

Abstract
In the last 25 years, a number of animal models, mainly rodents, have been generated with the goal to mimic cholestatic liver injuries and, thus, to provide in vivo tools to investigate the mechanisms of biliary repair and, eventually, to test the efficacy of innovative treatments. Despite fundamental limitations applying to these models, such as the distinct immune system and the different metabolism regulating liver homeostasis in rodents when compared to humans, multiple approaches, such as surgery (bile duct ligation), chemical-induced (3,5-diethoxycarbonyl-1,4-dihydrocollidine, DDC, α-naphthylisothiocyanate, ANIT), viral infections (Rhesus rotavirustype A, RRV-A), and genetic manipulation (Mdr2, Cftr, Pkd1, Pkd2, Prkcsh, Sec63, Pkhd1) have been developed. Overall, they have led to a range of liver phenotypes recapitulating the main features of biliary injury and altered bile acid metabolisms, such as ductular reaction, peribiliary inflammation and fibrosis, obstructive cholestasis and biliary dysgenesis. Although with a limited translability to the human setting, these mouse models have provided us with the ability to probe over time the fundamental mechanisms promoting cholestatic disease progression. Moreover, recent studies from genetically engineered mice have unveiled ‘core’ pathways that make the cholangiocyte a pivotal player in liver repair. In this review, we will highlight the main phenotypic features, the more interesting peculiarities and the different drawbacks of these mouse models.
Keywords: Cholangiocyte, Biliary injury, Altered bile acid metabolism, Experimental models
Cholestatic liver disorders are chronic disease conditions, resulting from a functional impairment of bile formation and/or bile flow due to defective secretion by hepatocytes or cholangiocytes, or to mechanical obstruction of bile flow through intra- or extrahepatic bile ducts [1–3]. From a clinical point of view, a common end-point of many forms of cholestasis is the progression to biliary fibrosis with development of portal hypertension not necessarily associated with cirrhosis and, eventually, to end-stage liver disease independently of etiology [2–4]. In the last 25 years, a number of different animal models have been established and have laid the basis for the development of novel pathogenetic concepts and, consequently, for testing in vivo new treatment strategies for this group of liver diseases. Among several vertebrate animals that have been variably considered, rodents, and particularly mice, have drawn special interest from basic and clinical researchers because of the many experimental advantages they can offer. They are small, have short life span and gestational period, and are therefore easy to maintain and breed in captivity [5,6]. Furthermore, the remarkable genetic similarity between mice and humans, combined with the possibility and convenience of genetic manipulation, provide us with the possibility to study the implication of specific genes/signaling pathways and relatively, to identify potential therapeutic targets [7]. Unfortunately, given the multiple differences between human and mouse, including liver morphology, no single mouse model can strictly reproduce all the features of human cholestatic liver diseases to date. Indeed, large variations in responses to noxious agents exist between humans and mice regarding pathogenicity, timing, and immuno-inflammatory reactions [5–8].
In the present review, we will systematically summarize the most commonly used rodent models available to study cholestatic liver disease and, in particular, the cholangiopathies (Table 1). In the first part of this review, we will briefly highlight the main features and the more interesting peculiarities of the current animal models in use for biliary injuries and altered bile acid metabolism. In the second part, we will focus on the genetically modified mice that target specific gene products affecting cholangiocyte functions essential for biliary repair. The fundamental histological biliary lesions variably reproduced by the different mouse models are summarized in Fig. 1.
Table 1.
Rodent models of biliary injury*.
| Disease model | Rodent model | Clinical phenotype | |
|---|---|---|---|
| Surgical | |||
| Bile Duct Ligation | Obstructive cholestasis | Surgical bile duct ligation and dissection rats or mice | Jaundice, rapid portal fibrosis |
| Chemical | |||
| DDC | Chronic cholestasis (PSC) | 3,5-Diethoxycarbonyl-1,4-dihydrocollidine treated mice | Segmental bile duct obstruction, pericholangitis involving extrahepatic bile ducts |
| ANIT | Acute and chronic cholestasis (PBC) | α-Naphtylisothiocyanate-treated rats or mice | Cholangitis confined to intrahepatic bile ducts with hepatocellular damage |
| Infective | |||
| RRV-A | BA | Rhesus rotavirus type A (RRV)-treated mice | Progressive extrahepatic ductal obstruction |
| Genetic | |||
| Mdr2 | PFIC-3, ICP, PSC | Abcb4−/− | Non-purulent cholangitis, biliary cirrhosis |
| BSEP(Spgp) | PFIC-2, BRIC2 | Abcb11− | None |
| CFTR | CFLD | CFTRtm1UNC | Ductal plugging, focal biliary cirrhosis |
| F508del | |||
| PKD1 | PLD-ADPKD | Pkd1(flox/):pCxCreER | Liver cysts non communicating with the biliary tree |
| PKD2 | Pkd2(flox/):pCxCreER | ||
| PRKCSH | ADPLD | Prkcsh(flox/):pCXCreER | Liver cysts non communicating with the biliary tree without kidney involvement |
| SEC63 | Sec63(flox/): pCXCreER | ||
| PKHD1 | ARPKD,CHF, Caroli syndrome |
Pkhd1del4/del4 PCK |
Progressive bile duct enlargement with portal fibrosis; renal involvement only in PCK rat |
PSC, Primary sclerosing cholangitis; PBC, Primary biliary cholangitis; BA, Biliary atresia; PFIC-3, Progressive familial intrahepatic cholestasis type 3; ICP, Intrahepatic cholestasis of pregnancy; PFIC-2, Progressive familial intrahepatic cholestasis type 2; BRIC2, Benign recurrent intrahepatic cholestasis type 2; CFLD, Cystic fibrosis liver disease; PLD-ADPKD, Polycystic liver disease associated with autosomal dominant polycystic kidney disease; ADPLD, autosomal dominant polycystic liver disease; ARPKD, Autosomal recessive polycystic kidney diseases; CHF, Congenital hepatic fibrosis.
The table includes only the animal models discussed in this review.
Fig. 1.
