

SUMMARY
During Salmonella-induced gastroenteritis, mucosal inflammation creates a niche that favors the expansion of the pathogen population over the microbiota. Here, we show that S. Typhimurium infection was accompanied by dysbiosis, decreased butyrate levels, and substantially elevated lactate levels in the gut lumen. Administration of a lactate dehydrogenase inhibitor blunted lactate production in germ-free mice, suggesting that lactate was predominantly of host origin. Depletion of butyrate-producing Clostridia, either through oral antibiotic treatment or as part of the pathogen-induced dysbiosis, triggered a switch in host cells from oxidative metabolism to lactate fermentation, increasing both lactate levels and Salmonella lactate utilization. Administration of tributyrin or a PPARγ agonist diminished host lactate production and abrogated the fitness advantage conferred to Salmonella by lactate utilization. We conclude that alterations of the gut microbiota, specifically a depletion of Clostridia, reprogram host metabolism to perform lactate fermentation, thus supporting Salmonella infection.
eTOC Blurb
Intestinal infection with Salmonella Typhimurium results in inflammation-induced dysbiosis. Gillis et al. demonstrate that depletion of commensal Clostridia reduces butyrate availability and subsequently alters host metabolism to produce lactate. Lactate oxidation by Salmonella enhances fitness in the gut and allows the pathogen to outcompete the microbiota.
INTRODUCTION
In the anaerobic environment of the intestine, commensal bacteria have evolved sophisticated strategies to compete for limiting amounts of complex polysaccharides to support growth. Bacterial pathogens invading this ecosystem are likely to have evolved unique metabolic adaptations to circumvent nutritional competition with commensal microbes.
The invasive enteric pathogen Salmonella enterica Typhimurium (S. Tm) is a common cause of bacterial food-borne gastroenteritis (reviewed in (Monack, 2013; Rivera-Chavez and Baumler, 2015; Santos, 2014)). In murine models of S. Tm infection, two spatially distinct but cooperative populations of the pathogen exist in the gut (Ackermann et al., 2008). One population invades and replicates in the gut mucosa, a process that depends on two distinct type three secretion systems (T3SS-1 and T3SS-2) (Galan and Curtiss, 1989; Hensel et al., 1995; Tsolis et al., 1999; Zhang et al., 2002). Tissue invasion and replication trigger inflammatory host immune responses that keep the infection localized to the intestinal tract. The induction of a potent host inflammatory response is essential for effective colonization of the gastrointestinal tract (Barman et al., 2008; Sekirov et al., 2010; Stecher et al., 2007). A subpopulation of S. Tm in the gut lumen relies on mucosal inflammation to fundamentally change the environment of the gut. The creation of a novel niche in the lumen of the intestinal tract fosters an outgrowth of S. Tm at the expense of the microbiota (Barman et al., 2008; Stecher et al., 2007).
Recent studies have identified nutrients that specifically enhance growth of S. Tm during gut inflammation. For example, production of reactive oxygen and nitrogen species generates the alternative electron acceptors tetrathionate and nitrate (Lopez et al., 2015; Lopez et al., 2012; Winter et al., 2010). Inflammation-associated changes in epithelial metabolism allow molecular oxygen to diffuse from the tissue into the gut lumen (Rivera-Chavez et al., 2016). Respiration facilitates the utilization of poorly fermentable carbon sources such as ethanolamine, 1,2-propanediol, fructose-asparagine, and succinate (Ali et al., 2014; Faber et al., 2017; Spiga et al., 2017; Thiennimitr et al., 2011). Furthermore, oxidation of sugars by nitric oxide gives rise to the aldaric acids galactarate and glucarate (Faber et al., 2016). Collectively, these studies suggest that inflammation-driven changes in the nutritional environment of the gut lumen are critical for S. Tm niche creation and competition with the microbiota.
A survey of more than 300 compounds demonstrated that S. Tm is able to utilize over 70 structurally diverse compounds as carbon sources in vitro (Gutnick et al., 1969). Comprehensive analyses of the S. Tm metabolism during systemic infection revealed significant redundancy in nutrient acquisition (Becker et al., 2006; Steeb et al., 2013). Thus, it is conceivable that during gut colonization S. Tm relies on a variety of carbon sources, the identity and source of which are unknown.
Here, we performed metabolic profiling to identify nutrients that are accessible to S. Tm in the niche of the inflamed gut lumen. We discovered that luminal lactate levels are increased during S. Tm infection. Lactate availability in the gut was dependent on both the gut microbiota and host metabolism. Alterations in the gut microbiota led to decreased butyrate levels, prompting host cell metabolism to switch from an oxidative metabolism to lactate fermentation. Utilization of host-derived lactate enhanced fitness of S. Tm. These results identify lactate as a connection between host and microbial metabolism.
RESULTS
Lactate levels in the gut lumen are increased during S. Tm infection
To better understand how the metabolic landscape of the intestine changes during S. Tm infection, we performed semi-quantitative profiling of extracellular metabolites in the gut lumen. Groups of Swiss Webster mice were intragastrically inoculated with the S. Tm wild-type strain or mock-treated with LB broth. Seven days after infection, the cecal content was collected and subjected to gas chromatography mass spectrometry (GC/MS) (Table S1). This analysis identified several extracellular metabolites with differential abundance in the cecal contents of infected mice compared to mock-treated mice. In this profiling, lactate was the most abundant metabolite during S. Tm infection while only small amounts of this compound were detected in mock-treated mice. To validate this finding, we developed a quantitative gas chromatography tandem mass spectrometry-based assay (GC/MS/MS) to measure lactate concentrations in the intestinal content (Fig. 1A). Compared to mock-treated animals, cecal lactate levels were significantly increased during S. Tm infection, confirming the results of the metabolic profiling. This finding led us to hypothesize that lactate may be a nutrient for S. Tm during infection.
Figure 1. The host is a source of lactate during S. Tm infection.
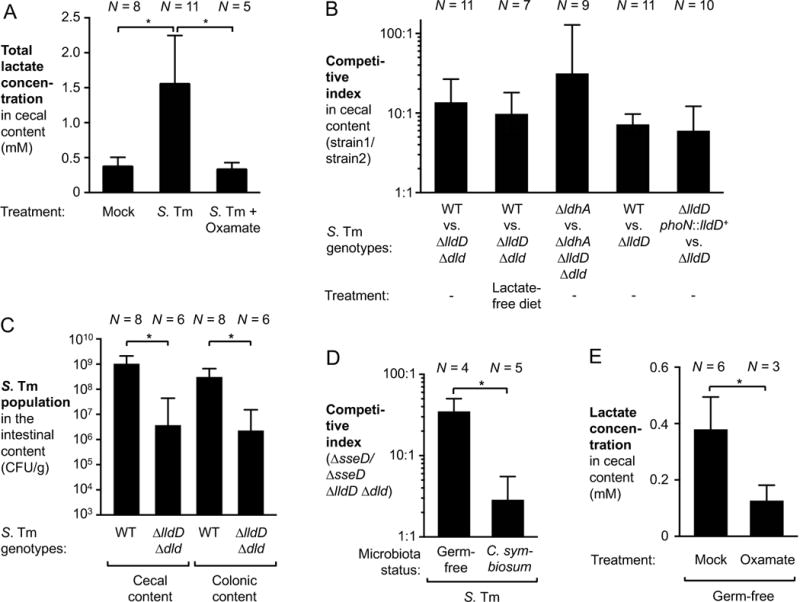
(A) Groups of Swiss Webster mice were inoculated with the S. Tm wild-type strain (WT) or mock-treated with LB broth. One group of S. Tm-infected mice received 0.5 % sodium oxamate in their drinking water for the duration of the experiment. After 8 days of infection the mice were sacrificed and lactate concentration in the cecal content analyzed by GC/MS/MS. (B) Groups of Swiss Webster mice were intragastrically inoculated with a 1:1 ratio of the indicated S. Tm strains. Samples were collected 8 days after infection. Competitive indices were calculated using the relative abundance of each strain in the cecal content, corrected by the ratio in the inoculum. (C) Two groups of Swiss Webster mice were intragastrically infected with the S. Tm wild-type strain or the ΔlldD Δdld mutant, respectively. The S. Tm populations in the cecal and colonic content (CFU/g) was determined after 8 days. (D) Groups of germ-free mice were pre-colonized with C. symbiosum or were mock-treated. After 3 days, both groups were intragastrically inoculated with an equal mixture of a ΔsseD and a ΔsseD ΔlldD Δdld mutant. Cecal content was collected 10 days after infection to determine the CI. (E) Germ-free Swiss Webster mice were treated with oxamate in their drinking water for 2 days or were mock-treated. Lactate concentrations in the cecal content were quantified by GC/MS/MS.
Bars represent geometric means ± standard error. *, P < 0.05. The number of animals per group (N) is indicated above each bar. See also Fig. S1 – S4.
