

Abstract
Antimicrobial resistant infections are a serious public health threat worldwide. Whole genome sequencing approaches to rapidly identify pathogens and predict antibiotic resistance phenotypes are becoming more feasible and may offer a way to reduce clinical test turnaround times compared to conventional culture-based methods, and in turn, improve patient outcomes. In this study, we use whole genome sequence data from 1668 clinical isolates of Klebsiella pneumoniae to develop a XGBoost-based machine learning model that accurately predicts minimum inhibitory concentrations (MICs) for 20 antibiotics. The overall accuracy of the model, within ±1 two-fold dilution factor, is 92%. Individual accuracies are ≥90% for 15/20 antibiotics. We show that the MICs predicted by the model correlate with known antimicrobial resistance genes. Importantly, the genome-wide approach described in this study offers a way to predict MICs for isolates without knowledge of the underlying gene content. This study shows that machine learning can be used to build a complete in silico MIC prediction panel for K. pneumoniae and provides a framework for building MIC prediction models for other pathogenic bacteria.
Introduction
Klebsiella pneumoniae infections are a major cause of morbidity and mortality worldwide. Over the past several years, the emergence of antimicrobial resistant (AMR) K. pneumoniae strains has been increasing at an alarming rate, with reports of pan-resistant strains appearing in the literature and lay press1–3. Reports of hospital-based outbreaks are particularly concerning, and recent evidence suggests that AMR K. pneumoniae clones are circulating in the community3–5. As antimicrobial resistance increases, fewer effective antibiotics are available for physicians to treat these life-threatening infections. In response, the World Health Organization recently listed carbapenem and third generation cephalosporin resistant Enterobacteriaceae (including K. pneumoniae) among the most critical organisms for antimicrobial drug development6.
When a patient is diagnosed with an infection, it is critically important to prescribe appropriate antimicrobial therapy as quickly as possible. Rapid pathogen identification and appropriate antimicrobial therapy administration significantly decreases mortality, improves patient outcomes, reduces health care costs, and decreases the use of ineffective or inappropriate antibiotics7–9. For bloodstream infections, mortality increases every hour that appropriate therapy is delayed9. The conventional clinical microbiology laboratory evaluation of a suspected infection requires inoculation of the specimen on primary culture media and incubation until there is sufficient growth to perform taxonomic identification and minimal inhibitory concentration (MIC) determination. In many cases, subcultures are needed to purify mixed cultures containing more than one organism or generate sufficient colonial material for testing. Depending on the growth rate of the organism and the MIC testing procedures used, the multiple culture steps can add one or more days to the laboratory workup10,11.
Compared to conventional culture-based methods, rapid molecular assays may significantly reduce turnaround times by eliminating one or more subculture steps. The most common sequence-based methods for predicting the AMR phenotypes of an organism identify the presence of genes implicated in resistance using PCR, microarrays or whole genome sequencing12–14. Well-designed gene-based detection methods are capable of providing an accurate prediction of susceptibility or resistance for the genes tested, but and there are several limitations to this approach. First, it relies on well-curated databases of AMR genes, which can be difficult to maintain15–18. For example, the commonly used databases of AMR genes are currently excellent at cataloging well-studied AMR genes like β-lactamases19, but often lack data for diverse efflux mechanisms18. Second, similarity-based matching strategies to determine AMR gene content may incorrectly assign AMR functions to paralogous non-AMR genes. Gene based approaches may also fail to identify critical mutations in intergenic regions, including regulatory and promoter sequences, leading to a false-negative susceptibility prediction. Also, PCR based methods use primers for amplification, which may not anneal if mutations are present in the complementary region, again rendering an incorrect result. Finally, since these methods are based on preexisting knowledge of AMR conferring genomic regions, they are not able to predict resistance if the molecular mechanism is unknown or multifactorial.
The public sharing of whole genome sequence data with clinical AMR metadata has enabled the use of machine learning (ML) methods that predict AMR phenotypes without relying on a database of preexisting AMR genes or mutations. Two recent studies have used this approach to obtain accurate predictions of susceptibility or resistance in organisms with no a priori information about the gene content of the organisms20,21. To do this, they used short nucleotide k-mers as features and the laboratory derived AMR phenotypes as labels. Other studies have successfully used AMR genes, SNPs, and whole genome sequence data (or a combination thereof) to build ML classifiers with good accuracies22–27. Recent examples of gene-based and whole genome-based classification approaches for Klebsiella were reported by Stoesser et al.27, Long et al.3, and Pesesky et al.24.
To date, most AMR prediction methods have focused on classifying “susceptible” and “resistant” phenotypes. While simple and oftentimes sufficient, this approach can be error prone because it relies on the clinical interpretations of break point values. Also, intermediate phenotypes do not fit within this classification scheme. A small number of studies have attempted to predict MICs based on gene content28–30. One notable recent publication used an ML algorithm trained on the SNPs from several key AMR genes to successfully predict MICs for Neisseria gonorrhoeae 29.
In this study, we present an ML approach for predicting MICs for K. pneumoniae. Our strategy requires no a priori knowledge of the underlying gene content. The current model offers MIC prediction for 20 antibiotics. To our knowledge, this is the largest MIC prediction study for a human pathogen to date. We discuss the strengths and limitations of our approach and the necessary steps required to implement in silico MIC prediction using whole genome sequence data for K. pneumoniae in the clinical laboratory.