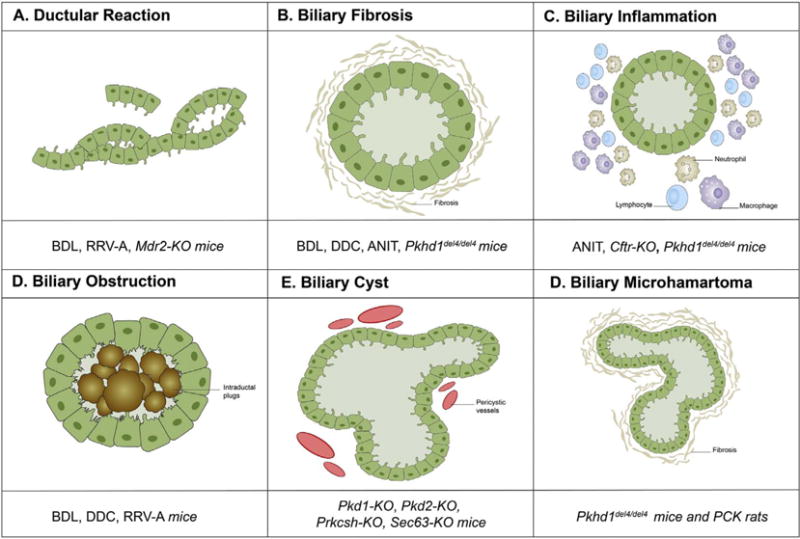
Fundamental phenotypes of biliary injury reproduced by a range of mouse models of different origin. The diagram illustrates the diverse biliary lesions recapitulated by each animal model discussed in the review, behaving as critical determinant of disease progression in cholestasis and cholangiopathies.
1. Bile duct ligation (BDL)
Bile duct ligation (BDL) is the most widely and the longest used experimental model for cholestasis because of its high reproducibility. Originally developed in rats, this model has been then successfully adapted for mice [9,10]. This technique requires a midventral laparotomy and isolation of the common bile duct above the duodenum, followed by double ligation of the bile duct and dissection between the ligatures, thus generating a model of obstructive cholestasis [11]. The structural and functional changes induced by BDL have been extensively analyzed and largely described in the literature. Briefly, surgical BDL induces strong proliferation of cholangiocytes in conjunction with variable activation of oval cells, i.e. hepatic progenitor cells, depending upon additional liver injuries, resulting in extensive ductular reaction, cholestasis, portal inflammation and rapid establishment of biliary fibrosis [9,11,12]. Although experimentally convenient, the flipside of the fast time course of biliary lesions is the fact that development of liver cirrhosis after only 30 days is clearly at odds with the slow progression of most chronic liver diseases, particularly cholangiopathies, usually spanning over decades [13]. More faithfully, this model reproduces acute obstructive biliary lesions, which, however, are rarely seen in human pathology, and are related to specific clinical settings, such as biliary atresia and choledocolithiasis [9,11,12,14]. Therefore, the BDL model has been predominantly used in studies evaluating cholangiocyte proliferation, apoptosis and portal fibrosis due to extrahepatic cholestasis. It is also useful in studies assessing the therapeutic effects of biliary drainage on hepatic blood flow and portal pressure [9]. Finally, additional issues are the relatively high mortality rates due to bile leakage and rupture of a biliary cyst (or gallbladder in mice) that may occur when performing BDL [11].
2. 3,5-Diethoxycarbonyl-1,4-dihydrocollidine (DDC)
Based on these considerations, to study the early pathological alterations occurring in chronic cholestatic diseases, such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), is essential to rely on an animal model with slow development of biliary injury [15–17]. Chronic DDC-feeding in mice is a well-established model of cholestatic liver injury originally proposed to study Mallory-Denk body formation, which is specifically associated with metabolic liver injury, as observed in alcoholic and nonalcoholic steatohepatitis, based on DDC ability to induce chronic oxidative cell stress [18–20]. Conversely, cholestatic effects exerted by DDC depends upon the ability to stimulate biliary porphyrin secretion that after a 4-week treatment leads to the generation of intraductal pigment plugs. Due to these properties, chronic DDC feeding has been proposed also as a model for xenobiotic-induced cholangiopathy, featuring ductular proliferation, intense pericholangitis associated with onion skin-type periductal fibrosis, which slowly progressed over time, leading to portal-portal bridging, and extending to the large bile ducts, thus recapitulating specific pathological hallmarks of human PSC [15,19]. This type of progressive biliary injury is initially characterized by specific transporter abnormalities, involving canalicular expression of Sodium/Taurocholate Cotransporter (Ntcp), organic anion transporting polypeptide (Oatp4), and Multidrug Resistance-Associated Protein 2 (Mrp2), responsible for reduced biliary excretion of glutathione (GSH) and phospholipids, occurring before phenotypic changes of cholangiocytes (‘reactive cholangiocyte’) [19]. Reactive cholangiocytes show increased expression of profibrogenic and proinflammatory cytokines, including osteopontin and tumor necrosis factor-alpha (TNF-α), associated with infiltration of neutrophils, and then with activation of periductal myofibroblasts, around both large and small bile ducts [21]. Recent morphological evidence showed that DDC-induced liver injury also leads to hepatocellular necrosis and, consequently, activation of Kupffer cells and compensatory hepatocyte proliferation [22–24]. Therefore, it is largely recognized the usefulness of this model to investigate the mechanisms of chronic cholestasis targeting both cholangiocytes and hepatocytes, and to test novel therapeutic approaches for these conditions [19,21].
3. α-Naphthyl-isothiocyanate (ANIT)
Similar to DDC, ANIT-feeding in mice provides a valuable model to investigate the molecular mechanisms of chemically induced cholangiopathy, with both acute and chronic injury [25]. ANIT is metabolized by hepatocytes and after conjugation with GSH, secreted via MRP2 into bile, whereby it exerts its toxic effects on cholangiocytes. Since ANIT-GSH complex is unstable in the bile, free ANIT undergoes absorption and metabolism recycling cycles, leading to progressively higher biliary concentrations, which become toxic [26]. Persistent exposure to toxic concentrations of unstable ANIT conjugates results in both cholangiocyte and hepatocyte damage and necrosis [27]. Low dose administration of ANIT over the course of 8 weeks in mice leads to bile duct proliferation, periportal inflammation, coupled with progressive biliary fibrosis and mild hepatocellular injury with increased transaminase levels [28–31]. On the other hand, administration of a single, high dose of ANIT (300 mg/kg body weight) to mice leads to rapid (15 to 24 h) cholestasis caused by severe destruction of cholangiocytes often extending to periportal hepatocytes [32]. TGF-β and αVβ6 integrin are intracellular signaling pathways involved in the mediation of obstructive cholestatic injury [29,33,34]. Notably, as compared with the DDC model, large extrahepatic bile ducts mostly remained unaffected, thus the biliary phenotype in ANIT-treated rodents resembles more the pathological findings observed in human PBC.