Lactate utilization provides a fitness advantage in the inflamed gut
The S. Tm chromosome is predicted to encode two respiratory lactate dehydrogenases, LldD and Dld (McClelland et al., 2001). To investigate the function of the S. Tm enzymes, we performed competitive growth assays in vitro (Fig. S1). To this end, we generated a mutant lacking both lldD and dld (ΔlldD Δdld mutant). Mucin broth was inoculated with 1:1 mixture of the wild-type strain (AJB715) and a ΔlldD Δdld mutant, and incubated for 18 h. The relative titer of each strain was enumerated on selective agar and the ratio of wild-type strain to mutant bacteria, corrected by the ratio in the inoculum, was used to calculate the competitive index (Fig. S1A). Both strains were recovered in similar quantities in the absence of lactate suggesting that there was no general anaerobic growth defect for the ΔlldD Δdld mutant. When the media was supplemented with both lactate and exogenous electron acceptors, the wild-type strain outcompeted the ΔlldD Δdld mutant (Fig. S1A and B), a finding that is consistent with the respiratory function of membrane-bound LldD and Dld.
We then evaluated whether respiratory lactate dehydrogenases provide a fitness advantage to S. Tm in a model of infection. We intragastrically inoculated conventionally-raised Swiss Webster mice with equal amounts of both the S. Tm wild-type strain and a ΔlldD Δdld mutant. Eight days after infection, host responses in cecal and colonic tissue as well as luminal bacterial populations were analyzed. Infection with S. Tm resulted in substantial pathological changes in the cecal mucosa (Fig. S2A – D), while infrequent and limited changes were observed in the colon (data not shown). The wild-type strain was recovered from the cecal content at significantly higher numbers than the ΔlldD Δdld mutant (13-fold; P < 0.05 when comparing the individual wild-type and ΔlldD Δdld populations) (Fig. 1B). To determine whether lactate oxidation provides a fitness advantage in competition with the gut microbiota, we intragastrically inoculated groups of Swiss Webster mice with the S. Tm wild-type strain and the ΔlldD Δdld mutant, respectively (single infection). Eight days after infection, colonization of the cecal and colonic lumen by the ΔlldD Δdld mutant was significantly reduced compared to the wild-type strain (Fig. 1C). Respiratory lactate dehydrogenase activity did not significantly enhance S. Tm fitness during systemic infection (Fig. S2E). Collectively, these data indicated that during gut colonization, S. Tm accessed the lactate pool and that lactate degradation provided a fitness advantage for the pathogen.
Lactate utilized by S. Tm is host-derived
Next, we investigated the source of lactate in the gut lumen. As a simple fermentation end product, lactic acid can be generated by virtually all members in the tree of life, including gut bacteria (Belenguer et al., 2007) and mammalian cells (Augoff et al., 2015). To determine if lactate originated from the mouse diet, we repeated the S. Tm competition experiment in mice that were fed a defined, lactate-free diet (Fig. 1B). In this experiment, the wild-type strain outcompeted the lactate utilization deficient strain to a similar extent as with our standard rodent diet (9-fold, P < 0.05 when comparing the individual wild-type and ΔlldD Δdld populations), indicating that dietary lactate only plays a minor role during S. Tm infection.
The S. Tm genome encodes a third lactate dehydrogenase, ldhA, which is involved in fermentative lactate production. It is conceivable that cross-feeding of lactate could occur between two S. Tm subpopulations in the gut lumen. To explore this possibility, we determined the competitive fitness of a ΔldhA mutant and a ΔldhA ΔlldD Δdld mutant. In the absence of ldhA, respiratory lactate dehydrogenase activity still provided a fitness advantage in colonizing the cecal lumen (Fig. 1B), demonstrating that lactate does not originate from S. Tm.
Given that many members of the microbiota can produce lactate (Belenguer et al., 2007), we tested the hypothesis that the microbiota was the predominant source of lactate during S. Tm infection using a gnotobiotic mouse model. Mice reared in a germ-free environment rapidly succumb to S. Tm infection due to uncontrolled systemic replication. To circumvent this limitation, we generated a S. Tm mutant that lacks a functional T3SS-2 (ΔsseD mutant) and thus is unable to replicate at systemic sites (Hensel et al., 1995). Germ-free mice were intragastrically inoculated with an equal mixture of the ΔsseD mutant and an isogenic ΔsseD ΔlldD Δdld mutant. Significant cecal inflammation was observed ten days after infection (Fig. S3 and S4). Importantly, the ΔsseD mutant was recovered in higher numbers than the isogenic ΔsseD ΔlldD Δdld mutant (35-fold, P < 0.05 when comparing the individual wild-type and ΔlldD Δdld populations; Fig. 1D), suggesting that lactate utilization still provides a fitness advantage for S. Tm even in the absence of the microbiota.
We next investigated whether the host could be a major source of lactate. As a proof of principle experiment, germ-free Swiss Webster mice were either mock-treated or received sodium oxamate, an inhibitor of fermentative lactate dehydrogenases, in the drinking water for 2 days. We measured the concentration of lactate in the cecal content using GC/MS/MS (Fig. 1E). Lactate was abundant in the cecal lumen of germ-free mice (~ 0.38 mM). Oxamate treatment significantly reduced lactate levels, suggesting that host lactate dehydrogenases are a major source of lactate in germ-free animals. Also, oxamate treatment of conventionally-raised, S. Tm-infected mice significantly decreased lactate concentrations (Fig. 1A), an observation that is consistent with the notion that increases in lactate levels during S. Tm infection were likely of host origin.
S. Tm can utilize both L-(+)- and D-(−)- lactate in vitro (Fig. S1B). The lactate-degrading, respiratory lactate dehydrogenases LldD and Dld allow for the utilization of L-(+)- and D-(−)- lactate, respectively (Fig. S1B). Lactate-generating, mammalian lactate dehydrogenases solely generate the L-enantiomer while fermenting microbes in the gut microbiota produce both enantiomers. Based on our hypothesis that the majority of lactate during S. Tm infection is host-derived, we predicted that S. Tm would predominantly utilize L-lactate during infection. A mutant lacking only LldD activity (ΔlldD mutant) recapitulated the fitness defect of the ΔlldD Δdld double mutant (Fig. 1B), supporting this hypothesis. This fitness advantage is also seen when we competed a genetically complemented strain (ΔlldD phoN::lldD+) against the L-lactate utilization deficient mutant (ΔlldD) (Fig. 1B). A mutant lacking only Dld activity did not display a competitive colonization defect (data not shown). Collectively, these experiments suggest that utilization of host-derived lactate enhances S. Tm fitness in the inflamed gut.
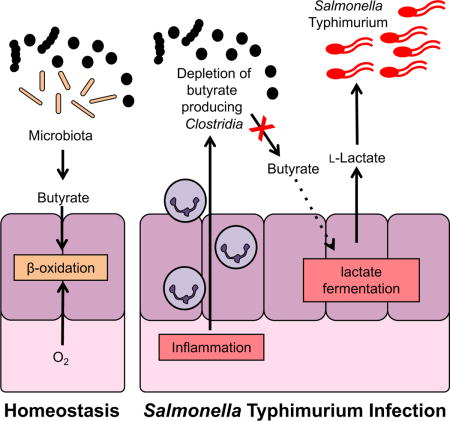
Depletion of microbial butyrate changes epithelial cell metabolism to lactate fermentation
Under homeostatic conditions, fermentation of host-inaccessible complex polysaccharides by members of the microbiota, primarily Clostridia clusters IV and XIVa, gives rise to the short chain fatty acid butyrate (Barcenilla et al., 2000; Pryde et al., 2002). In intestinal epithelial cells, butyrate is consumed through β-oxidation and energy is generated by oxidative phosphorylation. In the absence of butyrate, e.g. in germ-free mice, intestinal epithelial cells change their metabolism from fatty acid oxidation to glucose fermentation with lactate as the main fermentation end product (Donohoe et al., 2012). In animal models of infectious and non-infectious colitis, a depletion of members in the class Clostridia and concomitant decreases in butyrate availability have been reported to alter colonocyte metabolism (Ahmad et al., 2000; Byndloss et al., 2017; Kelly et al., 2015; Rivera-Chavez et al., 2016). Thus, we hypothesized that ablation of Clostridia through antibiotic treatment would recapitulate these changes in the absence of a pathogen and result in lactate accumulation in the gut lumen. To test this idea, conventional C57BL/6 mice were orally treated with a single dose of streptomycin (Fig. 2). Streptomycin treatment decreased Clostridia populations by more than 4 orders of magnitude, with Clostridia populations returning to normal levels after 3 to 4 days (Fig. 2A). Concomitant with the ablation of Clostridia, a significant decrease in butyrate concentrations in the cecal content was observed (Fig. 2B). In contrast, lactate levels were significantly increased 2, 3, and 4 days after streptomycin-induced perturbation (Fig. 2C), mirroring the decline in butyrate levels. To determine whether exogenous supplementation with butyrate was sufficient to suppress the spike in lactate levels, streptomycin-treated mice were orally treated with tributyrin (glyceryl tributyrate) (Fig. 2D). This exogenous supplementation with a butyrate source significantly decreased the cecal lactate concentration three days after streptomycin treatment.
Figure 2. Perturbation of the gut microbiota by oral streptomycin treatment results in elevated lactate levels.
