

Abstract
The design and engineering of secondary metabolite gene clusters that are characterized by complicated genetic organization, require the development of collections of well-characterized genetic control elements that can be reused reliably. Although a few intrinsic terminators and RBSs are used routinely, their translation and termination efficiencies have not been systematically studied in Actinobacteria. Here, we analyzed the influence of the regions surrounding RBSs on gene expression in these bacteria. We demonstrated that inappropriate RBSs can reduce the expression efficiency of a gene to zero. We developed a genetic device – an in vivo RBS-selector – that allows selection of an optimal RBS for any gene of interest, enabling rational control of the protein expression level. In addition, a genetic tool that provides the opportunity for measurement of termination efficiency was developed. Using this tool, we found strong terminators that lead to a 17–100-fold reduction in downstream expression and are characterized by sufficient sequence diversity to reduce homologous recombination when used with other elements. For the first time, a C-terminal degradation tag was employed for the control of protein stability in Streptomyces. Finally, we describe a collection of regulatory elements that can be used to control metabolic pathways in Actinobacteria.
Introduction
For decades, metabolic engineers and synthetic biologists have been attempting to develop principles for biological engineering from the “ground up” to allow the rational construction of complex circuits and systems with balanced expression of genes using suitable building blocks. Such synthetic circuits and systems should exhibit temporal and spatial control and should be orthogonal, not restricted to the regulatory machinery of a host cell. Genetic information in the cell is transferred from DNA to RNA and from RNA to protein via transcription and translation, respectively. Many structural elements, such as promoters, ribosomal binding sites (RBSs), terminators, and 5′- and 3′-untranslated regions (UTRs), influence the efficiency of these processes. In addition, a myriad of regulatory proteins and small non-coding RNAs (e.g., riboswitches, ribozymes) govern the expression of a gene. Thus, a precise understanding of the regulation of gene expression at the above mentioned levels and the interaction of regulatory genetic elements is needed. Towards this goal, libraries of natural and synthetic controlling elements that influence gene expression on different levels in Escherichia coli and Saccharomyces cerevisiae have been studied, and numerous synthetic control modules are being developed1–5. However, there is a deficiency of the building blocks described above for less-studied but highly industrially important bacteria, such as Actinobacteria.
With the advent of next-generation sequencing techniques, it became obvious that the biosynthetic potential of Actinobacteria has been underestimated, because their genomes contain a hidden wealth of silent secondary metabolite gene clusters6–8. The problem of the existence of dormant or unexpressed gene clusters is mainly related to the intricate and tight regulatory networks that precisely orchestrate metabolite production in bacteria and respond to various environmental and intracellular signals9,10. One of the strategies for overcoming this obstacle is the decoupling of metabolite biosynthesis from the regulatory networks that exist in the cell by placing genes in a cluster under the control of constitutive or orthogonal inducible promoters. A lot of effort has been made to develop engineering elements that govern the expression of genes at the transcriptional level11–19. However, controlling elements, such as RBSs, terminators and degradation tags, that influence the remaining aspects of the process and may be used to balance expression, and thus control output, have remained overlooked in Actinobacteria. The complexity of secondary metabolite gene clusters20,21 results from the existence of bottlenecks that require the expression of multiple gene products from individually controlled gene sets; hence, there is demand for libraries of promoters, RBSs and terminators. In addition, sequences with minimal identity are needed to avoid homologous recombination with synthetic operons and consequent genetic instability. One additional hurdle that should be considered during the design of metabolite gene clusters is the genetic context, which may influence the activity of elements such as promoters and RBSs22,23. Therefore, there is an urgent need for diverse regulatory elements, such as promoters, RBSs, terminators, and genetic tools that will allow researchers to measure, characterize and select appropriate controlling elements for a gene of interest in Actinobacteria.
Herein, we report the development of a genetic tool – an in vivo RBS-selector – for the selection of an optimal synthetic RBS for any gene of interest, enabling rational control of the protein expression level. The designed methodology allows a single experiment to test the activity of a huge number of randomly synthesized RBSs and choose the best RBS with the required activity that is suitable for a certain gene. Furthermore, this tool provides the possibility of selecting an optimal RBS for a gene of interest in combination with any promoter, due to the fact that the genetic context of the latter element may influence the activity of the former. Using this tool, we generated a library of synthetic RBSs for the gusA gene24, resulting in a number of reporters with different expression strengths and, consequently, a flexible dynamic range. In addition, a genetic tool for the reliable measurement of termination efficiency and a library of intrinsic terminators that play an important role in transcriptional regulation were developed for Streptomyces. For the first time, an 11-amino acids C-terminal degradation tag was used for the regulation of gene expression at a posttranslational level in Streptomyces, and several variants of the unstable GusA reporter were constructed. In conclusion, the combination of the control elements described above provides the possibility of spatial, temporal and quantitative regulation of gene expression at transcriptional and/or translational levels, thereby facilitating the ability to overcome bottlenecks and obtain greater amounts of compounds as well as toxic and non-detrimental proteins. The described features make these tools very promising for metabolic engineering and biotechnology of Streptomyces and other Actinobacteria.
Results
The nucleotides surrounding the Shine-Dalgarno domain dictate the efficiency of translation initiation
Engineering and development of systems that control the expression of genes are in most cases focused on the transcriptional level13,14,16,17. However, in E. coli and some other bacteria, translation initiation is the rate-limiting step in the translation process and can strongly influence protein synthesis25–27. Changes in the sequence, shape and structure of the translation initiation region (TIR) alter mRNA folding, which in turn affects the thermodynamic energy barrier that ribosomes must cope with to form a stable initiation complex and initiate translation1,26,28,29. Therefore, there is a need to engineer efficient genetic elements that control the expression of genes on posttranscriptional or translational levels, as these are additional critical steps for the production of proteins.