4. Infected RRV type A mice
In contrast to PBC and PSC, biliary atresia (BA), a multifaceted pediatric colangiopathy of heterogeneous pathogenesis, shows a more rapid progression to biliary cirrhosis due to a fibro-inflammatory obstruction of the extrahepatic bile ducts, associated to a marked proliferation of the intrahepatic ductal component developing together with variable inflammation and fibrosis at the portal level [35]. Several experimental models, including invertebrate animals (sea lampreys and zebrafish) have been proposed to investigate BA [36]. However, to date, mice infected with Rhesus rotavirus type A (RRV) in the first two postnatal days (exploiting the time-restricted susceptibility of cholangiocytes to infections after birth) represent the most accepted model since it captures several fundamental features of the human condition [37–39]. In particular, intrahepatic bile duct hyperplasia associated with portal inflammation dominated by a type I cell infiltrate, are faithfully reproduced in this model [40,41]. Thus, the RRV model provides unique opportunities: a) to probe the progression of extrahepatic bile duct obstruction over the course of the disease, and b) to unravel the role of the immune cell response in promoting epithelial injury and ductal obstruction [40–42]. However, given the early mortality of these mice at 2 weeks of age (thus before cirrhosis may develop), this model is unfitting to study mechanisms linking biliary injury to peribiliary fibrosis.
5. Mdr2-KO mice
The Mdr2 knockout (Mdr2-KO) mouse is one of the best-characterized models of fibrosing cholangiopathy (although caused primarily by hepatocyte dysfunction), generated by Smit and colleague [43,44] which in the last few years has gained considerable interest as surrogate model of PSC [15,16]. The multidrug resistance gene (Mdr2 in rodents/MDR3 in humans) belongs to the group of ABC transporters (ATP binding cassette subfamily B member 4 or Abcb4) [45] and encodes a canalicular flippase (translocase) expressed by hepatocytes, mediating the transport of biliary phospholipids, mainly phosphatidylcholine, into the outer leaflet of the canalicular cell membrane, which facilitates their subsequent excretion into the bile [44,46]. When Mdr2 is defective, lack of phospholipids in the bile profoundly affects its chemical composition, with increased biliary concentration of non-micellar-bound free bile acids, which exert detergent effects on the cell membrane, mainly at the cholangiocyte level [43,45,46]. Therefore, disruption of tight junctions and of the basement membrane beneath the biliary epithelium causes bile leakage to the portal tract, which induces inflammation, brisk ductular proliferation, ultimately leading to peribiliary deposition of fibrotic tissue. Obviously, the Mdr2-KO mouse has important implications for understanding the pathophysiology of human diseases derived from MDR3 defects, which are involved in a wide spectrum of cholestatic liver disorders, ranging from progressive familial intrahepatic cholestasis type 3 (PFIC-3), intrahepatic cholestasis of pregnancy (ICP), to adult biliary cirrhosis [47–19]. Moreover, as a highly reproducible biliary fibrosis model, with well-established and easily detectable readouts, the Mdr2-KO mouse has been often used to test innovative pharmacological strategies aimed at hindering scarring progression. From this point of view, nor-ursodeoxycholic acid (norUDCA) or UDCA treatment as well as integrin blockade have already proven to ameliorate fibrosis in Mdr2-KO mice, though some concerns have been raised with respect to their applicability to the human context [50–53]. Interestingly, at the age of 4–6 months, liver disease related to Mdr2 defects evolve to macroscopically visible tumor nodules which, quite surprisingly, are histologically compatible with hepatocellular carcinoma, rather than cholangiocarcinoma [22,54,55]. Thus, the Mdr2-KO mouse represents also a valuable model to investigate the long-term pro-tumorigenic functions played by a peribiliary chronic inflammatory response induced by cholestasis.
6. BSEP(Spgp)-KO mice
The bile salt export pump (BSEP) or sister of P-glycoprotein (Spgp) encoded by ABCB11, is the primary efflux transporter (member of the ATP binding cassette-ABC superfamily) of bile acids localized at the canalicular membrane of hepatocytes [56]. When BSEP function is impaired, defective bile acid secretion leads to progressive severe cholestasis. BSEP deficiency is associated with progressive familial intrahepatic cholestasis type 2 (PFIC-2), benign recurrent intrahepatic cholestasis type 2 (BRIC2), and several forms of acquired cholestasis [49,57–59]. Interestingly, rodent models lacking Bsep do not develop severe cholestasis probably due to the replacement of the bile acid pool with more hydrophilic bile acids, including muricholic acid and atypical bile acid species, which instead are not produced in humans [19,56,60]. Consequently, these animals do not show any histopathologic sign of liver injury unless treated with cholic acid [61].
Overall, the above-mentioned animal models have some major drawbacks in terms of translatability into clinic, particularly with respect to the stark differences in severity and celerity from human cholestasis. Therefore, mice harboring genetic inactivation of functional proteins normally expressed by cholangiocytes may offer a more fascinating prospect to unravel the complex scenario of biliary injury as it really occurs in humans. Moreover, targeting specific proteins involved in biliary ontogenesis enables us to probe selective morphogenetic pathways that are recapitulated in biliary repair.
7. Genetic mouse models of cholangiocyte dysfunction
7.1. Cftr-KO mice
Cystic fibrosis liver disease (CFLD) is a severe chronic cholangiopathy characterized by progressive peribiliary fibrosis that develops in less than 5% of patients with cystic fibrosis (CF) but, if present, represents the major cause of morbidity/mortality in these patients. Since the defective protein, cystic fibrosis transmembrane conductance regulator (CFTR), is a membrane channel selectively expressed by ductal epithelia, including cholangiocytes [62–64] where it regulates bile flow, Cl− and HCO3− secretion, vesicle-mediated fluid secretion and apical release of ATP, CFLD has been classically considered a consequence of the impaired bile flow and biliary alkalinization [63,65]. Almost 2000 different CFTR mutations have been identified and are grouped into five classes based on their functional defect and decreased severity [66,67]. Among them, deletion of a phenylalanine residue at position 508 (F508del, class II) is the most common mutation being present in 80% to 90% of patients with CF [66] and generation of F508del mice provide new tools to identify genes that are associated with residual ΔF508CFTR activity, leading to new therapeutic approaches [68–70].Adifferent mutation, S489X, blocking Cftr transcription, is harbored by congenic C57BL/6J-Cftrtm1Unc (Cftr-KO) mice. Studies performed in these mice have highlighted the concept that loss of CFTR function is not sufficient by itself to cause liver disease [69,71–73]. Thus, other factors, including modifier genes or environmental factors, might play a pathogenic role. Indeed, Cftr-KO mice express the secretory defect but develop liver disease under certain specific inbreeding conditions, or very late in life. More recent studies showed that in CF cholangiopathy the genetic defect is linked with a ‘second hit’ generated by an altered biliary innate immunity after exposure to potentially hepatotoxic conditions [74–76]. Indeed, dextran sodium sulfate (DSS)-feeding in Cftr-KO mice induced biliary damage and portal inflammation [75]. Noteworthy, despite its channel function, CFTR regulates TLR-mediated pro-inflammatory responses, by negatively controlling the activation of Src kinase, the non-receptorial tyrosine kinase responsible for phospho-TLR4 (Tyr674). When CFTR function is defective in cholangiocytes, Src is free to target TLR4 and increase its response to endotoxins, i.e. LPS [75,77]. In addition, aberrant activation of Src decreases the epithelia barrier function by destabilizing cell junctional complexes [77,78].