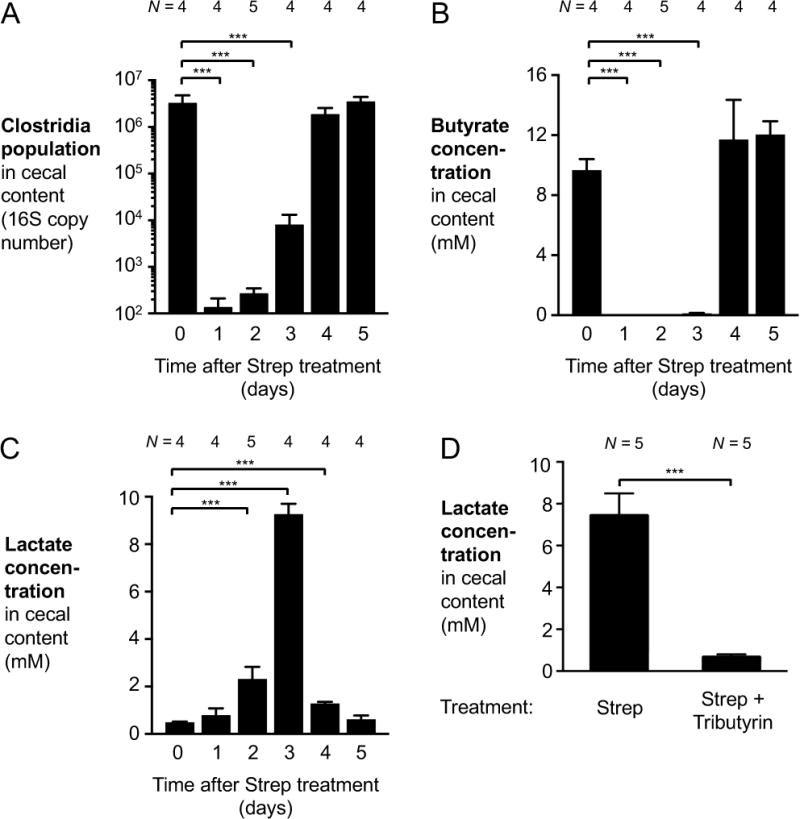
(A–C) Groups of C57BL/6 mice received a single oral dose of streptomycin (Strep). Samples of cecal content were taken 1 to 5 days after streptomycin administration. Mock-treated controls represent the 0-day time point. (A) qPCR analysis of DNA extracted from the cecal content to assess Clostridia populations in the microbiota. (B and C) The concentration of butyrate (B) and lactate (C) in the cecal content was quantified by GC/MS. (D) Groups of C57BL/6 mice were treated with a single dose of streptomycin. Animals then received oral tributyrin treatment through gavage and fortified chow, or were mock-treated. Lactate levels in the cecal content were measured 3 days after streptomycin treatment.
Bars represent geometric means ± standard error. ***, P < 0.001. The number of animals per group (N) is indicated above each bar.
In germ-free mice, colonocytes perform lactate fermentation due to a lack of microbial short chain fatty acids (Donohoe et al., 2012). We reasoned that colonization of gnotobiotic mice with a butyrate-producing bacterial strain would decrease availability of lactate. To test this idea, we repeated the competitive infection experiment with the ΔsseD and the ΔsseD ΔlldD Δdld mutant in germ-free mice that had been mono-associated with Clostridium symbiosum 3 days prior to infection. The fitness advantage conferred by S. Tm lactate dehydrogenases in the absence of other microbes was significantly reduced in the presence of C. symbiosum (Fig. 1D). No significant differences in cecal inflammation between the germ-free mice and those that had been pre-colonized with C. symbiosum prior to infection were noted (Fig. S3 and S4). Taken together, these experiments indicate that the lack of microbial butyrate induces a shift in host metabolism to lactate fermentation.
Lactate utilization during post-antibiotic expansion of the S. Tm population
Antibiotic therapy predisposes to infection with non-typhoidal Salmonella serovars in both immune-competent and immune-suppressed individuals (Pavia et al., 1990). In murine models, oral pre-treatment with streptomycin decreases colonization resistance and leads to a pronounced post-antibiotic expansion of the Salmonella population in the gut lumen (Stecher et al., 2007). Given our findings on streptomycin-induced perturbations on lactate production by the epithelium, we investigated the role of S. Tm lactate metabolism in streptomycin pre-treated mice. Conventional C57BL/6 mice were intragastrically treated with one dose of streptomycin. One day later, these animals were intragastrically inoculated with S. Tm. Five days after infection, the cecal contents were collected for 16S qPCR analysis and measurements of lactate and butyrate concentrations (Fig. 3; Fig. S5E). Compared to healthy control mice, streptomycin-treated and S. Tm infected mice exhibited a significant decrease in Clostridia and Bacteroidetes populations and a bloom of the Enterobacteriaceae population (Fig. 3A). Cecal butyrate levels were significantly decreased while lactate levels were elevated in streptomycin-treated and S. Tm infected mice compared to mock-treated controls (Fig. 3B and C). Respiratory lactate dehydrogenases (5.4-fold), in particular the L-lactate dehydrogenase LldD (5.1-fold), provided a fitness advantage in colonizing the murine cecum since the S. Tm wild-type strain was recovered in higher numbers than isogenic ΔlldD Δdld and ΔlldD mutants in competitive infection experiments (Fig. 3D). Similarly, lactate utilization provided a fitness advantage in the absence of T3SS-1 and T3SS-2-induced inflammation (ΔinvA ΔspiB mutant background) (Fig. 3D – E; Fig. S5A – C).
Figure 3. S. Tm utilizes lactate during post-antibiotic expansion.
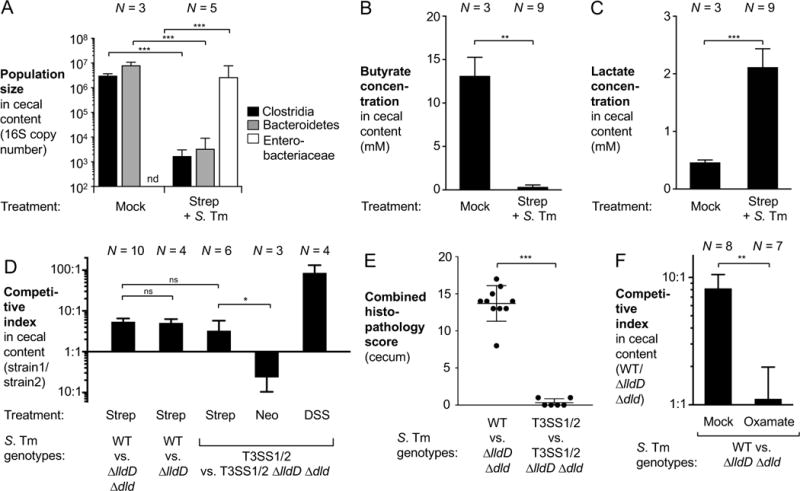
(A–C) Groups of C57BL/6 mice, pretreated with a single dose of oral streptomycin (Strep) were infected with the S. Tm wild-type strain (WT) or were mock-treated. Five days after infection, cecal content was collected for analysis. (A) 16S qPCR analysis of Clostridia, Bacteroidetes, and Enterobacteriaceae populations in the cecal microbiota. (B – C) Concentrations of butyrate (B) and lactate (C) in the cecal content were measured by targeted GC/MS. (D–E) C57BL/6 mice were pretreated with a single dose of streptomycin (Strep), neomycin (Neo), or 4 days of dextran sulfate sodium (DSS) treatment and were subsequently infected with an equal mixture of the indicated S. Tm strains through the intragastric route. DSS treatment was continued throughout the duration of the experiment, as indicated. Five days after infection, samples were collected for analysis. T3SS1/2, ΔinvA ΔspiB mutant. See also Fig. S5E. (D) Competitive indices of the indicated strains in the cecal content. (E) Combined histopathology score of cecal tissue. Each dot represents one animal while lines represent the average ± standard deviation. See also Fig. S5A – C. (F) Streptomycin-pretreated mice, infected with the S. Tm wild-type strain and the ΔlldD Δdld mutant, were treated with sodium oxamate in the drinking water or mock treated beginning one day after infection until the end of the experiment. The competitive index in the cecal content was determined 4 days after infection. See also Fig. S5E.
Bars represent geometric means ± standard error. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significant; nd, not detected. The number of animals per group (N) is indicated above each bar.
Streptomycin is effective at removing Gram-positive Clostridia (Fig. 2A). In contrast, oral pre-treatment with neomycin, an antibiotic that primarily targets Gram-negative bacteria (Macdonald and Beck, 1983), did not enable lactate utilization by S. Tm (Fig. 3D). We therefore concluded that streptomycin-induced perturbation of the microbiota is sufficient for lactate accumulation to occur.
To evaluate whether inhibition of fermentative host lactate dehydrogenases is sufficient to decrease lactate availability during S. Tm infection, groups of streptomycin pre-treated mice were intragastrically inoculated with the S. Tm wild-type strain and the ΔlldD Δdld mutant and were either mock-treated or received sodium oxamate in the drinking water and determined the competitive index 4 days after infection. Administration of sodium oxamate abolished the fitness advantage conferred by lactate oxidation in S. Tm in vivo (Fig. 3F). Sodium oxamate did not directly inhibit respiratory LldD and Dld activity (Fig. S1C) in vitro. Taken together, the experiments provide further support for the idea that during S. Tm infection, increased lactate levels are caused by altered host metabolism.