There are currently no available data on the efficiency of translation initiation in Streptomyces. Therefore, we decided to estimate how changes in the sequence of 5′ untranslated regions (UTRs) can influence translation in these bacteria and in S. lividans TK24 (Table 1) in particular. A library of synthetic RBSs for the gusA reporter gene was constructed (Table S1). For this purpose, the weakest known constitutive promoter, P72, from our library of semi-synthetic promoters16 was fused with the randomly synthesized RBSs and the gusA reporter gene to allow validation of the translation efficiency based on glucuronidase activity. The generated synthetic RBSs contain a consensus Shine-Dalgarno domain, “GGAGG”, which was fixed to a length of 5 bp because it has been shown that expression levels in E. coli decrease when the sequence is expanded to more than six nucleotides30. The nucleotides surrounding the SD region, up to 10 bp upstream and 6 bp downstream, were randomly synthesized (Fig. 1a). Two types of degenerate primers were used in this work (Fig. 1a). One type of primer contained N-type random nucleotides, where N is any base (A, T, G or C). The other type of primer contained W-type random nucleotides, where W is either an A or T base. Two types of primers were used, since it was shown that AT-rich RBSs provide better level of translation in E. coli31,32. The weakest P72 promoter was employed so that even very small changes in translation could be observed. The use of degenerate XbaRBSForw and NdeNRBSRev or XbaRBSForw and NdeWRBSRev primer pairs (Table 2), respectively, and an amplified hygromycin resistance gene fused to outward oriented P72 promoter facilitated the rapid and easy cloning of a variety of different synthetic RBSs upstream of the gusA gene (for details see Materials and Methods). The obtained library of synthetic RBSs was sequenced, and 70 different variants were chosen for further investigation. An additional plasmid without an RBS between the P72 promoter and the gusA gene was constructed. In this plasmid, the reporter gene was directly fused with the promoter. All 70 plasmids carrying different RBS regions were transferred to the Streptomyces lividans TK24 strain via tri-parental conjugation with E. coli. Exconjugants were selected on the basis of their resistance to apramycin and hygromycin. Consequently, 70 recombinant strains that harbored different RBSs were obtained. All of these strains were grown for 2 days in liquid TSB medium and then subjected to quantitative measurement of glucuronidase activity for the indirect analysis of RBS activity. The results of the analysis are depicted in Fig. 1b. As shown in the figure, we obtained couple RBSs (W5, W4, W20, N2 and N16) that were approximately 2–3 times stronger than the control, and there were also several that were much weaker. Four of the 70 RBSs, namely N4, N20, N24 and N37, could severely impair translation efficiency by decreasing the translation level to 0. The sequences of the strongest RBSs (Fig. 2) that we succeeded in creating exhibited greater AT richness, which is in accord with the previously reported data from E. coli31,32. Summing up, the sequences of the nucleotides located up- and downstream from the SD domain can clearly strongly affect translation efficiency.
Table 1.
Strains and plasmids used in this study.
| Bacterial strains and plasmids | Description | Source or reference |
| S. albus J1074 | Isoleucine and valine auxotrophic derivative of S. albus G (DSM 40313) lacking SalI-restriction activity | Salas J., Oviedo, Spain |
| S. lividans TK24 | Derivative of S. lividans TK21 that contains mutation in the rpsL gene and is resistant to spectinomycin | 41 |
| E. coli DH5α | Routine cloning | MBI Fermentas |
| E. coli ET12567 (pUZ8002) | Conjugative transfer of DNA | 41 |
| pGUS | Promoter probe vector containing promoterless gusA | 24 |
| pUC19 | Apr, general cloning vector | MBI Fermentas |
| pSET152 | Amr; φC31-based integrative vector | 41 |
| pGUSHL4aadA | pTESa-based vector for translational fusion with gusA | 24 |
| pGUSbezRBS | Derivative of pGUS containing the gusA gene without promoter and RBS | This work |
| pGUSP72bezRBS | Derivative of pGUS containing the gusA gene with the P72 promoter but without RBS | This work |
| pGUSRBSProg1 | Derivative of pGUSbezRBS containing synthetic RBS generated using RBS calculator | This work |
| pGUSNRBS-8-3 | Series of pGUSbezRBS derivatives containing NRBS-2 that differ in the size of the insert separating the SD from the ATG | This work |
| pSETP82Ap | Derivative of pSET152 containing ampicillin resistance gene fused with the P82 promoter | This work |
| pGUSNRBSEGFP | Derivative of pGUSHL4aadA containing randomly generated RBSs fused with first 60 bp of the egfp gene and gusA gene | This work |
| pEGFPN-9 | Derivative of pGUS containing the egfp gene fused with the P82 promoter and RBSN-9 | This work |
| pEGFPN-131 | Derivative of pGUS containing the egfp gene fused with the P82 promoter and RBSN-131 | This work |
| pGUSSPL21Termin | Derivative of pGUS containing six different terminators inserted downstream of the P21 promoter | This work |
| pGCymRP21 | Derivative of pGUS containing the gusA gene under the control of the P21-cmt promoter | 18 |
| pGCymRP21-LVA | Derivative of pGCymRP21 containing gusA gne fused to the C-terminal degradation tag | This work |
| pGCymRP21-ASV | Derivative of pGCymRP21 containing gusA gne fused to the C-terminal degradation tag | This work |
| pGCymRP21-AAV | Derivative of pGCymRP21 containing gusA gne fused to the C-terminal degradation tag | This work |
Figure 1.

Scheme of random RBSs construction and estimation of their activity. (a) Schematic representation of the RBS containing plasmids used in the study. gusA, reporter gene; P72, promoter; Hygr, Spr and Amr, hygromycin, spectinomycin and apramycin resistance genes, respectively; int, integrase gene; N – A, T, G or C; W – A or T. (b) Glucuronidase activity in cell lysates from recombinant S. lividans strains containing gusA under the control of the P72 promoter fused to different synthetic RBSs. The strains were grown in TSB medium for 2 days. Error bars indicate the standard deviations of three independent experiments.
Table 2.
Primers used in this study.