7.2. Pkd1- and Pkd2-KO mice and mice harboring other genetic defects related to polycystic liver disease
Polycystic liver disease associated with autosomal dominant polycystic kidney disease (PLD-ADPKD) is part of a spectrum of inherited cystic diseases that also includes autosomal dominant polycystic liver disease (ADPLD). Patients with PLD-ADPKD develop bilateral fluid-filled cysts in the kidney accompanied, in approximately 90% of cases, by multiple cysts scattered throughout the liver parenchyma without connection with the biliary tree. PLD-ADPKD is caused by mutations in either PKD1 or PKD2, the genes encoding for polycystin-1 (PC1) and polycystin-2 (PC2), respectively [79–82]. PC1 and PC2 are membrane proteins localized in the ductal epithelia, both in kidney and liver, where they regulate signaling pathways involved in epithelial cell morphogenesis, differentiation and proliferation [80,81]. Liver disease caused by defects in PC1 and PC2 are characterized by the formation of a large number of bile duct-derived liver cysts throughout the liver parenchyma without connection with the biliary tree, that may reach remarkable sizes and lead to major complications (mass effect, rupture, bleeding) [81,82]. Using the tamoxifen-induced Cre-mediated recombination for gene inactivation, Pkd1-KO and Pkd2-KO mice develop a liver phenotype closely resembling the human PLD-ADPKD [79,81,83,84]. In these mice, bile ducts are normal before the tamoxifen-induced gene inactivation, subsequently develop cystic lesions evident 4 weeks after induction, which progressively enlarge through maturation [85]. The mechanisms underpinning biliary cystogenesis have been recently elucidated. In Pkd2-KO mice, the cystic epithelium was shown to produce the vascular endothelial growth factor (VEGF) and to express the cognate receptor VEGFR2, leading to an autocrine loop characterized by cAMP/PKA-dependent activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway that, by stimulating the cystic epithelium proliferation, sustains the progressive cyst enlargement [85,86]. Likewise, VEGF secreted by the cystic biliary epithelium exerts a paracrine effect on the pericystic vasculature, further supporting the cyst growth, like observed in human patients [87,88]. Of note, the perturbation of the cAMP signaling observed in the cystic epithelia associates to changes in the intracellular Ca2+ homeostasis and to the involvement of Ca2+-inhibited adenylate cyclase isoform 5 (AC5) via the activation of an alternative pathway (store-operated cAMP production-SOcAMP) [89–91].
Liver cysts developing in ADPLD manifest a clinical phenotype indistinguishable from that of ADPKD. The two known disease causative genes for ADPLD are PRKCSH and SEC63, which encode for non-cilial, ER-localized proteins involved in protein biogenesis and post-translational modifications [92–94]. PRKCSH, also known as hepatocystin, encodes the non-catalytic β-subunit of glucosidase II (GIIβ). Glucosidase II acts in the calnexin-calreticulin cycle of folding and quality control for integral membrane and secreted proteins passing through the ER membrane [93,95]. The SEC63 gene product, SEC63p, is a 83-kDa protein that works in concert with the SEC61/62 ER translocon and BiP to facilitate the co-translational transport process across the endoplasmic reticulum (ER) membrane of nascent polypeptides destined to become either secreted or membrane-inserted proteins [92,96]. Mouse models based on tissue selective and inducible expression of Cre have been generated as orthologous gene models for human ADPLD. Induction of PDL genes inactivation in kidney and liver in adult Prkcshflox/flox; pCX-CreER mice (Prkcsh-KO) and Sec63flox/flox; pCX-CreER mice (Sec63-KO) results in the formation of either bile duct or kidney tubule cysts (unlike the human condition where no renal phenotype has been reported) [97]. Prkcsh- and Sec63-KO mice have been used to unravel the role of the integral ciliary proteins involved in the pathogenesis of ADPKD (i.e. PC1 and PC2) and autosomal recessive polycystic kidney disease (ARPKD) (i.e. PKHD1, see below) [97]. Studies from Fedeles and coll. Showed that loss of either GIIβ or Sec63p results in reduced levels of functional PC1-PC2 complex, with PC1 acting as the rate-limiting factor that halts the severity of the polycystic phenotype [98]. Furthermore, PC1 levels can be increased following proteasome inhibition leading to significant amelioration (as increase in apoptosis and decrease in proliferation of the cyst-lining epithelia) of the cystic phenotype of a model of ADPLD with Prkcsh inactivation [97,98].
7.3. Pkhd1del4/del4 mice and PCK rat
ARPKD, Caroli’s disease and congenital hepatic fibrosis (CHF) are rare diseases of the renal tubular and bile duct epithelia, caused by mutations in the PKHD1 gene [99,100]. PKHD1 encodes for fibrocystin (FPC), also known as polyductin, a plasma membrane protein located in the primary cilium that has a morphogenetic role in tubulogenesis and in maintaining the architecture of the epithelial duct lumen (‘planar cell polarity’) [100,101]. An experimental CHF mouse model has been generated by disrupting the exon 4 on Pkhd1 (Pkhd1del4/del4 mice) [102]. Pkhd1del4/del4 mouse develops intrahepatic bile duct dysgenesia (‘biliary microhamartoma’), which progressively evolves to a cyst-like configuration, but no renal involvement. Dysgenetic bile ducts can be observed as early as 2 weeks after birth, and their progression to cysts is paralleled by a progressive peribiliary fibrogenesis, detectable after 3 months, whilst hepatic function is normally preserved for up to 12 months. The presence of splenomegaly in more than 50% of the mice already at 6 months of age indicates clinically relevant portal hypertension, a feature mimicking the human disease behaviour, where portal hypertension represents the main determinant of the disease complications [102]. This model has been suitable to uncover some crucial aspects of biliary fibrogenesis in CHF. FPC-defective cholangiocytes displayed a hectic pro-inflammatory phenotype with abundant chemokine secretion (CXCL1, CXCL10, CXCL12), dependent upon an over-activation of the β-catenin signaling, enabling them to recruit macrophages and to respond to macrophage-derived cytokines (TNF-α, TGF-β) by up-regulating αvβ6 integrin expression. αvβ6 integrin is the local activator of latent TGF-β1, the most potent fibrogenic mediator in the liver [103,104]. Biliary fibrosis sped up when portal myofibroblasts were also recruited in the portal infiltrate, and this phase associated with development of portal hypertension. Accordingly, inhibition of macrophage infiltration by clodronate was relevant to impede progression of portal fibrosis [103].