S. Tm-induced dysbiosis is characterized by a depletion of butyrate-producing Clostridia
To investigate microbiota changes induced by S. Tm, groups of conventional Swiss Webster mice were intragastrically inoculated with the S. Tm wild-type strain or mock-treated. After 8 days, the cecal content of these mice was collected for 16S rDNA sequencing (Fig. 4A – C). Unweighted principal coordinate analysis of microbiota composition showed distinct clustering of the microbiota of mock treated mice compared with that of infected mice (Fig. 4A). Taxonomic analysis of microbiota composition of mock treated mice revealed that during S. Tm infection, the phyla Bacteroidetes and Firmicutes were significantly decreased (Fig. 4B). In particular, populations of the class Clostridia were significantly diminished (3-fold; P < 0.05). In contrast, a bloom of Gammaproteobacteria, the class to which S. Tm belongs, was observed (Fig. 4C). In agreement with our initial profiling (Table S1), targeted measurements of butyrate by GC/MS demonstrated that mock-treated mice had nearly 10-fold higher levels of butyrate in their cecal content compared to that of S. Tm infected mice (Fig. 4D) and a concomitant increase in lactate concentrations (Fig. 1A). The fitness advantage conferred by lactate oxidation was time-dependent and correlated with the magnitude of cecal inflammation (Fig. S6). In a model of chemically-induced colitis (dextran sulfate sodium-induced colitis model; Fig. S5E), lactate oxidation provided a fitness advantage for Salmonella in the absence of bacterial virulence factors, consistent with the idea that gut inflammation is sufficient to increase luminal lactate levels (Fig. 3D). Collectively, these data suggest that Salmonella-induced inflammation is associated with a depletion of butyrate-producing Clostridia, which in turn correlates with increased availability of lactate.
Figure 4. Gut microbiota changes during S. Tm infection.
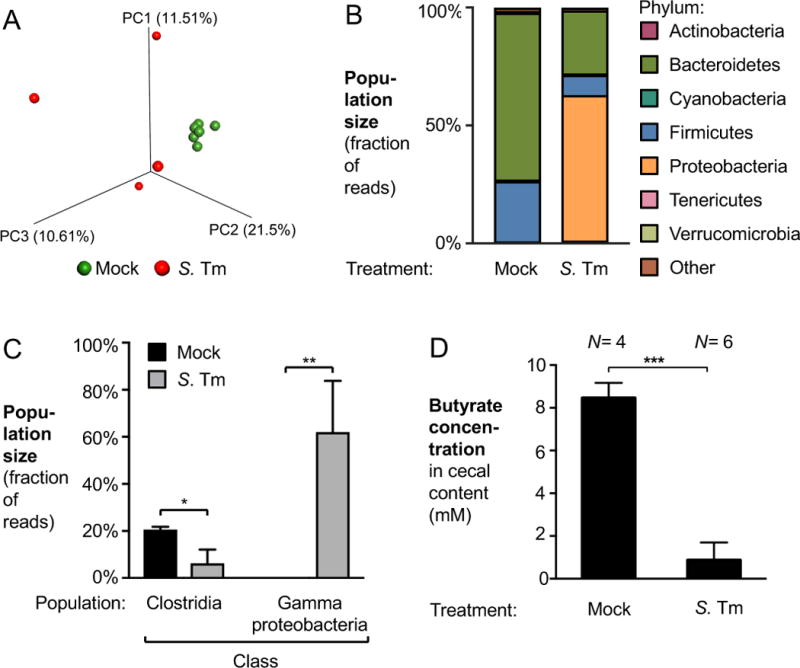
(A – C) Groups of Swiss Webster mice were intragastrically infected with the S. Tm wild-type strain (WT) or mock-treated. Eight days after infection, cecal content was collected for 16S rDNA profiling. (A) Principal coordinate (PC) plot of unweighted UniFrac distances generated from cecal microbial communities. Mock treatment, green spheres; S. Tm infection, red spheres. (B) Cecal microbiota composition at the phylum level. (C) Abundance of the classes Clostridia and Gammaproteobacteria in the ceca of mock-treated (black bars) and S. Tm-infected mice (gray bars). In mock-treated animals, Gammaproteobacteria were detected at very low abundance (less than 0.15%) (D) Swiss Webster mice were infected as described above. Butyrate concentrations in the cecal contents were determined 8 days after infection using GC/MS. See also Fig. 1A.
Bars represent geometric means ± standard error. *, P < 0.05; **, P < 0.01; ***, P < 0.001. The number of animals per group (N) is indicated above each bar.
Manipulation of PPARγ signaling influences lactate production during S. Tm infection
PPARγ is a transcription factor that controls fatty acid-based metabolism in the host. PPARγ is expressed in intestinal epithelial cells (Adachi et al., 2006) and orchestrates the switch from fermentation of glucose to β-oxidation in this cell type (Byndloss et al., 2017) (Fig. 5A). We therefore hypothesized that lactate production by the intestinal epithelium could be prevented by pharmacological activation of PPARγ signaling. We administered the PPARγ agonist rosiglitazone or a vehicle control via intraperitoneal injection to streptomycin-treated C57BL/6 mice to drive epithelial β-oxidation (Fig. 5B and C). As expected, rosiglitazone did not interfere with the streptomycin-induced depletion of butyrate in the cecal content (Fig. 5B). Interestingly, cecal lactate levels were significantly reduced during rosiglitazone treatment (Fig. 5C). Administration of rosiglitazone in the streptomycin-treated mouse model of Salmonella infection decreased the fitness advantage conferred by L-lactate utilization (Fig. 5D). These experiments support the idea that PPARγ controls the metabolic switch that is responsible for host epithelial lactate production during S. Tm infection.
Figure 5. Effect of PPARγ signaling on lactate availability during S. Tm colitis.
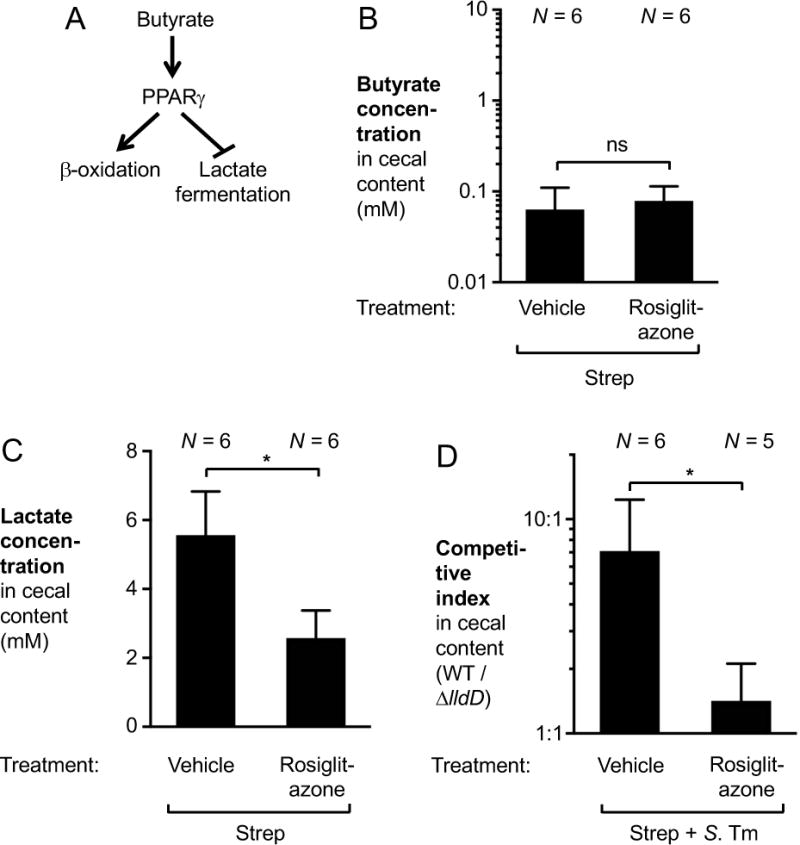
(A) Schematic model of the effect of PPARγ on host cell metabolism. (B and C) Groups of C57BL/6 mice were given a single oral dose of streptomycin (Strep). The following day, rosiglitazone or vehicle (50 % DMSO in PBS) was administered intraperitoneally. Concentrations of butyrate (B) and lactate (C) in the cecal content were measured by GC/MS the day after. (D) Groups of streptomycin-pretreated C57BL/6 mice were infected with an equal mixture of the S. Tm wild-type strain and a ΔlldD mutant for 5 days. Rosiglitazone or vehicle control was administered intraperitoneally for the last 3 days of infection.
Bars represent geometric means ± standard error. *, P < 0.05; ns, not statistically significant. The number of animals per group (N) is indicated above each bar.