| Primers | Sequence 5′-3′ | Purpose |
|---|---|---|
| XbagusAForw | AAAATCTAGATACGCATATGCTGCGGCCCGTCGAAACC | Cloning the gusA gene without RBS and promoter |
| EVgusARev | AAAAGATATCTGCTTCCCGCCCTGCTGCGG | |
| XbaRBSForw | AAAAGATATCGAAATCACTCCCAATTAATCTAG | Cloning randomly synthesized RBSs upstream of the gusA gene |
| NdeNRBSRev | AAAACATATGNNNNNNCCTCCNNNNNNNNNNTTTCTCATCCTAAAGAATCTCTC | |
| NdeWRBSRev | AAAACATATGWWWWWWCCTCCWWWWWWWWWWTTTCTCATCCTAAAGAATCTCTC | |
| NdeP72bezRBS | AAAACATATGTTTCTCATCCTAAAGAATCTCTC | Cloning the gusA gene with the P72 promoter but without RBS |
| RBSProg1Rev | AAAACATATGAATGAACCTCCTTCTTTCTTTTTCTCATCCTAAAGAATCTCTC | Cloning the gusA gene fused to in silico designed RBS |
| NdeRBS8Nrev | AAAACATATGTGTTTCCTCCAACGGTTCATTTTCTCATCCTAAAGAATCTCTC | Cloning gusA fused to synthetic RBSs |
| NdeRBS7Nrev | AAAACATATGTGTTCCTCCAACGGTTCATTTTCTCATCCTAAAGAATCTCTC | |
| NdeRBS6Nrev | AAAACATATGTGTCCTCCAACGGTTCATTTTCTCATCCTAAAGAATCTCTC | |
| NdeRBS5Nrev | AAAACATATGTGCCTCCAACGGTTCATTTTCTCATCCTAAAGAATCTCTC | |
| NdeRBS3NRev | AAAACATATGCCTCCAACGGTTCATTTTCTCATCCTAAAGAATCTCTC | |
| ApForw | TTTTTCTAGTTATATGAGTAAACTTGGTCT | Fusion the Apr gene with the P82 promoter |
| ApP82Rev | GCTACAATCCTACTTGAAGAATCCTAATTTTAGCCTCAGGAGACGAAAGGGCCTCGTGATA | |
| EGFPNRBSRev | TTTTGATATCGAACAGCTCCTCGCCCTTGCTGGATCGGGATCCTTTTTCGAACTGCGGGTGGCTCCACATNNNNNNCCTCCNNNNNNNNNGCTACAATCCTACTTGAAGAATC | Cloning randomly synthesized RBSs upstream of the egfp gene |
| EGFPNRBS-9For | TTTTTCTAGATCCTGAGGCTAAAATTAGGATTCTTCAAGTAGGATTGTAGCTGACTAATGGGAGGCGTCTGATGTGGAGCCACCCGCAGTTC | EGFP fusion with NRBS-9 |
| EGFPNRBS-131For | TTTTTCTAGATCCTGAGGCTAAAATTAGGATTCTTCAAGTAGGATTGTAGCATCGTAGGAGGAGGCAAAACATGTGGAGCCACCCGCAGTTC | EGFP fusion with NRBS-131 |
| EGFPRev | GATATCTTACTTGTACAGCTCGTCCATGC | |
| GusARevLVA | AAAAGATATCTTATCAGGCTACGAGGGCGAAGGCCTGCTGGGAGGAATCGCGC | gusA fusion with degradation tags |
| GusARevAAV | TTGGTGTTGGCCTGCTTCCCGCCCTGCTGCGGAAAAGATATCTTATCAAACGGCAGCGGCGAAGGCCTGCTGGGAGGAATCGCGCTTGGTGTTGGCCTGCTTCCCGCCCTGCTGCGG | |
| GusARevASV | AAAAGATATCTTATCATACGGAAGCGGCGAAGGCCTGCTGGGAGGAATCGCGCTTGGTGTTGGCCTGCTTCCCGCCCTGCTGCGG | |
| GusASpeForw | AAAAACTAGTCGAGCAACGGAGGTAC |
Figure 2.

The strongest RBSs obtained in this work. (a) Sequences of the strongest RBSs and their activity. (b) WebLogo analysis of the strongest RBSs. WebLogo was generated using WebLogo 3 (http://weblogo.threeplusone.com/).
There are two widely used online programs (UTR Designer (https://omictools.com/utr-designer-tool) and RBS Calculator (https://www.denovodna.com/software/reverse)) that allow the translation initiation rate from a certain RBS to be predicted1,29. To estimate the reliability of these models, the translation initiation rate from the 70 generated RBSs was calculated using the programs mentioned above and compared to the measured GusA activity driven from these RBSs. From the data depicted in Table S1, it is clear that not all of the measured GusA expression levels corresponded to the predicted levels. For example, the N2 RBS region yielded the strongest GusA expression level, while both programs calculated the expression driven by this region to be even weaker than the control (Table S1). There was also sometimes a discrepancy in the predictions of the programs. For instance, RBS Calculator predicted that the W5 RBS should be the strongest, whereas UTR Designer predicted that the W5 RBSs should be 3 times weaker on average compared with the other strong RBSs in our library (Table S1). Considering our experimental evidence and the theoretical predictions regarding RBS activity together, we postulate that they are not correlated in all cases. The coefficient of correlation was approximately 0.32 for RBS Calculator and 0.27 for UTR Designer.
Taking into account the ability of RBS Calculator to not only calculate but also generate an optimal RBS for a certain gene of interest, we used this program to generate an optimal RBS for the gusA reporter gene. This RBS was fused with the P72 weak promoter and gusA, and its activity was compared with the best RBS region that we succeeded in obtaining experimentally for this gene. This comparison was made based on glucuronidase activity. From the obtained data (Fig. 3) it is clear that the selected N2 RBS region was 1.5 times stronger than the programmed RBS on average. Therefore, when reliable gene expression is necessary, it is important to generate and test several RBS-surrounding regions to choose the most suitable sequence. Thus, our tool is an alternative to the programs and provides the ability to get at once plenty of RBSs with different activities for a gene of interest. Utilizing the developed tool one will get the activity of RBSs in the conditions that are required, since genetic content and the cell environment, which are not considered by in silico tools, can severely influence gene expression.
Figure 3.

Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the P72 promoter fused to N2 (TK24 NRBS-2+) and programmed using RBS Calculator RBSs (TK24 RBSProgr+). The strains were grown in TSB medium for 2 days. Error bars indicate the standard deviations of three independent experiments.
The distance between the SD domain and the start codon is critical
One of the additional parameters influencing the translation initiation rate is the distance between the SD domain and the start codon33. Minimal SD-AUG spacing is required, probably because the 16S rRNA and fMet-tRNA sequences must be located a certain distance apart, due to configurational limitations. Optimal spacers for E. coli are considered to range from 5 to 9 nucleotides34. To verify whether the same is true for Streptomyces, we constructed 5 RBSs that differed in the number of nucleotides located between the SD sequence and the start codon. All of these RBSs were derivatives of the strongest N2 RBS (Figs 1 and S1) that we succeeded in obtaining for gusA. Each of the generated RBSs was fused to the P72 promoter upstream of the gusA reporter gene. Subsequently, these mutant RBSs were transferred to the S. lividans TK24 strain, in which glucuronidase activity was measured (Fig. 4). Based on the measurement data (Fig. 4a), we suggest that a spacer region comprising 6 or 9 nucleotides is optimal for efficient translation in Streptomyces. However, we cannot rule out the influence of other factors, such as changes in mRNA stability or the mRNA folding energy. Latter is the energy required to unfold the RNA secondary structures in order to make it accessible to regulatory molecules (proteins, micro-RNAs). Less stable structure contributes to the increase of mRNA expression level25. We calculated the RNA folding energy for all five mutant RBSs (Fig. 4b) using NUPACK35 and determined that RBS-3 and RBS-5 presented among the highest RNA folding energies, similar to that of RBS2-(9 bp). However, the glucuronidase activity associated with RBS-2 was the highest and was nearly the same as that for RBS-6, which displayed an approximately 2 times lower RNA folding energy. In addition, the RNA folding energy of RBS-7 was similar to that of RBS-6, although the translation efficiency from the former was 3 times lower. These data confirmed that the observed changes in glucuronidase activity were not simply related to changes in the secondary structure of RNA but, rather, were caused by the difference in the number of nucleotides located in the spacer region. In conclusion, it is not only the RNA sequence, structure and folding energy but also the distance between the SD sequence and the start codon that plays an important role in translation initiation in Streptomyces.
