healthcare, Logan, UT) and supplemented with gentamicin (Gibco, Carlsbad, CA) and vancomycin (Sigma Aldrich, St. Louis, MO) as previously described (Recuero-Checa et al., 2016; Somboonna et al., 2011).
Ct reference strain L2/434 was propagated in HeLa cells in DMEM medium with 10% heat-inactivated FBS, harvested and purified as described (Recuero-Checa et al., 2016). Briefly, cells were grown in various-sized well plates (E&K Scientific, Santa Clara, CA) with or without glass coverslips (Electron Microscopy Sciences, Inc., Hatfield, PA) and infected at an MOI of 1 for 2 h when fresh DMEM was added. The cells were incubated until the end of each experiment.
4.2 Quantitation of C. trachomatis infectious progeny
Quantitation of infectious progeny was performed as previously described (Recuero-Checa et al., 2016). Briefly, infected cells were transferred at indicated time points into fresh media as above, lysed by sonication, and 10-fold serial dilutions were used to infect fresh monolayers of HeLa cells. Inclusions were visualized using Pathfinder® Chlamydia Culture Confirmation System (Bio-Rad, Hercules, CA). Hoechst 33258 (Life Technologies, Carlsbad, CA) was used to detect nuclei and bacterial DNA. Inclusion forming units (IFUs) were expressed as IFU/mL.
4.3 Microscopy
Cells were grown and infected on 12-mm glass coverslips (Electron Microscopy Sciences) in 24 well plates (E&K Scientific) as described (Recuero-Checa et al., 2016). Briefly, cells were fixed with 4% formaldehyde in PBS, permeabilized with 0.02% saponin, and incubated with primary Ct MOMP-specific antibodies (Virostat, Portland, ME). Cells were incubated with Alexa Fluor secondary antibodies (Life Technologies, Carlsbad, CA) using manufacturer's dilutions. Samples were incubated with BODIPY 493/503 (Life Technologies) for co-staining of neutral lipids and LDs, and with Hoechst 33258 (Life Technologies) to detect nuclei and bacterial DNA. Images were collected on a Zeiss LSM710 confocal inverted microscope. Image processing was performed using Huygens Essential (Scientific Volume Imaging, The Netherlands) and Imaris (Bitplane AG, Switzerland) software program s.
4.4 High throughput and time-lapse microscopy
High throughput microscopy was performed using a Nikon Eclipse Ti-E inverted microscope (Nikon Instruments Inc., NY, USA) with a High Content Analysis system and image acquisition using NIS Elements (Nikon Instruments Inc.) for IFU determinations and to calculate inclusion areas after manually marking Regions Of Interests (ROI). Approximately 500 inclusions were measured to calculate the average inclusion area for each sample for three independent experiments.
WT cells grown on a glass bottom 24-well plates (Greiner Bio-One) were infected with GFP-L2 in the presence or absence of OA. Time-lapse imaging was performed using a Nikon Eclipse Ti-E inverted microscope (Nikon Instruments Inc.) with a High Content Analysis system and image acquisition using NIS Elements. Images were taken every 30 minutes from 8 until 24 hpi.
4.5 Lipid droplet (LD) formation and abrogation
LD formation was induced with 100 μM OA (Sigma-Aldrich) as described (Recuero-Checa et al., 2016). 20 μM of T863 (Sigma-Aldrich) was used to abrogate LD formation in SKO cells as described (Cao et al., 2011).
WT and SKO treated T863 cells were treated with 5 μM of Triacsin C (Santa Cruz Biotechnologies) 16 h before infection and then treated with 100 μM OA or left untreated at the time of infection with Ct. Infections with Ct were performed as above.
4.6 Transmission Electron Microscopy (TEM)
Cells were infected with Ct for 24 or 48 h and fixed with 2.5 % glutaraldehyde for 2 h at RT. Fixed cells were prepared for EM as described (Dacosta et al., 1990; Recuero-Checa et al., 2016). Thin sections were post-stained with uranyl acetate and lead citrate for examination on a Tecnai BioTwin (FEI Company, Hillsboro, OR, USA) electron microscope at 80 or 120 kV.
4.7 Western Blot immunodetection
Western blots were prepared as previously described (Recuero-Checa et al., 2016). Briefly, cells were grown in 24-well plates, infected with Ct as described, and harvested at designated times pi. The primary antibodies included β-Actin and Ct-specific HSP60 (Santa Cruz Biotechnologies, Santa Cruz, CA); the secondary antibodies were peroxidase-conjugated (Santa Cruz Biotechnologies) diluted as per the manufacturer's protocol. Detection was performed with ECL Western Blot Detection Reagents (GE Healthcare, Lafayette, CO).