Oxygen is the terminal electron acceptor for lactate utilization
We next wanted to determine the identity of the terminal electron acceptor for lactate utilization. Under homeostatic conditions, β-oxidation consumes most of the oxygen available at the gut mucosal interface (Kelly et al., 2015). During S. Tm infection, unconsumed oxygen leaks into the gut lumen where it enables respiration by the facultative anaerobic pathogen (Rivera-Chavez et al., 2016). In addition, S. Tm respires nitrate and tetrathionate, byproducts of the inflammatory oxidative burst (Lopez et al., 2015; Lopez et al., 2012; Winter et al., 2010). In vitro, lactate utilization occurs in fully aerated media and under microaerobic conditions (1 % oxygen) (Fig. S1A and 6A). Under anaerobic in vitro conditions, lactate dehydrogenases provide a growth advantage only in the presence of nitrate or tetrathionate (Fig. S1A). To test if anaerobic nitrate or tetrathionate respiration is required for lactate utilization, we created a mutant that lacked moaA. The moaA gene product catalyzes the first step of molybdopterin biosynthesis, an essential cofactor in the anaerobic tetrathionate and nitrate reductases. We then performed competition experiments in the streptomycin-treated C57BL/6 model (Fig. S5D). The fitness advantage conferred by lactate dehydrogenase genes was recapitulated in the absence of molybdopterin biosynthesis, i.e. in the moaA mutant background, suggesting that molybdenum cofactor-dependent anaerobic respiratory pathways are dispensable for lactate utilization. To test the idea that oxygen could be the terminal electron acceptor for lactate utilization, we created a mutant lacking cytochrome bd oxidase (CydAB) activity. CydAB is a terminal oxidase that is expressed under microaerobic conditions. Using the streptomycin-treated mouse model, we then determined the competitive fitness of the ΔcydA mutant against an isogenic strain that is unable to utilize L-lactate (ΔcydA ΔlldD). The fitness advantage conferred by LldD (Fig. 3D and 6B) was negated in the absence of CydAB activity (Fig. 6A and B), suggesting that oxygen is the preferred electron acceptor for oxidation of L-lactate during S. Tm infection.
Figure 6. Impact of aerobic respiration on lactate utilization in S. Tm.
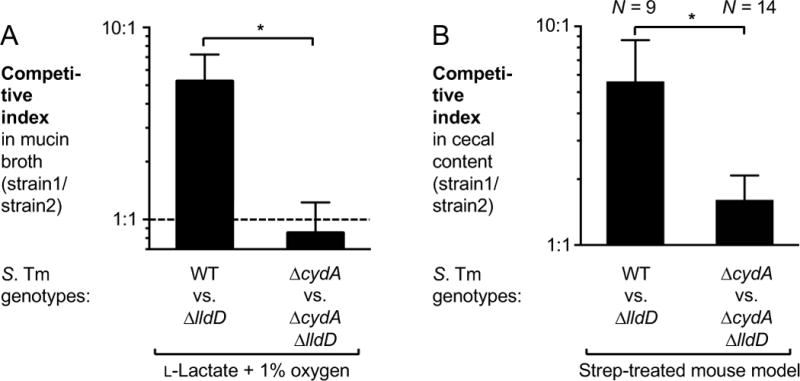
(A) Mucin broth supplemented with L-lactate was inoculated with an equal mixture of the S. Tm wild-type strain (WT) and a ΔlldD mutant or a ΔcydA and a ΔcydA ΔlldD mutant and incubated in a Coy chamber with 1% oxygen. Fitness was measured by determining the competitive index. The experiment was performed independently at least four times. (B) Groups of C57BL/6 mice were pre-treated with streptomycin (Strep) and subsequently infected with the indicated S. Tm strains. Five days after infection, cecal content was collected to determine the competitive index.
Bars represent geometric means ± standard error. *, P < 0.05. The number of animals per group (N) is indicated above each bar. See also Fig. S5D.
DISCUSSION
The nutritional mechanisms that support S. Tm outgrowth during inflammation, specifically the carbon sources that can be accessed by S. Tm in the inflamed intestine, remain poorly characterized. To address this question, we took an untargeted metabolic profiling approach to identify metabolites that become more abundant during S. Tm induced inflammation. This experiment identified lactate as a potential nutrient that increased in abundance in the infected cecum. Disruption of the normal microbiota composition through antibiotic treatment or the onset of mucosal inflammation can lead to accumulation of microbial metabolites, for example because consumers of small metabolites are depleted (Ferreyra et al., 2014; Hughes et al., 2017; Ng et al., 2013). We thus had initially hypothesized that most of the lactate in the gut lumen during S. Tm infection was of microbial origin. Contrary to this initial hypothesis, increased lactate levels were not primarily due to changes in gut microbial community composition or their metabolism. Instead, the increase in gut lactate levels was caused by a shift in the central metabolism of the host, most likely intestinal epithelial cells. While the microbiota was not directly responsible for increased lactate levels in our animal models, it played a major functional role by providing butyrate, the preferred carbon source of intestinal epithelial cells. Depletion of butyrate-producing Clostridia, either through streptomycin administration, DSS treatment, or during S. Tm induced dysbiosis, was sufficient to induce the production of host-derived lactate. The picture emerging from this study is that during S. Tm infection, butyrate and lactate link the metabolism of the gut microbiota, the host, and the pathogen at the mucosal interface.
Recent work by Rivera-Chavez et al. demonstrates how a change in intestinal epithelial cell metabolism from β-oxidation to fermentation results in the leakage of oxygen from the tissue into the intestinal lumen, supporting growth of facultative anaerobic S. Tm (Rivera-Chavez et al., 2016). In our study, we found that terminal oxidase activity (CydAB) was required for S. Tm to benefit from lactate utilization, indicating that oxygen was the terminal electron acceptor for lactate degradation in vivo. A respiratory metabolism allows S. Tm to be much more energetically competitive with the gut microbiota, which is largely comprised of strict anaerobes that solely rely on fermentative processes. While oxygen is required for L-lactate oxidation in vivo, luminal oxygenation is not sufficient for S. Tm to utilize lactate from other sources (e.g. microbiota), since inhibition of host lactate production with oxamate abolished the fitness advantage conferred by lactate oxidation. Our findings suggest that lactate dehydrogenases and terminal oxidases form a disease-specific metabolic module. Thus, manipulation of epithelial metabolism through S. Tm-induced dysbiosis produces both the electron donor and an electron acceptor for lactate oxidation.
S. Tm also exploits a corresponding shift in host cell metabolism during systemic infection. During persistent S. Tm infection, anti-inflammatory M2 macrophages represent a major replicative niche for the pathogen, especially during later stages of infection (Eisele et al., 2013; McCoy et al., 2012; Nix et al., 2007). M2 macrophages perform a fatty-acid oxidation based metabolism, which is controlled by the transcriptional regulator PPARδ. Reliance on β-oxidation for energy production liberates intracellular glucose for consumption by S. Tm, which supports replication inside of M2 macrophages (Eisele et al., 2013). In intestinal epithelial cells, reprogramming of central metabolism is controlled by PPARγ (Byndloss et al., 2017). Consistent with this recent report, we found that activation of PPARγ during S. Tm infection decreased availability of lactate in the gut lumen.
The inverse relationship between elevated fecal L-lactate and decreased butyrate levels during episodes of inflammation has been known for almost fifty years. Elevated levels of fecal L-lactate are frequently found in patients with non-infectious chronic diarrhea, especially during severe ulcerative colitis and in a subset of Crohn’s disease patients with colon involvement. Interestingly, in Crohn’s disease patients in which disease activity is restricted to the ileum, no such trend is observed (Bjerrum et al., 2015; Hove et al., 1995; Hove and Mortensen, 1995; Montgomery et al., 1968; Vernia et al., 1988a; Vernia et al., 1988b). In many of these patient samples a decrease in butyrate concentrations was noted (Vernia et al., 1988a; Vernia et al., 1988b). This metabolic switch has also been shown in germ-free mice (Donohoe et al., 2012) and in ex vivo-cultured colonocytes from DSS-treated mice (Ahmad et al., 2000), a chemical model of experimental colitis. The inverse correlation between elevated luminal lactate levels and severe colonic inflammation is consistent with our results showing that inflammation-associated dysbiosis induces a metabolic reprogramming (e.g. lactate production), and suggests that this mechanism could also be occurring in patients with chronic non-infectious diarrhea. In patients with inflammatory bowel disease, it was speculated that disease-associated increases in certain metabolites were due to malabsorption or altered microbiota metabolism; in contrast, our studies in animal models of S. Tm colitis raise the possibility that increases in lactate, and possibly other metabolites, are in fact generated by the host metabolism during disease. Overall, by combining bacterial genetics with targeted and untargeted metabolomics, our work revealed a metabolic connection between the gut microbiota, the host, and the enteric pathogen S. Tm and expands our understanding of how microbiota dysbiosis affects tissue homeostasis.
STAR METHODS
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sebastian Winter (Sebastian.Winter@UTSouthwestern.edu)
Experimental model and subject details
Mouse lines
Conventional Swiss Webster and C57BL/6 mice were bred in-house in barrier specific pathogen-free facilities at UT Southwestern. Mice were mixed and randomized 3 days prior to the beginning of each experiment. Male and female mice aged 6–8 weeks were used for all experiments and no differences between male and female mice were identified. Germ-free mice were reared in germ-free facilities at UT Southwestern and fed sterile food and water. All mice were on a 12-hour light/dark cycle and received food and water ad libitum. Mice that were euthanized early due to health concerns and mice that were insufficiently colonized (<10 colonies in 100 μL of undiluted sample; competitive infections only) were excluded from analysis. All mouse experiments were performed in accordance with the Institutional Animal Care and Use Committee at UT Southwestern (APN#2014-0061 and 2016-101801).