Figure 4.

Analysis mutant RBSs that differ in the size of the insert separating the SD sequence from the ATG codon. (a) Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the P72 promoter fused to different mutant RBSs. The strains were grown in TSB medium for 2 days. Error bars indicate the standard deviations of three independent experiments. (b) RNA folding energy of mutant RBSs, calculated with NUPACK.
Development of a genetic tool – an in vivo RBS-selector – for the selection of an optimal RBS for any gene of interest
Taking into account the various lines of evidence described above and previously showing that the 5′-UTR sequence can strongly modulate the translation initiation rate as a result of the shape and structure of mRNA and therefore plays a central role in posttranscriptional regulation, along with the fact that theoretical predictions do not always correlate with experimental data, we decided to generate a genetic tool that would allow the design and selection of an optimal RBS for any gene of interest in vivo. According to our predictions, this tool should reflect the influence of all major RNA components on the translation initiation rate. Through systematic analysis of the translation efficiency in E. coli, it was shown that in addition to the nucleotides surrounding the SD domain and spacer region between the SD sequence and the start codon, the first 30–60 nucleotides of the gene of interest can also strongly influence the mRNA folding energy and, consequently, ribosome access and binding to mRNA29,36. Therefore, an optimal RBS that is selected for one gene can be completely inefficient for another. Considering these data, the genetic tool that we designed contained the following components: P72, which is the weakest constitutive promoter and thus allows the determination of even very small changes in translation; randomly generated nucleotides surrounding the constitutive SD domain; the first 60 nucleotides, including the start codon, of any gene of interest; and the gusA reporter gene, to which all of these components were fused via a special linker so that the translation efficiency could be calculated indirectly (Fig. 5). Using this tool, we performed selection of an optimal RBS for egfp mRNA37. This gene was involved in the study since after the selection of the several RBSs based on glucuronidase activity it will be easy to estimate their efficiency after direct fusion with egfp based on fluorescence. The utilization of degenerate primers covering the first 60 nucleotides of the egfp gene fused with a randomly synthesized RBS and an amplified hygromycin resistance gene facilitated the rapid and easy cloning of a variety of different synthetic RBSs upstream of the gusA reporter gene. The obtained library of synthetic RBSs was transferred to the Streptomyces albus J1074 strain (Table 1) via conjugation, and the selection of an optimal RBS was performed based on glucuronidase activity (Figure S2). As a result, two RBSs (the weakest (NRBS-9) and the strongest (NRBS-131)) were chosen for further analysis (Fig. 6a). The egfp gene was placed under the control of the individual selected RBSs, namely NRBS-9 or NRBS-131, giving two plasmids pEGFPN-9 and pEGFPN-131 (Table 1). These plasmids were transferred into S. albus by means of conjugation. As a result, two strains S. albus pEGFPN-9+ and S. albus pEGFPN-131+ were constructed. Then relative EGFP fluorescence was measured in these strains (Fig. 6b) and compared with glucuronidase activity under the control of these RBSs. An excellent correlation between the two activities was observed. These data confirmed that we succeeded in creating a genetic tool, referred to as an in vivo RBS-selector, that provides the ability to perform selection of an optimal RBS for any gene of interest and enables rational control over the protein expression level. This tool requires only three steps: RBS library generation, transfer to the appropriate strain and indirect assessment of RBS activity based on the GusA assay (Figure S2).
Figure 5.
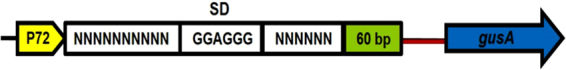
Schematic representation of the key elements of the genetic tool for the selection of RBSs for genes of interest. SD –Shine-Dalgarno domain; P72 – synthetic promoter; gusA – reporter gene; N – randomly synthesized nucleotides (A, T, C, G), 60 bp – first 60 bp of any gene of interest, including its start codon. The red line denotes the linker between the proximal region of the gene of interest and gusA.
Figure 6.

Glucuronidase activity and EGFP fluorescence in cell lysates of recombinant S. albus strains. (a) Glucuronidase activity in cell lysates of recombinant S. albus strains containing gusA under the control of the P72 promoter fused to different mutant RBSs. (b) EGFP fluorescence in cell lysates of recombinant S. albus strains containing egfp under the control of the P72 promoter fused to two different mutant RBSs. The strains were grown in TSB medium for 2 days. NRBS-9 – is the weakest RBS obtained in the study and NRBS-131 is the strongest one. Error bars indicate the standard deviations of three independent experiments.
A library of factor-independent terminators for S. lividans TK24
Another important type of element significantly influencing the expression of genes in prokaryotes and eukaryotes is terminators. Although the impact of terminators is relatively small compared with promoters and RBS-surrounding regions, they constitute an important component of genetic circuits. In Actinobacteria in particular, secondary metabolite gene clusters are organized into numerous operons facing each other that are controlled independently. The transcription of genes in operons is governed by different promoters and should be terminated efficiently. There are two type of terminators that function in prokaryotes: factor-dependent terminators, which rely on the special regulatory protein Rho, and factor-independent terminators, which do not require any additional protein cofactors for the termination of transcription38–41. The main role of terminators in operons is to terminate transcription and prevent read-through from different promoters. Because the number of sequenced genomes and, consequently, interesting gene clusters increases every year and heterologous expression of gene clusters under the control of artificial promoters is the key strategy for obtaining new metabolites, there is an urgent need for well-defined and effective terminators. There are only a few terminators that are routinely used in Streptomyces42. Thus, we decided to expand the repertoire of strong terminators for Streptomyces and, in particular, for S. lividans. To accomplish this aim, we chose four terminators (U, V, T4 lang and T4 kurz) from an online database (WebGeSTer Database), another terminator (ttsbiB) that is widely used in Mycobacteria43, and a sixth (I) that was generated in-house using an online tool. To assess the efficiency of transcription termination with the above mentioned terminators and to determine their most efficient combinations, the terminators were placed between strong constitutive promoter (P21) from our library of promoters16 and the gusA reporter gene (Figure S3). In this case, glucuronidase activity should be inversely correlated with the strength of a terminator. To test different combinations of terminators, a set of 18 plasmids was generated (Figure S3). All of the constructs were transferred to the S. lividans strain via conjugation, and the efficacy of termination was assessed based on glucuronidase activity. As a control, a plasmid carrying the gusA gene cloned downstream of the P21 promoter was used. The obtained results are depicted in Figs 7 and S4. GusA activity in the control strain is denoted as 100%. According to our data (Fig. 7), the ttsbiB terminator was the strongest, as the resultant read-through was less than 4%. The U and V terminators were a bit weaker, with a read-through of less than 14%. The T4 lang terminator showed less efficiency, resulting in read-through in average 31%. The T4 kurz and I terminators were the weakest. In conclusion, we expanded the number of strong terminators for S. lividans and developed a genetic device that allows the assessment of terminator efficacy.