4.8 Extracellular Flux Analysis
A titration experiment was performed to optimize the number of cells per well for experiments run in the Seahorse Xf96 Analyzer (Agilent Technologies, Santa Clara, CA); 2, 5, 10, 20 or 30 K MEF cells were used per well. Mixing, incubation, and measurement times were 0.5, 2, and 3 min, respectively. Oligomycin was utilized to inhibit ATP synthase and Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) was used as a mitochondrial uncoupling agent to allow maximal O2 consumption under a given condition. Rotenone was employed as a mitochondrial respiratory chain complex 1 inhibitor. The Seahorse Xf Cell Mito Stress Test was performed according to the manufacturer's protocol with an Xf96 Analyzer; 20 K cells gave optimal results and were used in all experiments. For OCR measurements, immediately after infection, SKO cells with and without T863 were incubated at 35°C until assay measurement, according to the manufacturer's protocol.
4.9 Quantitation of DNA Copy number by qPCR
WT and SKO cells with and without T863 treatment were infected at 24, 36 and 48 h time points in the presence or absence of 100 μM OA as indicated. Cells were lysed and total nucleic acid was isolated using MasterPure™ Complete DNA and RNA purification Kit (Epicentre, Wisconsin, USA) following the manufacturer's protocol. Genomic DNA copy number was determined by qPCR using primers and thermocycling parameters with the ompA plasmid for generating the standard curve and no template controls as we have described (Gomes, 2006).
4.10 Transfection of MEFs with DGAT2
SKO cells were transfected with a pGFP-DGAT2 plasmid (OriGene, Cat. No. MG218673, Rockville, MD) or control plasmid, using a 10 μl tip Neon transfection system (Invitrogen), according to the manufacturer's protocol and the following optimized electroporation settings: pulse voltage, 1,550 V; pulse width, 20 ms; and pulse number, 1.
4.11 Statistical analysis
Statistical analysis was performed using GraphPad Prism (GraphPad Software, La Jolla, CA). Statistical significance between groups was determined by ANOVA or two-tailed Student's t-test depending on the number of groups. A p value of less than 0.05 was considered to be statistically significant. Data are shown as the mean ± standard deviation of n independent experiments.
Supplementary Material
Acknowledgments
We would like to thank Dr. Robert Farese for providing the DGAT1/2-/- and DGAT2-/- MEF cell lines, Dr. Amber Jolly for providing helpful comments on the manuscript and David Robert Lester, Weirui Xiao and Sid Chittaranjan for help with experiments. This work was supported in part by Public Health Service Grant R01 AI098843 from NIH (to DD).
References
- AbdelRahman YM, Belland RJ. The chlamydial developmental cycle. FEMS Microbiology Reviews. 2005;29(5):949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Aeberhard L, Banhart S, Fischer M, Jehmlich N, Rose L, Koch S, et al. The Proteome of the Isolated Chlamydia trachomatis Containing Vacuole Reveals a Complex Trafficking Platform Enriched for Retromer Components. PLoS Pathogens. 2015;11(6):e1004883. doi: 10.1371/journal.ppat.1004883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastidas RJ, Elwell CA, Engel JN, Valdivia RH. Chlamydial intracellular survival strategies. Cold Spring Harbor perspectives in medicine. 2013;3 doi: 10.1101/cshperspect.a010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beld J, Lee DJ, Burkart MD. Fatty acid biosynthesis revisited: structure elucidation and metabolic engineering. Mol BioSyst. 2015;11(1):38–59. doi: 10.1039/c4mb00443d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Zhou Y, Peng H, Huang X, Stahler S, Suri V, et al. Targeting acyl-CoA:Diacylglycerol Acyltransferase 1 (DGAT1) with small molecule inhibitors for the treatment of metabolic diseases. Journal of Biological Chemistry. 2011;286(48):41838–41851. doi: 10.1074/jbc.M111.245456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capmany A, Damiani MT. Chlamydia trachomatis intercepts Golgi-derived sphingolipids through a Rab14-mediated transport required for bacterial development and replication. PLoS ONE. 2010;5(11):e14084. doi: 10.1371/journal.pone.0014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabeo RA, Mead DJ, Hackstadt T. Golgi-dependent transport of cholesterol to the Chlamydia trachomatis inclusion. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(11):6771–6776. doi: 10.1073/pnas.1131289100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. Sexually Transmitted Disease Surveillance, 2015. Atlanta, GA: Department of Health and Human Services; 2016. [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proceedings of the National Academy of Sciences of the United States of America. 2008:1059379–9384. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchiaro JL, Valdivia RH. New insights into Chlamydia intracellular survival mechanisms. Cellular Microbiology. 2009:111571–1578. doi: 10.1111/j.1462-5822.2009.01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JV, Naher N, Abdelrahman YM, Belland RJ. Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cellular Microbiology. 2012;14(June):1497–1512. doi: 10.1111/j.1462-5822.2012.01823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacosta B, Ryter A, Mounier J, Sansonetti P. Immunodetection of lipopolysaccharide in macrophages during the processing of non invasive Shigella dysenteriae. Biology of the Cell. 1990;69(C):171–178. doi: 10.1016/0248-4900(90)90343-2. [DOI] [PubMed] [Google Scholar]