Bacterial strains
All strains used in this study are listed in the Key Resources Table. All S. Tm and E. coli strains were cultured in LB broth (10 g/L tryptone, 5 g/L yeast extract, 10 g/L sodium chloride) or on LB plates (LB broth, 15 g/L agar) and incubated at 37 °C. Nalidixic acid (Nal), carbenicillin (Carb), Kanamycin (Kan), and Chloramphenicol (Cm) were added to LB broth and LB agar plates at a concentration of 50 mg/mL, 100 mg/mL, 100 mg/mL, and 15 mg/mL, respectively, as needed. Clostridium symbiosum was grown on thioglycollate plates for 2 days at 37 °C under anaerobic conditions and in pre-reduced chopped meat media for 3 days at 37 °C under anaerobic conditions. To distinguish between S. Tm strains, the chromogenic substrate 5-Bromo-4-chloro-3-indolyl phosphate (X-phos) was used to detect the activity of the acidic phosphotase PhoN. All plasmids and primers used in this study are listed in the key resources table. All suicide plasmids were constructed using the Gibson Assembly Cloning kit. To generate plasmids pCG1, pCG2, and pCG89, upstream and downstream regions of the gene were amplified using the Q5 Hot Start High Fidelity DNA Polymerase. PCR products were ligated into SphI-digested pRDH10 during the Gibson Assembly reaction. For pMW1, the upstream and downstream regions of sseD were amplified and cloned into digested pGP706 as described above. The lldD promoter region and coding regions were then cloned into SphI digested pSW327 to generate pCG142. The insertion in all newly constructed plasmids except for pCG142 were sequenced. The cloning strain for all plasmids was DH5α λpir. For the construction of CG1, CG2, CG89, CG124, CG171 and MW285, suicide plasmids were grown in DH5α λpir and extracted before transformation into S17-1 λpir, which served as a donor strain for conjugation with S. Tm. Single crossover events were selected by using LB plates containing the appropriate antibiotics. Counterselection was performed to identify second crossover events using sucrose plates (5 % sucrose, 15 g/L agar, 8 g/L nutrient broth base). This results in the creation of a clean and unmarked deletion. For the construction of CG142, pCG142 was conjugated into IR715 using S17-1 λpir. A single crossover event was selected for using Carb resistant plates followed by picking and patching onto Nal and X-phos plates. Integration into the phoN gene was identified by the white color of the colonies on X-phos plates. A P22 HT int-105 phage lysate was then prepared from the exconjugates, which transduced the mutation into CG2. CG9 was generated by transducing ΔlldD using phage P22 HT int-105 into CG2. phoN::Kanr was similarly transduced into CG9 to generate CG110. pMW1 was conjugated into CG9 via S17 λpir to generate MW292. ΔldhA was transduced into CG9 to generate CG111. phoN::Kanr was also transduced into CG89 to generate CG116. Δdld and ΔlldD were sequentially transduced into SPN487 to generate CG12 and CG30. phoN::Cmr was transduced into MW285 to generate MW287. To generate SW1401, phoN::Kanr was transduced into SPN487. SW1401 was generated by transducing phoN::Kanr into SPN487. To generate CG127 and CG131, ΔlldD and phoN::Kanr were each transduced into CG124. To generate CG115, Δdld and ΔlldD were sequentially transduced into FF283. To generate CG107 phoN::Kanr was transduced into FF283.
Method Details
S. Tm infections in conventional Swiss Webster mice
Six to eight-week-old male and female Swiss Webster mice were gavaged with 1 × 109 CFU of S. Tm strains. For competition experiments, mice were administered 5 × 108 CFU of each strain. After 8 days of infection, mice were sacrificed and colonic content, cecal content, and tissue were collected. Colonic and cecal contents were placed into sterile Phosphate Buffered Saline (PBS; pH = 7.4) and were serially diluted on selective agar plates to determine CFU/g of each S. Tm strain. For the ΔlldD and WT competition time course experiment, mice were also sacrificed on day 4 and day 6 after infection and samples were taken as described for the 8-day infection. Competitive indices were calculated by dividing the CFU/g of wild-type S. Tm recovered over the CFU/g of mutant S. Tm recovered, which was then divided by the same ratio in the inoculum. In the lactate-free diet experiment, mice received lactate-free food for all 8 days of the experiment. For single infection experiments, groups of Swiss Webster mice received 1 × 109 CFU of either WT S. Tm or the isogenic ΔlldD Δdld mutant. Eight days after infection, cecal content was collected and plated to determine the CFU/g of each strain. To evaluate the contribution of the host lactate dehydrogenase, groups of S. Tm infected mice were treated with 0.5 % sodium oxamate, or mock (sterile water) in their drinking water for the duration of the 8-day experiment. The exact number of mice used in each group is indicated above each graph or in the figure legend.
S. Tm infections in C57BL/6 mice
Seven-week-old C57BL/6 male and female mice were intragastrically pre-treated with 20 mg of sterile streptomycin sulfate or neomycin trisulfate hydrate one day prior to infection. For all experiments, except for the sodium oxamate administration in the streptomycin-treated C57BL/6 model, mice were inoculated with 1 × 105 CFU of S. Tm strains. During competition experiments, mice were administered 5 × 104 CFU of each strain. After 5 days of infection, mice were euthanized and samples were collected as described above. Rosiglitazone-treated, S. Tm infected mice received 200 μL of 0.75 mg/mL rosiglitazone solution or vehicle control (50% DMSO in PBS) by intraperitoneal injection for the last 3 days of the infection. Dextran sulfate sodium (DSS)-treated mice received sterile 2.7% DSS in their drinking water for 4 days prior to infection with 1 × 105 CFU of S. Tm strains. After 5 days of infection, the competitive index in the cecal content was determined as described above. For sodium oxamate administration in the streptomycin-treated C57BL/6 model, mice were infected with 1 × 109 CFU of S. Tm strains. (5 × 108 CFU of each competitor strain). These mice received 0.5 % sodium oxamate in their drinking water or mock (sterile water) beginning the day after infection and were sacrificed 4 days after infection. Sample collection was done as described for other experiments to determine the competitive index. For intraperitoneal infections with S. Tm, C57BL/6 mice were injected intraperitoneally with 1 × 104 CFU of S. Tm strains (5 × 103 CFU of each competitor strain). After 3 days, mice were sacrificed and the spleen, liver and mesenteric lymph nodes were collected for analysis. Organs were homogenized and plated on selective agar to determine the competitive index as described above. The exact number of mice used in each group is indicated above each graph or in the figure legend.
Treatments in Streptomycin-treated C57BL/6 mice
Mice treated only with streptomycin received 20 mg of intragastric streptomycin sulfate and were sacrificed 1 to 5 days after treatment. Tributyrin treated mice received 5 % tributyrin fortified chow and 100 μL of undiluted sterile tributyrin by gavage every day after streptomycin treatment. Mice were sacrificed 3 days after streptomycin treatment. Rosiglitazone-treated mice received 200 μL of 0.75 mg/mL rosiglitazone solution or vehicle control by intraperitoneal injection for one day following streptomycin treatment. Cecal content and tissue was collected from these experiments for analysis 2 days after streptomycin treatment. The exact number of mice used in each group is indicated above each graph or in the figure legend.
S. Tm infections in germ-free Swiss Webster mice
Germ-free Swiss Webster mice were gavaged with 1 × 105 CFU of S. Tm strains. During competition experiments, mice were administered 5 × 104 CFU of each strain. Germ-free mice pre-colonized with C. symbiosum were gavaged with 3 × 109 CFU of C. symbiosum three days prior to a 10-day infection with S. Tm. For sodium oxamate experiments, germ-free Swiss Webster mice received sterile 0.5 % sodium oxamate in their drinking water or mock treatment (sterile water) for two days. The exact number of mice used in each group is indicated above each graph or in the figure legend.
Gas Chromatography Mass Spectroscopy metabolic profiling and measurements of lactate and butyrate
Cecal content was collected, resuspended in sterile PBS, and placed on ice. Samples were then vortexed for two minutes and centrifuged at 6,000 × g for 15 minutes at 4 °C. The supernatant was aliquoted and mixed with 5 μM of deuterated lactate (sodium L-lactate-3,3,3-d3), succinate (succinic-2,2,3,3,-d4 acid) and butyrate (sodium butyrate-d7) as internal standards. Samples were centrifuged in a vacuum-centrifuge until dry and stored at −80 °C. Dried samples were resuspended in pyridine, sonicated for 1 minute, and incubated at 80 °C for 20 minutes. Samples were then derivatized in N-tert-butyldimethylsilyl-N-methylfluorotriacetamide with 1 % tert-Butyldimethylchlorosilane and incubated at 80 °C for 1 hour. The samples were then centrifuged for 1 minute at 16,000 × g and the supernatant was transferred to autosampler vials for analysis. All experiments were conducted using a Shimadzu TQ8040 triple quadrupole GC-MS. The injection temperature was 250 °C and the in jection split ratio was set to 1:100 with an injection volume of 1 μL. The oven temperature started at 50 °C for 2 min, increasing to 100 °C at 20 °C per min and to 330 °C at 40 °C per min with a final hold at this temperature for 3 min. Flow rate of the helium carrier gas was kept constant at a linear velocity of 50 cm/s. The column used was a 30 m × 0.25 mm × 0.25 μm Rtx-5Sil MS. The interface temperature was 300 °C. The electron impact ion source temperature was 200 °C, with 70 V ionization voltage and 150 μA current. For qualitative experiments, Q3 scans (ranging from 50–550 m/z to 1000 m/z per second) were performed and putative compounds identified by searching the NIST/EPA/NIH Mass Spectral Library (Standard Reference Database v14). The retention times for lactate and deuterated lactate were 9.898 and 9.891 minutes, respectively. To measure butyrate, Q3 scans were performed as described above, and the retention times for butyrate and deuterated butyrate were 5.818 and 5.748 minutes, respectively. Multiple reaction monitoring was then used to quantitatively measure lactate and deuterated lactate. Lactate had a target ion of m/z 261>233 and a reference ion of m/z 261>189. Deuterated lactate had a target ion of m/z 264>236 and a reference ion of m/z 264>189. The target and reference ions for butyrate and were m/z 145 and m/z 75>146. The target and reference ions for deuterated butyrate were m/z 152 and m/z 76>153, respectively.