Figure 7.

Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the P21 promoter fused to different terminators. The strains were grown in TSB medium for 2 days. Error bars indicate the standard deviation of three independent experiments. K – stands for the control strain, that expresses gusA under the control of the P21 promoter.
Degradation tags for controlling gene expression at the posttranslational level
It has been shown for E. coli and some other bacteria that specific N- or C-terminal oligopeptide sequences can make stable proteins susceptible to degradation by certain intracellular, tail-specific proteases44–46. This feature has widely been exploited for the construction of unstable variants of reporter proteins such as an EGFP and luciferase47–49 in these bacteria. However, this strategy has never been used for regulating protein stability in Actinobacteria. Therefore, we decided to determine whether it is possible to use protein degradation tags to regulate gene expression at the posttranslational level and to construct unstable variants of reporter genes that might be used in the future for the investigation of temporal gene expression in Actinobacteria. For this purpose, the well-described ssrA degradation tag50 from E. coli was employed, because this type of system has also been described for Actinobacteria51. The ssrA gene encodes tm-RNA, which functions as both an mRNA and a tRNA; it also recognizes incomplete or damaged proteins and attaches a peptide tag with the sequence AANDENYALAA to the C-termini of these proteins via co-translational switching51. Moreover, variations in the last 3 amino acids of the peptide tag result in proteins of varying stability48.
To assess the efficiency of protein degradation in the presence of the above mentioned specific C-terminal oligopeptide extensions, these peptides were fused to GusA (Figure S5). As a result, three different plasmids, namely pGCymRP21-LVA, pGCymRP21-ASV and pGCymRP21-AAV (Table 1), were constructed. In these plasmids transcription of the gusA gene was under the control of the P21-cmt promoter18. The plasmids were transferred to S. lividans strains. The efficacy of degradation was assessed by measuring glucuronidase activity in the presence and absence of the inducer. As a control, a pGCymRP21 plasmid in which the gusA gene was cloned downstream of the P21-cmt promoter was employed18. The strains were grown in TSB medium for 48 hours in the presence or absence of the inducer (cumate). As shown by the results depicted in Fig. 8, the strain containing unmodified GusA exhibited the highest level of glucuronidase activity in the induced stage, while in the three other S. lividans strains harboring the reporter derivatives, GusA activity was 2–182-fold lower in the on (induced) state. Furthermore, S. lividans strains containing GusA with the LVA C-terminal amino acids displayed no detectable glucuronidase activity in the off stage (Fig. 8), indicating rapid degradation of the protein.
Figure 8.

Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the inducible P21-cmt promoter fused to different degradation tags. GUS wt – strain that contains wild type gusA gene; AAV - strain that contains GusA fused with AAV C-terminal tag; LVA – strain with GusA containing LVA C-terminal tag and ASV – strain with GusA fused to ASV C-terminal tag. The strains were grown in TSB medium for 2 days in the presence or absence of the inducer.
To test the stability of the wild-type and mutant variants of GusA, we collected the biomass that accumulated after 48 hours of growth in the presence of the inducer, which was then washed twice with water, transferred it to fresh TSB medium without an inducer, and allowed to grow for 48 hours. Glucuronidase activity was measured after 2, 6, 19, 24 and 48 hours (Fig. 9). Variants of the GusA protein that contain C-terminal degradation tags were rendered unstable, while GusA with the AAV tag degraded approximately 15 times faster than the wild-type protein, and GusA-LVA and GusA-ASV degraded 2 and 3 times faster, respectively, on average. These data are in agreement with the data previously described for E. coli, Mycobacteria and Pseudomonas putida47,48 and again confirm the dependence of protein stability on the sequence of the amino acids in the C-terminal tag.
Figure 9.
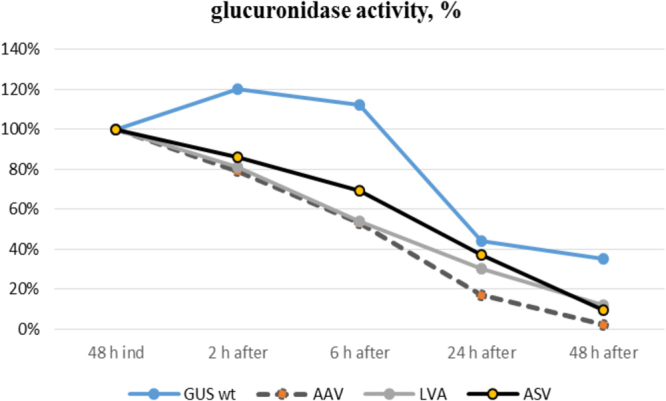
Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the inducible P21-cmt promoter fused to different degradation tags. GUS wt – strain that contains wild type gusA gene; AAV - strain that contains GusA fused with AAV C-terminal tag; LVA – strain with GusA containing LVA C-terminal tag and ASV – strain with GusA fused to ASV C-terminal tag. The strains were grown in TSB medium for 2 days in the presence of the inducer, after which the biomass was collected, washed and grown in fresh TSB medium without the inducer for 2 days.
To determine whether it was possible to restore gusA gene expression after GusA was degraded, we again induced gusA transcription with cumate for 2 days and measured GusA activity. In all strains, glucuronidase activity was restored to the maximal level observed after direct induction of the culture (Fig. 10).
Figure 10.

Glucuronidase activity in cell lysates of recombinant S. lividans strains containing gusA under the control of the inducible P21-cmt promoter fused to different degradation tags. GUS wt – strain that contains wild type gusA gene; AAV - strain that contains GusA fused with AAV C-terminal tag; LVA – strain with GusA containing LVA C-terminal tag and ASV – strain with GusA fused to ASV C-terminal tag. The glucuronidase activity of the wild type GusA in the induced stage was denoted as 100%. The strains were grown in TSB medium for 2 days in the presence of the inducer, after which the biomass was collected, washed and grown in fresh TSB medium without the inducer for 2 days; at this point, the inducer was added again, and the strains continued to grow for 2 more days.