Other rodent models have been fundamental in understanding the cellular alterations in cystic epithelia and in evaluating treatment efficacy. Along with the Pkhd1del4/del4 mouse, the PCK rat carrying a spontaneous splicing mutation in the Pck gene (PKHD1 in human), represents a well-recognized animal model of ARPKD [105,106]. Sanzen and coll. First described the liver phenotype in the PCK rats up to 4 months of age, characterized by progressive liver enlargement due to multiple saccular and segmental dilatations of the intrahepatic bile ducts. Further experimental studies on PCK rats highlighted the cyst disconnection from the biliary system with advancing age, consistent with the human disease features [107]. However, in contrast with the Pkhd1del4/del4 mouse, the PCK rat shows some critical differences, including a more pronounced development of the hepatic cysts, the renal involvement with cyst lesions affecting the outer medullary-collecting ducts, and the mild degree of portal fibrosis, without formation of fibrous septa or development of portal hypertension [108,109]. Overall, these characteristics make the PCK rat a less coherent model of the human CHF, at least with respect to the liver phenotype.
8. Conclusions
Generation of experimental animal models has provided fundamental clues to unveil many pathophysiological mechanisms triggered by biliary and cholestatic injury as well as to validate their clinical correlates. In the last decades, the main steps forward in modeling choletastic liver diseases have been reached with the help of mouse models of intrahepatic cholestasis (endotoxin-induced and drug-induced cholestasis) and extrahepatic biliary obstruction (BDL) [110–112]. However, although extensively characterized, these animal models have left several gaps in knowledge, given the difficulties to target selectively the biliary epithelium without the interference of additional (confounding) factors and to reproduce the smoldering clinical course peculiar of these conditions. On the other hand, several morphogenetic pathways whereby cholangiocytes regulate liver repair are increasingly recognized [62,113,114]. From this perspective, congenital cholangiopathies may represent a valuable asset to understand how a single genetic defect may affect specific cholangiocyte functions essential to mount a correct response to damage. Indeed, thanks to a variety of genetically engineered mouse models, different phases of biliary injury, starting from the earliest stages, can be probed with high accuracy, then followed through their progression, and eventually targeted by novel therapeutic approaches. In humans, these tasks cannot be usually performed given the long course of the disease and the limited possibility to realize sequential studies by collecting samples at different time points. Moreover, when human tissue is obtained from liver explants at the time of transplant, histological alterations are too advanced and generally less specific as dominated by the extensive liver damage. Given the broad role played by cholangiocytes in liver repair, these experimental models might provide in the near future a wealth of information extending well beyond the boundaries of these rare diseases.
Footnotes
This article is part of a Special Issue entitled: Cholangiocytes in Health and Disease edited by Jesus Banales, Marco Marzioni, Nicholas LaRusso and Peter Jansen.
Transparency document
The Transparency document associated with this article can be found, in online version.
References
- 1.L. European Association for the Study of the, EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51:237–267. doi: 10.1016/j.jhep.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology. 2010;139:1481–1496. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Gossard AA, Talwalkar JA. Cholestatic liver disease. Med Clin North Am. 2014;98:73–85. doi: 10.1016/j.mcna.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Pollock G, Minuk GY. Diagnostic considerations for cholestatic liver disease. J Gastroenterol Hepatol. 2017 doi: 10.1111/jgh.13738. [DOI] [PubMed] [Google Scholar]
- 5.S B, Fox JG, Davisson MT, et al., editors. The Mouse in Biomedical Research. Elsevier, Place Published; Amsterdam: 2007. [Google Scholar]
- 6.Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9:993–999. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- 7.M. HCI. Building a Better Mouse: One hundred Years of Genetics and Biology. Place Published; 2007. [Google Scholar]
- 8.Perlman RL. Mouse models of human disease: an evolutionary perspective. Evol Med Public Health. 2016;2016:170–176. doi: 10.1093/emph/eow014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geerts AM, Vanheule E, Praet M, Van Vlierberghe H, De Vos M, Colle I. Comparison of three research models of portal hypertension in mice: macroscopic, histological and portal pressure evaluation. Int J Exp Pathol. 2008;89:251–263. doi: 10.1111/j.1365-2613.2008.00597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krstulovic B, Van D, Desmet VJ. Comparative histochemical study of rat liver in bile-duct ligation and in alpha-napthyl isothiocyanate (ANIT) intoxication. Am J Pathol. 1968;52:423–436. [PMC free article] [PubMed] [Google Scholar]
- 11.Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, Tacke F, Weiskirchen R. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015 doi: 10.3791/52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aller MA, Arias JL, Prieto I, Losada M, Arias J. Bile duct ligation: step-by-step to cholangiocyte inflammatory tumorigenesis. Eur J Gastroenterol Hepatol. 2010;22:651–661. doi: 10.1097/MEG.0b013e32832e0a2f. [DOI] [PubMed] [Google Scholar]
- 13.Marques TG, Chaib E, da Fonseca JH, Lourenco AC, Silva FD, Ribeiro MA, Jr, Galvao FH, D’Albuquerque LA. Review of experimental models for inducing hepatic cirrhosis by bile duct ligation and carbon tetrachloride injection. Acta Cir Bras. 2012;27:589–594. doi: 10.1590/s0102-86502012000800013. [DOI] [PubMed] [Google Scholar]
- 14.Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, Krahenbuhl S, Krahenbuhl L, Schuppan D. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G323–G334. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fickert P, Pollheimer MJ, Beuers U, Lackner C, Hirschfield G, Housset C, Keitel V, Schramm C, Marschall HU, Karlsen TH, Melum E, Kaser A, Eksteen B, Strazzabosco M, Manns M, Trauner M, P.S.C.S.G. International Characterization of animal models for primary sclerosing cholangitis (PSC) J Hepatol. 2014;60:1290–1303. doi: 10.1016/j.jhep.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollheimer MJ, Fickert P. Animal models in primary biliary cirrhosis and primary sclerosing cholangitis. Clin Rev Allergy Immunol. 2015;48:207–217. doi: 10.1007/s12016-014-8442-y. [DOI] [PubMed] [Google Scholar]
- 17.Vierling JM. Animal models for primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2001;15:591–610. doi: 10.1053/bega.2001.0207. [DOI] [PubMed] [Google Scholar]