Analysis of microbiota composition by qPCR
Cecal content was flash frozen in liquid nitrogen and stored at −80 °C. Bacterial DNA was then isolated using the PowerFecal DNA Isolation Kit. Microbiota composition was determined using SYBR Green based qPCR. Two μL of template DNA and 250 nM of each primer were used, with a final reaction volume of 11 μL. Primer sequences are listed in Supplementary Table 3. Data was obtained through the QuantStudio 6 Flex Instrument. Serial dilutions of plasmids containing the gene of interest cloned into the TOPO cloning vector were also analyzed to generate a standard curve and calculate absolute counts of the gene of interest.
16S rDNA sequencing and analysis
Cecal content was collected and the DNA was extracted from fecal samples using the MoBio PowerFecal kit per the recommendations of the manufacturer. The 16sV4 Region was amplified with the 515f–806R primer pair, and barcoded prior to sequencing. The barcoded amplicons were purified and quantified on a 2200 TapeStation. The libraries were sequenced to generate 250bp paired end reads using an Illumina Miseq system. A standard workflow for processing and quality assessment was applied to process the raw 16S sequence data (Caporaso et al., 2010). The QIIME (Quantitative Insights into Microbial Ecology) open source software package (Version 1.91) (Caporaso et al., 2010) was employed to perform sequence alignment, operational taxonomic units (OTUs) picking against the Silva database (Version 128, released on 02/06/2017) (Quast et al., 2013) clustering, phylogenetic and taxonomic profiling, ANOSIM and beta diversity analysis on the demultiplexed sequences. The analysis of beta diversity (principle coordinate analysis) was visualized using Emperor (Vazquez-Baeza et al., 2013).
Cytokine mRNA quantification from intestinal tissue
Flash frozen tissue was homogenized using a Mini-BeadBeater and RNA was extracted using the TRI Reagent manufacturer protocol. RNA preparations were also subjected to DNase I treatment prior to use. cDNA was made using TaqMan reverse transcription reagents. qPCR analysis of Nos2, Cxcl1, and Tnf, was then conducted as described previously, using 2 μL of cDNA as a template.
Histopathology analysis
Tissue was fixed in 10 % phosphate buffered formalin, followed by storage in 70 % ethanol. Samples were embedded in paraffin, cut into 5 μm sections and stained with hematoxylin and eosin. Blinded samples were then scored for signs of inflammation, including neutrophil and mononuclear cell infiltration, edema, epithelial damage, and exudate as listed in Supplementary Table 2 (Winter et al., 2013).
Growth competitions in mucin broth
Sterile hog mucin (0.5% [w/v]) was dissolved in No-carbon E (NCE) medium (0.2 g/L MgSO4*7 H2O, 3.9 g/L KH2PO4, 5.0 g/L anhydrous K2HPO4, and 3.5 g/L NaNH4HPO4*4 H2O) (Berkowitz et al., 1968; Vogel and Bonner, 1956). Sodium D,L-lactate, D-lactate, or L-lactate was added to designated samples at a final concentration of 40 mM. Sodium nitrate and potassium tetrathionate were added to a final concentration of 20 mM. Where indicated, sodium oxamate was added to mucin broth at a final concentration of 5% [w/v].1.8 mL of mucin broth was inoculated with a 1:1 ratio of the indicated strains and incubated in an anaerobic chamber, in a microaerobic chamber, or aerobically in a shaking culture (250 RPM) for 18 hours. The CFU/mL of each indicator strain was determined by plating serial dilutions on indicator agar. Each experiment was performed with at least 4 independent biological replicates.
Quantification and Statistical analysis
Data analysis was performed in Microsoft Excel and GraphPad Prism v7.0. Histopathology scores and PMN counts were analyzed using the non-parametric Mann-Whitney U-test. All other data was transformed by the natural logarithm before inferential and descriptive statistical analysis. The log-transformed data was normally distributed, as determined by the D’Agostino-Pearson normality test for large groups and the Shapiro-Wilk normality test for smaller groups, except for lactate concentrations in mock-treated mice (Fig. 1A), butyrate concentrations measured day 2 after streptomycin treatment (Fig. 2B), the competitive index in the systemic sites (Fig. S2E), and the competitive index shown in Fig. S6B at the day 4 time point. No predicted statistical outliers were removed since the presence or absence of these potential statistical outliers did not affect the overall interpretation. Mice that were euthanized early due to health concerns and mice that were insufficiently colonized (<10 colonies in 100 μL of undiluted sample; competitive infections only) were excluded from analysis. A two-tailed, paired Student’s t-test was used to determine statistical differences between wild-type and mutant bacterial populations within the same animal (competitive infection experiments). A two-tailed, unpaired Student’s t-test was applied to the log-transformed data to determine statistical difference between groups of mice. A P value of less than 0.05 was considered significant. Unless otherwise stated, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically significant. In all mouse experiments, N refers to the number of animals from which samples were taken. Sample sizes (i.e. the number of animals per group) were not estimated a priori since effect sizes in our system cannot be predicted.
Data and software availability
16S rDNA sequencing data has been deposited in the European Nucleotide Archive under the accession number PRJEB21705 (https://www.ebi.ac.uk/ena/data/view/PRJEB21705).
Supplementary Material
Highlights.
Salmonella Typhimurium utilizes L-lactate as a nutrient during gut infection
Lactate used by S. Tm during infection is primarily host-derived
Depletion of Clostridia from the microbiota alters host metabolism to produce lactate
Oxygen is the terminal electron acceptor for L-lactate utilization by S. Tm
Acknowledgments
Work in SEW’s lab was funded by the NIH (AI118807, AI103248, AI128151), The Welch Foundation (I-1858) and a Research Scholar Grant (RSG-17-048-01-MPC) from the American Cancer Society. Work in LVH’s lab was funded by the NIH (DK070855), the Welch Foundation (I-1874) and the Howard Hughes Medical Institute. CCG was supported by a NSF Graduate Research Fellowship (1000194723) and a NIH Institutional Research Training Grant (GM109776). ERH was supported by NIH Training Grants (GM109776, AI007520). RC was supported by a NIH Training Grant (GM109776). WZ received a Research Fellows Award from the Crohn’s and Colitis Foundation of America (454921). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the funding agencies. We would like to thank Drs. Julie Pfeiffer, David Hendrixson, Ezra Burstein, and Vanessa Sperandio for helpful discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.E.W., C.C.G., L.V.H., and R.L.S. designed and conceived the study; C.C.G., E.R.H., L.S., M.G.W., W.Z., R.C., and C.L.B. performed all experiments. R.L.S. and T.F.C. performed the histopathology analysis. All authors contributed to data analysis and writing the manuscript.