In summary, we demonstrated the effectiveness of C-terminal degradation tags for the control of protein stability and used these tags to generate three rapidly degradable variants of the GusA protein, which will facilitate studies aimed at exploring dynamic changes in gene expression.
Conclusions
Regulation of gene expression at the transcriptional level is widely performed in Actinobacteria; however, the possibility of tuning gene expression at other levels, including the posttranscriptional, translational and posttranslational, has been overlooked. In this article, we described the development of a genetic tool referred to as an in vivo RBS-selector that allows the strength of RBSs to be estimated and an optimal RBS with predicted activity for any gene of interest to be selected. We showed, for the first time in Actinobacteria, that RBSs can strongly influence translation efficiency, decreasing it to 0 in certain cases, and that this type of controlling element might be used for tuning gene expression at the translational level. Furthermore, we demonstrated that there is only a moderate correlation between the strength of an RBS observed in experiments and its activity predicted using online tools, such as UTR Designer and RBS Calculator, which is in accord with the recently reported data from Streptomyces coelicolor52. In addition, an RBS selected for egfp based on glucuronidase activity was shown to be approximately 1.5 times stronger than the best RBS designed using RBS Calculator, indicating that genetic content and the cell environment can influence gene expression, which are not considered by in silico tools. We also constructed a library of RBSs for the gusA gene and, consequently, obtained numerous reporters with different sensitivities.
In addition, a genetic tool for the reliable estimation of terminator strength was developed and used for the assessment of termination efficiency in S. lividans. We employed this tool to characterize several well-defined strong terminators that cause 2–26-fold reductions of gene expression and are characterized by sufficient sequence diversity to reduce homologous recombination when used together with a synthetic operon. For the first time, the ssrA degradation tags were employed for the regulation of protein stability and, thus, control of gene expression at the posttranslational level in Actinobacteria. Three new variants of the unstable GusA protein were generated, which will facilitate dynamic monitoring of changes in gene transcription. The described features make the above-described tools very promising for metabolic engineering and biotechnology of S. lividans TK24 and other Actinobacteria.
Materials and Methods
Bacterial strains and growth conditions
The bacterial strains used in this study are listed in Table 1. E. coli strains were grown in Luria–Bertani (LB) broth medium. When required, antibiotics (Sigma, USA; Roth, Germany) were added to cultures at the following concentrations: 100 μg ml−1 ampicillin, 50 μg mlα kanamycin, 50 or 120 μg ml−1 hygromycin, 50 μg ml−1 apramycin.
For conjugation, Streptomyces albus, Streptomyces lividans strains were grown on oatmeal or mannitol soy (MS) agar42 for sporulation. For glucuronidase activity measurement strains were grown in liquid tryptic soy broth (TSB).
Recombinant DNA techniques
Chromosomal DNA from Streptomyces strains and plasmid DNA from E. coli were isolated using standard protocols42,53. Restriction enzymes and molecular biology reagents were used according to the recommendations of the supplier (Thermo Fisher Scientific, Germany, NEB, England).
Generation and cloning of the synthetic RBSs for the gusA reporter gene
A version of the gusA gene without promoter and RBS was synthesized using primers XbagusAForw and EVgusARev (Table 2). Obtained 1.8 kb fragment was digested with XbaI and EcoRV sites and ligated into respective sites of pGUS vector24. As a result, a pGUSbezRBS (Table 1) plasmid was constructed.
The library of synthetic RBSs was constructed using two types of degenerate primers, in which N is any of the four base pairs (A, T, G, C) and W – only adenine or thymine (A or T). Using primer pairs XbaRBSForw and NdeNRBSRev or XbaRBSForw and NdeWRBSRev (Table 2), respectively, the hygromycin resistance gene fused with the P72 weak promoter16 was amplified by PCR, digested with NdeI and XbaI and cloned into the respective sites of pGUSbezRBS vector, yielding two types of plasmids: pGUSNRBS and pGUSWRBS (Fig. 1a). Hygromycin gene fused only to the P72 promoter without RBS was amplified using primer pair XbaRBSForw and NdeP72bezRBS (Table 2). The obtained fragment was cut with XbaI/NdeI and cloned into the respective sites of pGUSbezRBS. As a result, plasmid pGUSP72bezRBS that contained gusA gene under the control of the P72 promoter, however without RBS, was constructed (Table 1). Plasmids pGUSP72bezRBS, pGUSNRBS or pGUSWRBS were introduced into the S. lividans TK24 strain via tri-parental conjugation. In this type of mating E. coli strain ET12567 x pUB307 was used as a helper to transfer the plasmids from E. coli strain DH5α into S. lividans.
To directly compare the activity of the synthetic RBSs designed for the gusA gene using the RBS calculator online tool (https://www.denovodna.com/software/reverse) with the RBSs we obtained on practice, former were fused with the P72 promoter and hygromycin resistance gene, and cloned into the pGUSbezRBS vector using the same algorithm. The forward primer XbaRBSForw in this case was always the same and the reverse primer included the sequence of in silico generated RBSs. In this way, Prog1 RBS was amplified using primers XbaRBSForw and RBSProg1Rev (Table 2). The obtained 1.3 kb fragment was digested with NdeI and XbaI and cloned into the respective sites of pGUSbezRBS, yielding pGUSRBSProg1 (Table 1).
Generation and cloning of synthetic RBSs with different spacing between the Shine-Dalgarno (SD) sequence and the start codon of the gusA reporter gene
To construct a series of plasmids that differ in the size of the insert separating the SD from the ATG, the strongest RBS, NRBS2, was used. For this purpose, the forward primer XbaRBSForw was always the same, and the reverse primer included the sequences of generated RBSs differing in the length of spacing. To synthesize an RBS with 8-bp spacing, the reverse primer NdeRBS8Nrev was used; for 7-bp spacing, NdeRBS7Nrev was used; for 6-bp spacing, NdeRBS6Nrev; for 5-bp spacing, NdeRBS5Nrev; and for 3-bp spacing, NdeRBS3Nrev (Table 2). The amplified fragments obtained using pairs of the abovementioned forward and reverse primers were digested with NdeI/XbaI and cloned into the respective sites of pGUSbezRBS. Consequently, the plasmids pGUSNRBS-8 bp, pGUSNRBS-7 bp, pGUSNRBS-6 bp, pGUSNRBS-5 bp, and pGUSNRBS-3 bp (Table 1) were constructed and used in further experiments.
Generation, cloning and selection of synthetic RBSs for the egfp gene based on the activity of gusA
To obtain a fusion of the ampicillin resistance marker with the P82 synthetic promoter16, the primer pair ApForw and ApP82Rev (Table 2) was used. The amplified 1.3-kb fragment was cloned into the EcoRV site of the pSET152 integrative vector, yielding pSETP82Ap (Table 1).