- 18.Fickert P, Trauner M, Fuchsbichler A, Stumptner C, Zatloukal K, Denk H. Bile acid-induced Mallory body formation in drug-primed mouse liver. Am J Pathol. 2002;161:2019–2026. doi: 10.1016/S0002-9440(10)64480-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fickert P, Stoger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, Tsybrovskyy O, Jaeschke H, Zatloukal K, Denk H, Trauner M. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 2007;171:525–536. doi: 10.2353/ajpath.2007.061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanada S, Strnad P, Brunt EM, Omary MB. The genetic background modulates susceptibility to mouse liver Mallory-Denk body formation and liver injury. Hepatology. 2008;48:943–952. doi: 10.1002/hep.22436. [DOI] [PubMed] [Google Scholar]
- 21.Fickert P, Thueringer A, Moustafa T, Silbert D, Gumhold J, Tsybrovskyy O, Lebofsky M, Jaeschke H, Denk H, Trauner M. The role of osteopontin and tumor necrosis factor alpha receptor-1 in xenobiotic-induced cholangitis and biliary fibrosis in mice. Lab Investig. 2010;90:844–852. doi: 10.1038/labinvest.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calvisi DF, Thorgeirsson SS. Molecular mechanisms of hepatocarcinogenesis in transgenic mouse models of liver cancer. Toxicol Pathol. 2005;33:181–184. doi: 10.1080/01926230590522095. [DOI] [PubMed] [Google Scholar]
- 23.French SW, Lee J, Zhong J, Morgan TR, Buslon V, Lungo W, French BA. Alcoholic liver disease - hepatocellular carcinoma transformation. J Gastrointest Oncol. 2012;3:174–181. doi: 10.3978/j.issn.2078-6891.2012.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, Tolba R, Trautwein C, Trebicka J, Weiskirchen R. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair. 2013;6:19. doi: 10.1186/1755-1536-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desmet VJ, Krstulovic B, Van Damme B. Histochemical study of rat liver in alpha-naphthyl isothiocyanate (ANIT) induced cholestasis. Am J Pathol. 1968;52:401–421. [PMC free article] [PubMed] [Google Scholar]
- 26.Carpenter-Deyo L, Marchand DH, Jean PA, Roth RA, Reed DJ. Involvement of glutathione in 1-naphthylisothiocyanate (ANIT) metabolism and toxicity to isolated hepatocytes. Biochem Pharmacol. 1991;42:2171–2180. doi: 10.1016/0006-2952(91)90353-7. [DOI] [PubMed] [Google Scholar]
- 27.Lleo A, Maroni L, Glaser S, Alpini G, Marzioni M. Role of cholangiocytes in primary biliary cirrhosis. Semin Liver Dis. 2014;34:273–284. doi: 10.1055/s-0034-1383727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Golbar HM, Izawa T, Ichikawa C, Tanaka M, Juniantito V, Sawamoto O, Kuwamura M, Yamate J. Slowly progressive cholangiofibrosis induced in rats by alpha-naphthylisothiocyanate (ANIT), with particular references to characteristics of macrophages and myofibroblasts. Exp Toxicol Pathol. 2013;65:825–835. doi: 10.1016/j.etp.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP. The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol. 2010;177:2837–2849. doi: 10.2353/ajpath.2010.100425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tjandra K, Sharkey KA, Swain MG. Progressive development of a Th1-type hepatic cytokine profile in rats with experimental cholangitis. Hepatology. 2000;31:280–290. doi: 10.1002/hep.510310204. [DOI] [PubMed] [Google Scholar]
- 31.Xu J, Lee G, Wang H, Vierling JM, Maher JJ. Limited role for CXC chemokines in the pathogenesis of alpha-naphthylisothiocyanate-induced liver injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G734–G741. doi: 10.1152/ajpgi.00300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Connolly AK, Price SC, Connelly JC, Hinton RH. Early changes in bile duct lining cells and hepatocytes in rats treated with alpha-naphthylisothiocyanate. Toxicol Appl Pharmacol. 1988;93:208–219. doi: 10.1016/0041-008x(88)90121-4. [DOI] [PubMed] [Google Scholar]
- 33.Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, Weinreb PH, Violette S, Sheppard D, Schuppan D, Popov Y. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. 2016;63:217–232. doi: 10.1002/hep.28274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, Kolb A, Friess H, Schuppan D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48:453–464. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 35.Bezerra JA. The next challenge in pediatric cholestasis: deciphering the pathogenesis of biliary atresia. J Pediatr Gastroenterol Nutr. 2006;43(Suppl. 1):S23–S29. doi: 10.1097/01.mpg.0000228197.28056.2f. [DOI] [PubMed] [Google Scholar]
- 36.Petersen C. Biliary atresia: the animal models. Semin Pediatr Surg. 2012;21:185–191. doi: 10.1053/j.sempedsurg.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Petersen C, Biermanns D, Kuske M, Schakel K, Meyer-Junghanel L, Mildenberger H. New aspects in a murine model for extrahepatic biliary atresia. J Pediatr Surg. 1997;32:1190–1195. doi: 10.1016/s0022-3468(97)90680-1. [DOI] [PubMed] [Google Scholar]
- 38.Petersen C, Kuske M, Bruns E, Biermanns D, Wussow PV, Mildenberger H. Progress in developing animal models for biliary atresia. Eur J Pediatr Surg. 1998;8:137–141. doi: 10.1055/s-2008-1071140. [DOI] [PubMed] [Google Scholar]
- 39.Riepenhoff-Talty M, Schaekel K, Clark HF, Mueller W, Uhnoo I, Rossi T, Fisher J, Ogra PL. Group a rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33:394–399. doi: 10.1203/00006450-199304000-00016. [DOI] [PubMed] [Google Scholar]
- 40.Hertel PM, Estes MK. Rotavirus and biliary atresia: can causation be proven? Curr Opin Gastroenterol. 2012;28:10–17. doi: 10.1097/MOG.0b013e32834c7ae4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233–242. doi: 10.1055/s-2007-985068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fabris L, Cadamuro M, Guido M, Spirli C, Fiorotto R, Colledan M, Torre G, Alberti D, Sonzogni A, Okolicsanyi L, Strazzabosco M. Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Am J Pathol. 2007;171:641–653. doi: 10.2353/ajpath.2007.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 44.Oude Elferink RP, Ottenhoff R, van Wijland M, Smit JJ, Schinkel AH, Groen AK. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95:31–38. doi: 10.1172/JCI117658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 46.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, Trauner M. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 47.de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, Desrochers M, Burdelski M, Bernard O, Oude Elferink RP, Hadchouel M. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A. 1998;95:282–287. doi: 10.1073/pnas.95.1.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis. 2007;27:77–98. doi: 10.1055/s-2006-960172. [DOI] [PubMed] [Google Scholar]
- 49.Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011;31:3–10. doi: 10.1055/s-0031-1272831. [DOI] [PubMed] [Google Scholar]