References
- Ackermann M, Stecher B, Freed NE, Songhet P, Hardt WD, Doebeli M. Self-destructive cooperation mediated by phenotypic noise. Nature. 2008;454(7207):987–990. doi: 10.1038/nature07067. [DOI] [PubMed] [Google Scholar]
- Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, Gumucio DL, Marin HE, Peters JM, Young HA, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55(8):1104–1113. doi: 10.1136/gut.2005.081745. Published online 2006/03/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad MS, Krishnan S, Ramakrishna BS, Mathan M, Pulimood AB, Murthy SN. Butyrate and glucose metabolism by colonocytes in experimental colitis in mice. Gut. 2000;46(4):493–499. doi: 10.1136/gut.46.4.493. Published online 2000/03/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MM, Newsom DL, Gonzalez JF, Sabag-Daigle A, Stahl C, Steidley B, Dubena J, Dyszel JL, Smith JN, Dieye Y, et al. Fructose-asparagine is a primary nutrient during growth of Salmonella in the inflamed intestine. PLoS Pathog. 2014;10(6):e1004209. doi: 10.1371/journal.ppat.1004209. Published online 2014/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augoff K, Hryniewicz-Jankowska A, Tabola R. Lactate dehydrogenase 5: an old friend and a new hope in the war on cancer. Cancer Lett. 2015;358(1):1–7. doi: 10.1016/j.canlet.2014.12.035. Published online 2014/12/22. [DOI] [PubMed] [Google Scholar]
- Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, Flint HJ. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. 2000;66(4):1654–1661. doi: 10.1128/aem.66.4.1654-1661.2000. Published online 2000/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, Salzman N. Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun. 2008;76(3):907–915. doi: 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D, Selbach M, Rollenhagen C, Ballmaier M, Meyer TF, Mann M, Bumann D. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;440(7082):303–307. doi: 10.1038/nature04616. [DOI] [PubMed] [Google Scholar]
- Belenguer A, Duncan SH, Holtrop G, Anderson SE, Lobley GE, Flint HJ. Impact of pH on lactate formation and utilization by human fecal microbial communities. Appl Environ Microbiol. 2007;73(20):6526–6533. doi: 10.1128/AEM.00508-07. Published online 2007/09/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz D, Hushon JM, Whitfield HJ, Jr, Roth J, Ames BN. Procedure for identifying nonsense mutations. J Bacteriol. 1968;96(1):215–220. doi: 10.1128/jb.96.1.215-220.1968. Published online 1968/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerrum JT, Wang Y, Hao F, Coskun M, Ludwig C, Gunther U, Nielsen OH. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics. 2015;11:122–133. doi: 10.1007/s11306-014-0677-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science. 2017;357(6351):570–575. doi: 10.1126/science.aam9949. Published online 2017/08/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. Published online 2010/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Wali A, Brylawski BP, Bultman SJ. Microbial Regulation of Glucose Metabolism and Cell-Cycle Progression in Mammalian Colonocytes. PLoS ONE. 2012;7(9):e46589. doi: 10.1371/journal.pone.0046589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, Mukundan L, Chawla A, Monack DM. Salmonella require the fatty acid regulator PPARdelta for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe. 2013;14(2):171–182. doi: 10.1016/j.chom.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F, Thiennimitr P, Spiga L, Byndloss MX, Litvak Y, Lawhon S, Andrews-Polymenis HL, Winter SE, Baumler AJ. Respiration of Microbiota-Derived 1,2-propanediol Drives Salmonella Expansion during Colitis. PLoS Pathog. 2017;13(1):e1006129. doi: 10.1371/journal.ppat.1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F, Tran L, Byndloss MX, Lopez CA, Velazquez EM, Kerrinnes T, Nuccio SP, Wangdi T, Fiehn O, Tsolis RM, et al. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature. 2016;534(7609):697–699. doi: 10.1038/nature18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreyra JA, Wu KJ, Hryckowian AJ, Bouley DM, Weimer BC, Sonnenburg JL. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe. 2014;16(6):770–777. doi: 10.1016/j.chom.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, Curtiss R., 3rd Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc Natl Acad Sci USA. 1989;86(16):6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutnick D, Calvo JM, Klopotowski T, Ames BN. Compounds which serve as the sole source of carbon or nitrogen for Salmonella typhimurium LT-2. J Bacteriol. 1969;100(1):215–219. doi: 10.1128/jb.100.1.215-219.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269(5222):400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- Hove H, Holtug K, Jeppesen PB, Mortensen PB. Butyrate absorption and lactate secretion in ulcerative colitis. Dis Colon Rectum. 1995;38(5):519–525. doi: 10.1007/BF02148853. [DOI] [PubMed] [Google Scholar]
- Hove H, Mortensen PB. Influence of intestinal inflammation (IBD) and small and large bowel length on fecal short-chain fatty acids and lactate. Dig Dis Sci. 1995;40(6):1372–1380. doi: 10.1007/BF02065554. Published online 1995/06/01. [DOI] [PubMed] [Google Scholar]
- Hughes ER, Winter MG, Duerkop BA, Spiga L, Furtado de Carvalho T, Zhu W, Gillis CC, Buttner L, Smoot MP, Behrendt CL, et al. Microbial Respiration and Formate Oxidation as Metabolic Signatures of Inflammation-Associated Dysbiosis. Cell Host Microbe. 2017;21(2):208–219. doi: 10.1016/j.chom.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17(5):662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Rivera-Chavez F, Byndloss MX, Baumler AJ. The Periplasmic Nitrate Reductase NapABC Supports Luminal Growth of Salmonella enterica Serovar Typhimurium during Colitis. Infect Immun. 2015;83(9):3470–3478. doi: 10.1128/IAI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Winter SE, Rivera-Chavez F, Xavier MN, Poon V, Nuccio SP, Tsolis RM, Baumler AJ. Phage-mediated acquisition of a type III secreted effector protein boosts growth of salmonella by nitrate respiration. MBio. 2012;3(3) doi: 10.1128/mBio.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RH, Beck M. Neomycin: a review with particular reference to dermatological usage. Clin Exp Dermatol. 1983;8(3):249–258. doi: 10.1111/j.1365-2230.1983.tb01777.x. Published online 1983/05/01. [DOI] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413(6858):852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- McCoy MW, Moreland SM, Detweiler CS. Hemophagocytic macrophages in murine typhoid fever have an anti-inflammatory phenotype. Infect Immun. 2012;80(10):3642–3649. doi: 10.1128/IAI.00656-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM. Helicobacter and salmonella persistent infection strategies. Cold Spring Harb Perspect Med. 2013;3(12):a010348. doi: 10.1101/cshperspect.a010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RD, Frazer AC, Hood C, Goodhart JM, Holland MR, Schneider R. Studies of intestinal fermentation in ulcerative colitis. Gut. 1968;9(5):521–526. doi: 10.1136/gut.9.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502(7469):96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix RN, Altschuler SE, Henson PM, Detweiler CS. Hemophagocytic macrophages harbor Salmonella enterica during persistent infection. PLoS Pathog. 2007;3(12):e193. doi: 10.1371/journal.ppat.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavia AT, Shipman LD, Wells JG, Puhr ND, Smith JD, McKinley TW, Tauxe RV. Epidemiologic evidence that prior antimicrobial exposure decreases resistance to infection by antimicrobial-sensitive Salmonella. J Infect Dis. 1990;161(2):255–260. doi: 10.1093/infdis/161.2.255. [DOI] [PubMed] [Google Scholar]
- Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217(2):133–139. doi: 10.1111/j.1574-6968.2002.tb11467.x. Published online 2002/12/14. [DOI] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. Database issue. Published online 2012/11/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Chavez F, Baumler AJ. The Pyromaniac Inside You: Salmonella Metabolism in the Host Gut. Annu Rev Microbiol. 2015;69:31–48. doi: 10.1146/annurev-micro-091014-104108. [DOI] [PubMed] [Google Scholar]
- Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, et al. Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe. 2016;19(4):443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RL. Pathobiology of salmonella, intestinal microbiota, and the host innate immune response. Front Immunol. 2014;5:252. doi: 10.3389/fimmu.2014.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I, Gill N, Jogova M, Tam N, Robertson M, de Llanos R, Li Y, Finlay BB. Salmonella SPI-1-mediated neutrophil recruitment during enteric colitis is associated with reduction and alteration in intestinal microbiota. Gut Microbes. 2010;1(1):30–41. doi: 10.4161/gmic.1.1.10950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiga L, Winter MG, Furtado de Carvalho T, Zhu W, Hughes ER, Gillis CC, Behrendt CL, Kim J, Chessa D, Andrews-Polymenis HL, et al. An Oxidative Central Metabolism Enables Salmonella to Utilize Microbiota-Derived Succinate. Cell Host Microbe. 2017;22(3):291–301 e296. doi: 10.1016/j.chom.2017.07.018. Published online 2017/08/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5(10):2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeb B, Claudi B, Burton NA, Tienz P, Schmidt A, Farhan H, Maze A, Bumann D. Parallel exploitation of diverse host nutrients enhances Salmonella virulence. PLoS Pathog. 2013;9(4):e1003301. doi: 10.1371/journal.ppat.1003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, Baumler AJ. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci USA. 2011;108(42):17480–17485. doi: 10.1073/pnas.1107857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolis RM, Adams LG, Ficht TA, Baumler AJ. Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect Immun. 1999;67(9):4879–4885. doi: 10.1128/iai.67.9.4879-4885.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2(1):16. doi: 10.1186/2047-217X-2-16. Published online 2013/11/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia P, Caprilli R, Latella G, Barbetti F, Magliocca FM, Cittadini M. Fecal lactate and ulcerative colitis. Gastroenterology. 1988a;95(6):1564–1568. doi: 10.1016/s0016-5085(88)80078-7. [DOI] [PubMed] [Google Scholar]
- Vernia P, Gnaedinger A, Hauck W, Breuer RI. Organic anions and the diarrhea of inflammatory bowel disease. Dig Dis Sci. 1988b;33(11):1353–1358. doi: 10.1007/BF01536987. [DOI] [PubMed] [Google Scholar]
- Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem. 1956;218(1):97–106. Published online 1956/01/01. [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467(7314):426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339(6120):708–711. doi: 10.1126/science.1232467. Published online 2013/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Santos RL, Tsolis RM, Stender S, Hardt WD, Baumler AJ, Adams LG. The Salmonella enterica serotype typhimurium effector proteins SipA, SopA, SopB, SopD, and SopE2 act in concert to induce diarrhea in calves. Infect Immun. 2002;70(7):3843–3855. doi: 10.1128/IAI.70.7.3843-3855.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.