A library of synthetic RBSs was constructed using degenerate primers, in which N is any of the four base pairs. Using the primers ApForw and EGFPNRBSRev, the ampicillin resistance gene fused with the P82 weak promoter16 and randomly generated RBSs was amplified via PCR, digested with EcoRV and XbaI and cloned into the respective sites of the pGUSHL4aadA vector24, yielding the pGUSNRBSEGFP plasmid (Table 1, Fig. 5). The pGUSNRBSEGFP plasmid was introduced into the S. albus strain via tri-parental conjugation.
Two synthetic RBSs (the strongest and the weakest) were selected based on gusA activity and used in further experiments. To fuse the egfp gene with these RBSs and the P82 promoter, two primer pairs were used: EGFPRev and EGFPNRBS-9For for the weakest RBS and EGFPRev and EGFPNRBS-131For for the strongest RBS. The obtained amplified fragments were digested with XbaI and cloned into an XbaI/EcoRV-hydrolyzed pGUS vector, yielding pEGFPN-9 and pEGFPN-131 (Table 1).
Construction of unstable variants of the GusA reporter
To translationally fuse the gusA gene with C-terminal degradation tags, the following reverse primers were used: GusARevLVA, GusARevAAV, and GusARevASV (Table 2). The forward primer was the same in all cases – GusASpeForw (Table 2). As a result of PCR amplification, three different 1.9-kb fragments were obtained, which were then digested with SpeI/EcoRV and cloned into the respective sites of the pGCymRP21 plasmid18, yielding plasmids pGCymRP21-LVA, pGCymRP21-ASV, and pGCymRP21-AAV (Table 1).
Calculation of terminator efficiency
In our reporter construct, terminators are placed between the strongest synthetic promoter (P21)16 and the gusA reporter gene. As a control, a plasmid containing the P21 promoter fused to the gusA gene, without terminators between them, was used. Presence of terminators between the P21 promoter and the gusA gene will reduce the fraction of gusA transcripts, preventing read-through and consequently reducing glucuronidase activity. Therefore, the ratio of glucuronidase activity in the presence of a terminator (GUSTerminator) to gusA activity in the control plasmid (GUSControl) was used for the calculation of terminator read-through (TR), which is reported as % values. Based on these values, terminator efficiency was estimated as follows:
TR (%) = GUSTerminator/GUSControl × 100
Assessment of RBS and terminator strength (GUS assay)
For direct detection of glucuronidase activity, 1- to 5-day plates were flooded with 5-bromo-4-chloro-3-indolyl glucuronide (X-Gluc) solution and incubated at 28 °C for 1–4 h. A 1 M X-Gluc stock solution was prepared in dimethyl sulfoxide. The final concentration of the X-Gluc solution used for flooding plates was 20 or 200 mM.
For the quantitative measurement of GusA activity, 1 ml of 24-h seed cultures of the S. lividans TK24 or S. albus J100 recombinant strains was inoculated into 25 ml of TSB. The cells were grown for 1 or 2 days. A 5-ml aliquot of the culture was harvested via centrifugation (6,000 × g for 10 min) and used for the measurement of glucuronidase activity, as described in Horbal et al., 2014. In case of all samples 2 ml of the culture broth was centrifuged, the supernatant was discarded and the biomass was dried for 2 days at 75 °C. All measurements were normalized to dry weight, and the presented results are from three independent experiments. Microsoft Excel was employed for statistical analysis.
Electronic supplementary material
Acknowledgements
This work was supported by the European Commission under the 7th Framework Program through the “Collaborative Project” action “STREPSYNTH” Grant no. 613877 and through the ERC starting Grant EXPLOGEN no. 281623 to AL. A part of this work was also supported by the DFG grant LU 1524/4-1.
Author Contributions
Study concept and design: A.L. and L.H.; performance of experiments: L.H. and T.S.; data interpretation: L.H. and A.L.; manuscript preparation: L.H. and A.L.; Manuscript approval: all authors.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-18846-1.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucks JB, Qi L, Mutalik VK, Wang D, Arkin AP. Versatile RNA sensing transcriptional regulators for engineering genetic networks. Proc. Natl. Acad. Sci. USA. 2011;108:8617–8622. doi: 10.1073/pnas.1015741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Egbert RG, Klavins E. Fine-tuning gene networks using simple sequence repeats. Proc. Natl. Acad. Sci. USA. 2012;109:16817–16822. doi: 10.1073/pnas.1205693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 2012;14:189–195. doi: 10.1016/j.ymben.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Mutalik VK, et al. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013;10:354–360. doi: 10.1038/nmeth.2404. [DOI] [PubMed] [Google Scholar]
- 6.Bentley SD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda H, et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 8.Ohnishi Y, et al. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J. Bacteriol. 2008;190:4050–4060. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu H, Sandiford SK, van Wezel GP. Triggers and cues that activate antibiotic production by actinomycetes. J. Ind. Microbiol. Biotechnol. 2014;41:371–386. doi: 10.1007/s10295-013-1309-z. [DOI] [PubMed] [Google Scholar]
- 10.van Wezel GP, McDowall KJ. The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat. Prod. Rep. 2011;28:1311–1333. doi: 10.1039/c1np00003a. [DOI] [PubMed] [Google Scholar]
- 11.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 12.Elowitz MB, Leibier S. A synthetic oscillatory network of transcriptional regulators. Nature. 2000;403:335–338. doi: 10.1038/35002125. [DOI] [PubMed] [Google Scholar]
- 13.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hammer K, Mijakovic I, Jensen PR. Synthetic promoter libraries tuning of gene expression. Trends Biotechnol. 2006;24:53–55. doi: 10.1016/j.tibtech.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Siuti P, Yazbek J, Lu TK. Synthetic circuits integrating logic and memory in living cells. Nature Biotechnol. 2013;31:448–452. doi: 10.1038/nbt.2510. [DOI] [PubMed] [Google Scholar]
- 16.Siegl T, Tokovenko B, Myronovkyi M, Luzhetskyy A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 2013;19:98–106. doi: 10.1016/j.ymben.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Shis DL, Hussain F, Meinhardt S, Swint-Kruse L, Bennett MR. Modular, multi-input transcriptional logic gating with orthogonal LacI/GalR family chimeras. ACS Synth. Biol. 2014;3:645–651. doi: 10.1021/sb500262f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horbal L, Fedorenko V, Luzhetskyy A. Novel and tightly regulated resorcinol and cumate-inducible expression systems for Streptomyces and other actinobacteria. Appl. Microbiol. Biotechnol. 2014;98:8641–8655. doi: 10.1007/s00253-014-5918-x. [DOI] [PubMed] [Google Scholar]