- 50.Fickert P, Wagner M, Marschall HU, Fuchsbichler A, Zollner G, Tsybrovskyy O, Zatloukal K, Liu J, Waalkes MP, Cover C, Denk H, Hofmann AF, Jaeschke H, Trauner M. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–481. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 51.Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, Marschall HU, Tsybrovskyy O, Zatloukal K, Denk H, Trauner M. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology. 2002;123:1238–1251. doi: 10.1053/gast.2002.35948. [DOI] [PubMed] [Google Scholar]
- 52.Halilbasic E, Fiorotto R, Fickert P, Marschall HU, Moustafa T, Spirli C, Fuchsbichler A, Gumhold J, Silbert D, Zatloukal K, Langner C, Maitra U, Denk H, Hofmann AF, Strazzabosco M, Trauner M. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2−/− mice. Hepatology. 2009;49:1972–1981. doi: 10.1002/hep.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fiorotto R, Spirli C, Fabris L, Cadamuro M, Okolicsanyi L, Strazzabosco M. Ursodeoxycholic acid stimulates cholangiocyte fluid secretion in mice via CFTR-dependent ATP secretion. Gastroenterology. 2007;133:1603–1613. doi: 10.1053/j.gastro.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 54.Katzenellenbogen M, Mizrahi L, Pappo O, Klopstock N, Olam D, Jacob-Hirsch J, Amariglio N, Rechavi G, Domany E, Galun E, Goldenberg D. Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol Cancer Res. 2007;5:1159–1170. doi: 10.1158/1541-7786.MCR-07-0172. [DOI] [PubMed] [Google Scholar]
- 55.Mu X, Espanol-Suner R, Mederacke I, Affo S, Manco R, Sempoux C, Lemaigre FP, Adili A, Yuan D, Weber A, Unger K, Heikenwalder M, Leclercq IA, Schwabe RF. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J Clin Invest. 2015;125:3891–3903. doi: 10.1172/JCI77995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lam P, Wang R, Ling V. Bile acid transport in sister of P-glycoprotein (ABCB11) knockout mice. Biochemistry. 2005;44:12598–12605. doi: 10.1021/bi050943e. [DOI] [PubMed] [Google Scholar]
- 57.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, Tanner MS, Kagalwalla AF, Nemeth A, Pawlowska J, Baker A, Mieli-Vergani G, Freimer NB, Gardiner RM, Thompson RJ. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 58.Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, Knisely AS, Houwen RH, Freimer NB. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 59.Paulusma CC, Groen A, Kunne C, Ho-Mok KS, Spijkerboer AL, Rudi de Waart D, Hoek FJ, Vreeling H, Hoeben KA, Marle Jvan, Pawlikowska L, Bull LN, Hofmann AF, Knisely AS, Oude Elferink RP. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44:195–204. doi: 10.1002/hep.21212. [DOI] [PubMed] [Google Scholar]
- 60.Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, Helgason CD, Ackerley C, Phillips MJ, Ling V. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A. 2001;98:2011–2016. doi: 10.1073/pnas.031465498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang R, Lam P, Liu L, Forrest D, Yousef IM, Mignault D, Phillips MJ, Ling V. Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatology. 2003;38:1489–1499. doi: 10.1016/j.hep.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 62.Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. J Clin Gastroenterol. 2005;39:S90–S102. doi: 10.1097/01.mcg.0000155549.29643.ad. [DOI] [PubMed] [Google Scholar]
- 63.Cohn JA, Strong TV, Picciotto MR, Nairn AC, Collins FS, Fitz JG. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology. 1993;105:1857–1864. doi: 10.1016/0016-5085(93)91085-v. [DOI] [PubMed] [Google Scholar]
- 64.Feranchak AP, Sokol RJ. Cholangiocyte biology and cystic fibrosis liver disease. Semin Liver Dis. 2001;21:471–488. doi: 10.1055/s-2001-19030. [DOI] [PubMed] [Google Scholar]
- 65.Colombo C, Battezzati PM, Strazzabosco M, Podda M. Liver and biliary problems in cystic fibrosis. Semin Liver Dis. 1998;18:227–235. doi: 10.1055/s-2007-1007159. [DOI] [PubMed] [Google Scholar]
- 66.Veit G, Avramescu RG, Chiang AN, Houck SA, Cai Z, Peters KW, Hong JS, Pollard HB, Guggino WB, Balch WE, Skach WR, Cutting GR, Frizzell RA, Sheppard DN, Cyr DM, Sorscher EJ, Brodsky JL, Lukacs GL. From CFTR biology toward combinatorial pharmacotherapy: expanded classification of cystic fibrosis mutations. Mol Biol Cell. 2016;27:424–433. doi: 10.1091/mbc.E14-04-0935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wilschanski M. Patterns of gastrointestinal disease associated with mutations of CFTR. Curr Gastroenterol Rep. 2008;10:316–323. doi: 10.1007/s11894-008-0062-3. [DOI] [PubMed] [Google Scholar]
- 68.Colledge WH, Abella BS, Southern KW, Ratcliff R, Jiang C, Cheng SH, MacVinish LJ, Anderson JR, Cuthbert AW, Evans MJ. Generation and characterization of a delta F508 cystic fibrosis mouse model. Nat Genet. 1995;10:445–452. doi: 10.1038/ng0895-445. [DOI] [PubMed] [Google Scholar]
- 69.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 70.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB, Jr, Capecchi MR, Welsh MJ, Thomas KR. Amouse model for the delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fisher JT, Zhang Y, Engelhardt JF. Comparative biology of cystic fibrosis animal models. Methods Mol Biol. 2011;742:311–334. doi: 10.1007/978-1-61779-120-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keiser NW, Engelhardt JF. New animal models of cystic fibrosis: what are they teaching us? Curr Opin Pulm Med. 2011;17:478–483. doi: 10.1097/MCP.0b013e32834b14c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Olivier AK, Gibson-Corley KN, Meyerholz DK. Animal models of gastrointestinal and liver diseases. Animal models of cystic fibrosis: gastrointestinal, pancreatic, and hepatobiliary disease and pathophysiology. Am J Physiol Gastrointest Liver Physiol. 2015;308:G459–G471. doi: 10.1152/ajpgi.00146.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blanco PG, Zaman MM, Junaidi O, Sheth S, Yantiss RK, Nasser IA, Freedman SD. Induction of colitis in cftr −/− mice results in bile duct injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G491–G496. doi: 10.1152/ajpgi.00452.2003. [DOI] [PubMed] [Google Scholar]
- 75.Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, Strazzabosco M. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141:1498–1508. 1508 e1491–1495. doi: 10.1053/j.gastro.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chuang YH, Lan RY, Gershwin ME. The immunopathology of human biliary cell epithelium. Semin Immunopathol. 2009;31:323–331. doi: 10.1007/s00281-009-0172-5. [DOI] [PubMed] [Google Scholar]
- 77.Scirpo R, Fiorotto R, Villani A, Amenduni M, Spirli C, Strazzabosco M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-gamma limits NF-kappaB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology. 2015;62:1551–1562. doi: 10.1002/hep.28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiorotto R, Villani A, Kourtidis A, Scirpo R, Amenduni M, Geibel PJ, Cadamuro M, Spirli C, Anastasiadis PZ, Strazzabosco M. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology. 2016;64:2118–2134. doi: 10.1002/hep.28817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, Kasiske BL, Odland D, Pei Y, Perrone RD, Pirson Y, Schrier RW, Torra R, Torres VE, Watnick T, Wheeler DC, P Conference Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2015;88:17–27. doi: 10.1038/ki.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Place Published; 1993. [Google Scholar]