- Dean D. Chlamydia trachomatis pathogenicity and disease. Chlamydial Infection: A Clinical and Public Health Perspective. 2013:25–60. [Google Scholar]
- Derré I, Swiss R, Agaisse H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathogens. 2011;7(6):e1002092. doi: 10.1371/journal.ppat.1002092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Engel JN. Lipid acquisition by intracellular Chlamydiae. Cellular Microbiology. 2012;14(April):1010–1018. doi: 10.1111/j.1462-5822.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Jiang S, Kim JH, Lee A, Wittmann T, Hanada K, et al. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLoS Pathogens. 2011;7(9):e1002198. doi: 10.1371/journal.ppat.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y, Onoduka J, Homma KJ, Yamaguchi S, Mori M, Higashi Y, et al. Long-chain fatty acids induce lipid droplet formation in a cultured human hepatocyte in a manner dependent of Acyl-CoA synthetase. Biol Pharm Bull. 2006:292174–2180. doi: 10.1248/bpb.29.2174. doi:JST.JSTAGE/bpb/29.2174 [pii] [DOI] [PubMed] [Google Scholar]
- Gomes J, Borrego MJ, Atik B, Santo I, Azevedo J, Brito de Sá A, et al. Correlating Chlamydia trachomatis infectious load with urogenital ecological success and disease pathogenesis. Microbes Infect. 2006;8:16–26. doi: 10.1016/j.micinf.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Fischer ER, Scidmore MA, Rockey DD, Heinzen RA. Origins and functions of the chlamydial inclusion. Trends in Microbiology. 1997;5(7):288–293. doi: 10.1016/S0966-842X(97)01061-5. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(11):4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris Ca, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, et al. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. Journal of lipid research. 2011;52(4):657–67. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herker E, Ott M. Emerging role of lipid droplets in host/pathogen interactions. The Journal of biological chemistry. 2012;287(4):2280–7. doi: 10.1074/jbc.R111.300202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH. The Obligate Intracellular Pathogen Chlamydia trachomatis Targets Host Lipid Droplets. Current Biology. 2006:161646–1651. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Ooij C Van, Kalman L, Ijzendoorn S Van, Nishijima M, Hanada K, Mostov K, Engel JN. Host cell-derived sphingolipids are required for the intracellular growth of Chlamydia trachomatis. Cellular Microbiology. 2000:2627–637. doi: 10.1046/j.1462-5822.2000.00077.x. [DOI] [PubMed] [Google Scholar]
- Rank RG, Whittimore J, Bowlin AK, Wyrick PB. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infection and Immunity. 2011;79(8):3291–3301. doi: 10.1128/IAI.00200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recuero-Checa MA, Sharma M, Lau C, Watkins PA, Gaydos CA, Dean D. Chlamydia trachomatis growth and development requires the activity of host Long-chain Acyl-CoA Synthetases (ACSLs) Scientific reports. 2016;623148 doi: 10.1038/srep23148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Thompson JW, Chen YS, Dubois LG, Haas JT, Moseley A, Valdivia RH. Chlamydia trachomatis Infection Leads to Defined Alterations to the Lipid Droplet Proteome in Epithelial Cells. Plos One. 2015;10(4):e0124630. doi: 10.1371/journal.pone.0124630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Valdivia R. Emerging Roles for Lipid Droplets in Immunity and Host- Pathogen Interactions. Annual Review of Cell and Developmental Biology. 2012;28(1):411–437. doi: 10.1146/annurev-cellbio-092910-153958. [DOI] [PubMed] [Google Scholar]
- Schiller H, Bensch K. De novo fatty acid synthesis and elongation of fatty acids by subcellular fractions of lung. Journal of lipid research. 1971;12(2):248–55. [PubMed] [Google Scholar]
- Somboonna N, Wan R, Ojcius DM, Pettengill MA, Joseph SJ, Chang A, et al. Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. mBio. 2011;2(3):e00045–11. doi: 10.1128/mbio.00045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar TM, Bohlin J, Binnewies TT, Ussery DW. Genome comparison of bacterial pathogens. Genome Dynamics. 2009;6:1–20. doi: 10.1159/000235759. [DOI] [PubMed] [Google Scholar]
- Wylie JL, Hatch GM, Mcclarty G. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. Journal of Bacteriology. 1997;179(23):7233–7242. doi: 10.1128/JB.179.23.7233-7242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Cherian PT, Frank MW, Rock CO. Chlamydia trachomatis relies on autonomous phospholipid synthesis for membrane biogenesis. Journal of Biological Chemistry. 2015;290(31):18874–18888. doi: 10.1074/jbc.M115.657148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Dodson VJ, Frank MW, Rock CO. Chlamydia trachomatis scavenges host fatty acids for phospholipid synthesis via an acyl-acyl carrier protein synthetase. Journal of Biological Chemistry. 2015;290(36):22163–22173. doi: 10.1074/jbc.M115.671008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomorodipour A, Andersson SGE. Obligate intracellular parasites: Rickettsia prowazekii and Chlamydia trachomatis. FEBS Letters. 1999;452(1–2):11–15. doi: 10.1016/s0014-5793(99)00563-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.