- 19.Rytter JV, et al. Synthetic promoter libraries for Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2014;98:2617–2623. doi: 10.1007/s00253-013-5481-x. [DOI] [PubMed] [Google Scholar]
- 20.Trefzer A, et al. Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob. Agents Chemother. 2002;46:1174–1182. doi: 10.1128/AAC.46.5.1174-1182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horbal, L. et al. The pathway-specific regulatory genes, tei15* and tei16*, are the master switches of teicoplanin production in Actinoplanes teichomyceticus. Appl. Microbiol. Biotechnol. 98, 9295–9309 (2014b). [DOI] [PubMed]
- 22.Lanza AM, Crook NC, Alper HS. Innovation at the intersection of synthetic and systems biology. Curr. Opin. Biotechnol. 2012;23:712–717. doi: 10.1016/j.copbio.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Young, E. & Alper, H. Synthetic biology: tools to design, build, and optimize cellular processes. J. Biomed. Biotechnol. 130781 (2010). [DOI] [PMC free article] [PubMed]
- 24.Myronovskyi, M., Welle, E., Fedorenko, V. & Luzhetskyy, A. Beta-glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl. Environ. Microbiol. 77, 5370–5383 (2011). [DOI] [PMC free article] [PubMed]
- 25.Kaberdin VR, Bläsi U. (2006) Translation initiation and the fate of bacterial mRNAs. FEMS Microbiol. Rev. 2011;30:967–979. doi: 10.1111/j.1574-6976.2006.00043.x. [DOI] [PubMed] [Google Scholar]
- 26.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bentele K, Saffert P, Rauscher R, Ignatova Z, Blüthgen N. Efficient translation initiation dictates codon usage at gene start. Mol. Syst. Biol. 2013;9:675. doi: 10.1038/msb.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 2006;24:1027–1032. doi: 10.1038/nbt1226. [DOI] [PubMed] [Google Scholar]
- 29.Seo SW, et al. Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab. Eng. 2013;15:67–74. doi: 10.1016/j.ymben.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Komarova AV, Tchufistova LS, Supina EV, Boni IV. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA. 2002;8:1137–1147. doi: 10.1017/S1355838202029990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gingold H, Pilpel Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 2011;7:481. doi: 10.1038/msb.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Espah Borujeni A, Channarasappa AS, Salis HM. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014;42:2646–2659. doi: 10.1093/nar/gkt1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinke-Appel J, et al. Contacts between 16S ribosomal RNA and mRNA, within the spacer region separating the AUG initiator codon and the Shine-Dalgarno sequence; a site-directed cross-linking study. Nucleic Acids Res. 1994;22:3018–3025. doi: 10.1093/nar/22.15.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Bjerknes M, Kumar R, Jay E. Determination of the optimal aligned spacing between the Shine Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res. 1994;22:4953–4957. doi: 10.1093/nar/22.23.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zadeh JN, et al. NUPACK: Analysis and design of nucleic acid systems. J. Comput. Chem. 2010;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 36.Marzi S, et al. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell. 2007;130:1019–1031. doi: 10.1016/j.cell.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Sun J, Kelemen GH, Fernández-Abalos JM, Bibb MJ. Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2) Microbiol. 1999;145:2221–2227. doi: 10.1099/00221287-145-9-2221. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds R, Bermúdez-Cruz RM, Chamberlin MJ. Parameters affecting transcription termination by Escherichia coli RNA polymerase I. Analysis of 13 rho-independent terminators. J. Mol. Biol. 1992;224:31–51. doi: 10.1016/0022-2836(92)90574-4. [DOI] [PubMed] [Google Scholar]
- 39.Wilson KS, von Hippel PH. Transcription termination at intrinsic terminators: the role of the RNA hairpin. Proc. Natl. Acad. Sci. USA. 1995;92:8793–8797. doi: 10.1073/pnas.92.19.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciampi MS. Rho-dependent terminators and transcription termination. Microbiol. 2006;152:2515–2528. doi: 10.1099/mic.0.28982-0. [DOI] [PubMed] [Google Scholar]
- 41.Cambray G, et al. Measurement and modeling of intrinsic transcription terminators. Nucleic Acids Res. 2013;41:5139–5148. doi: 10.1093/nar/gkt163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. Practical Streptomyces genetics, 2nd edn. John Innes Foundation, Norwich (2000).
- 43.Huff J, Czyz A, Landick R, Niederweis M. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene. 2010;468:8–19. doi: 10.1016/j.gene.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Billich S, et al. Synthetic peptides as substrates and inhibitors of human immune deficiency virus-1 protease. J. Biol. Chem. 1988;263:17905–17908. [PubMed] [Google Scholar]
- 45.Gonciarz-Swiatek M, et al. Recognition, targeting, and hydrolysis of the lambda O replication protein by the ClpP/ClpX protease. J. Biol. Chem. 1999;274:13999–14005. doi: 10.1074/jbc.274.20.13999. [DOI] [PubMed] [Google Scholar]
- 46.Gur E, Sauer RT. Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 2008;22:2267–2277. doi: 10.1101/gad.1670908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Triccas JA, Pinto R, Britton WJ. Destabilized green fluorescent protein for monitoring transient changes in mycobacterial gene expression. Res. Microbiol. 2002;153:379–383. doi: 10.1016/S0923-2508(02)01327-X. [DOI] [PubMed] [Google Scholar]
- 48.Andersen JB, et al. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 1998;64:2240–2246. doi: 10.1128/aem.64.6.2240-2246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen MS, Wilgus JR, Chewning CS, Sayler GS, Simpson ML. A destabilized bacterial luciferase for dynamic gene expression studies. Syst. Synth. Biol. 2007;1:3–9. doi: 10.1007/s11693-006-9001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Biol. 2000;7:449–455. doi: 10.1038/75843. [DOI] [PubMed] [Google Scholar]
- 51.Barends S, Kraal B, van Wezel GP. The tmRNA-tagging mechanism and the control of gene expression: a review. Wiley Interdiscip Rev RNA. 2011;2:233–246. doi: 10.1002/wrna.48. [DOI] [PubMed] [Google Scholar]
- 52.Yi JS, et al. A novel approach for gene expression optimization through native promoter and 5′ UTR combinations based on RNA-seq, ribo-seq, and TSS-seq of Streptomyces coelicolor. ACS Synth. Biol. 2017;6:555–565. doi: 10.1021/acssynbio.6b00263. [DOI] [PubMed] [Google Scholar]
- 53.Sambrook, J. & Russell, D. Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY (2001).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.