- 81.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 82.Strazzabosco M, Somlo S. Polycystic liver diseases: congenital disorders of cholangiocyte signaling. Gastroenterology. 2011;140:1855–1859. 1859 e1851. doi: 10.1053/j.gastro.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, Igarashi P, Somlo S. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific in-activation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo C, Yang W, Lobe CG. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 2002;32:8–18. doi: 10.1002/gene.10021. [DOI] [PubMed] [Google Scholar]
- 85.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology. 2010;138:360–371. e367. doi: 10.1053/j.gastro.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology. 2010;51:1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fabris L, Cadamuro M, Fiorotto R, Roskams T, Spirli C, Melero S, Sonzogni A, Joplin RE, Okolicsanyi L, Strazzabosco M. Effects of angiogenic factor over-expression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 88.Fabris L, Cadamuro M, Libbrecht L, Raynaud P, Spirli C, Fiorotto R, Okolicsanyi L, Lemaigre F, Strazzabosco M, Roskams T. Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development. Hepatology. 2008;47:719–728. doi: 10.1002/hep.22015. [DOI] [PubMed] [Google Scholar]
- 89.Spirli C, Locatelli L, Fiorotto R, Morell CM, Fabris L, Pozzan T, Strazzabosco M. Altered store operated calcium entry increases cyclic 3′,5′-ade-nosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55:856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 90.Spirli C, Mariotti V, Villani A, Fabris L, Fiorotto R, Strazzabosco M. Adenylyl cyclase 5 links changes in calcium homeostasis to cAMP-dependent cyst growth in polycystic liver disease. J Hepatol. 2017;66:571–580. doi: 10.1016/j.jhep.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spirli C, Morell CM, Locatelli L, Okolicsanyi S, Ferrero C, Kim AK, Fabris L, Fiorotto R, Strazzabosco M. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56:2363–2374. doi: 10.1002/hep.25872. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 92.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, Li A, Cai Y, Kamath PS, King BF, Azurmendi PJ, Tahvanainen P, Kaariainen H, Hockerstedt K, Devuyst O, Pirson Y, Martin RS, Lifton RP, Tahvanainen E, Torres VE, Somlo S. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 93.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33:345–347. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 94.Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, King BF, Torres VE, Somlo S. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72:691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Trombetta ES, Simons JF, Helenius A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem. 1996;271:27509–27516. doi: 10.1074/jbc.271.44.27509. [DOI] [PubMed] [Google Scholar]
- 96.Alder NN, Shen Y, Brodsky JL, Hendershot LM, Johnson AE. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J Cell Biol. 2005;168:389–399. doi: 10.1083/jcb.200409174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, Cai Y, Geng L, Crews CM, Somlo S. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet. 2011;43:639–647. doi: 10.1038/ng.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fedeles SV, Gallagher AR, Somlo S. Polycystin-1: a master regulator of intersecting cystic pathways. Trends Mol Med. 2014;20:251–260. doi: 10.1016/j.molmed.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Williams SS, Cobo-Stark P, James LR, Somlo S, Igarashi P. Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr Nephrol. 2008;23:733–741. doi: 10.1007/s00467-007-0735-4. [DOI] [PubMed] [Google Scholar]
- 100.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 101.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schoneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gallagher AR, Esquivel EL, Briere TS, Tian X, Mitobe M, Menezes LF, Markowitz GS, Jain D, Onuchic LF, Somlo S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol. 2008;172:417–429. doi: 10.2353/ajpath.2008.070381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, Popov Y, Scirpo R, De Matteis M, Amenduni M, Pietrobattista A, Torre G, Schuppan D, Fabris L, Strazzabosco M. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63:965–982. doi: 10.1002/hep.28382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Spirli C, Locatelli L, Morell CM, Fiorotto R, Morton SD, Cadamuro M, Fabris L, Strazzabosco M. Protein kinase A-dependent pSer(675) -beta-catenin, a novel signaling defect in a mouse model of congenital hepatic fibrosis. Hepatology. 2013;58:1713–1723. doi: 10.1002/hep.26554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 105.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Masyuk TV, Huang BQ, Ward CJ, Masyuk AI, Yuan D, Splinter PL, Punyashthiti R, Ritman EL, Torres VE, Harris PC, LaRusso NF. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–1310. doi: 10.1016/j.gastro.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 107.K H, Sanzen T, Yasoshima M, Kawamura Y, Ishibashi M, Nakanuma Y. Polycystic kidney rat is a novel animal model of Caroli’s disease associated with congenital hepatic fibrosis. Am J Pathol. 2001;158:1605–1612. doi: 10.1016/S0002-9440(10)64116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muff MA, Masyuk TV, Stroope AJ, Huang BQ, Splinter PL, Lee SO, Larusso NF. Development and characterization of a cholangiocyte cell line from the PCK rat, an animal model of autosomal recessive polycystic kidney disease. Lab Investig. 2006;86:940–950. doi: 10.1038/labinvest.3700448. [DOI] [PubMed] [Google Scholar]
- 109.Mason SB, Liang Y, Sinders RM, Miller CA, Eggleston-Gulyas T, Crisler-Roberts R, Harris PC, Gattone VH., II Disease stage characterization of hepatorenal fibrocystic pathology in the PCK rat model of ARPKD. Anat Rec (Hoboken) 2010;293:1279–1288. doi: 10.1002/ar.21166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P, Dooley S. Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol. 2013;304:G449–G468. doi: 10.1152/ajpgi.00199.2012. [DOI] [PubMed] [Google Scholar]
- 111.Osterreicher CH, Trauner M. Animal models of biliary tract injury. Curr Opin Gastroenterol. 2012;28:239–243. doi: 10.1097/MOG.0b013e32835264d9. [DOI] [PubMed] [Google Scholar]
- 112.Starkel P, Leclercq IA. Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:319–333. doi: 10.1016/j.bpg.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 113.Alvaro D, Mancino MG. New insights on the molecular and cell biology of human cholangiopathies. Mol Asp Med. 2008;29:50–57. doi: 10.1016/j.mam.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 114.Lazaridis KN, LaRusso NF. The Cholangiopathies. Mayo Clin Proc. 2015;90:791–800. doi: 10.1016/j.mayocp.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]