Results
Approach
For several years, the microbiology laboratory at Houston Methodist Hospital System has been banking clinical isolates of K. pneumoniae. We recently sequenced the genome of AMR K. pneumoniae strains recovered from patients between 2011 and 20153. Our goal is to use whole genome sequencing to detect the emergence of highly virulent clones, monitor the spread of AMR, and guide patient care decisions31,32. We routinely perform whole genome sequencing in our clinical laboratory. Importantly, as the cost and speed of whole genome sequencing continues to decrease, it increasingly becomes a viable option for routine microbial diagnostics.
Using the whole genome sequence data for our K. pneumoniae clinical isolates, we sought to build an ML model that accurately predicts the MIC for 20 antibiotics. We chose a strategy that uses the entire genome as input, rather than individual genes, since this approach requires no a priori knowledge of the underlying gene content, and could potentially use data from uncharacterized AMR genes, intergenic or polymorphic coding regions, or non-AMR genes that may indirectly effect the MIC. To accomplish this, we computed the counts of all overlapping 10-mer oligonucleotide k-mers and combined them with the clinical laboratory generated MIC data for each antibiotic to form one large matrix containing both the k-mers and antibiotics as features. After exploring the problem as both a multiclass classification problem and a regression problem and evaluating common ML algorithms, we chose an extreme gradient boosting regression model through the XGBoost library33 (Fig. 1). We then iteratively evaluated the available parameters of the algorithm to maximize the accuracy of the model (Figure S1, Materials and Methods).
Figure 1.
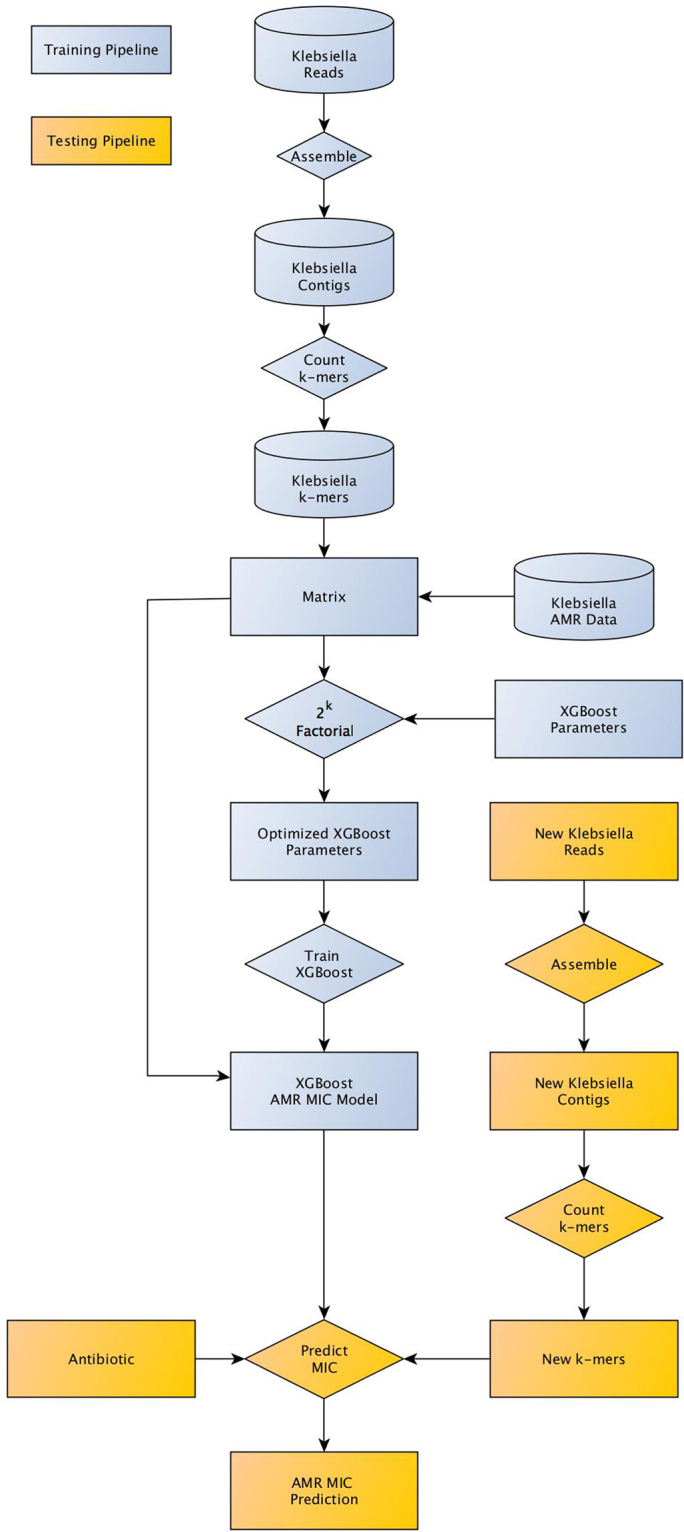
The pipeline used to optimize and train the XGBoost model using known data (blue), and to predict the MIC values for a new genome (yellow).
Model Accuracy
A 10-fold cross validation was used to access the overall stability and accuracy of the model. The raw accuracy of the model, defined as the ability to predict the exact laboratory derived MIC for a given genome and antibiotic (Supplemental Table S1), and accuracy within ±1 two-fold dilution factor (or 1-tier) of the actual MIC was measured (Table 1). Bounding the accuracy to within one two-fold dilution factor of the laboratory determined MIC is consistent with current FDA standards for diagnostic tools and conventional clinical microbiology practices34,35. We also evaluated the model based on the very major error (VME) rate, defined as a resistant isolate having a MIC that is predicted to be susceptible, and the major error rate (ME), defined as a susceptible isolate having a MIC that is predicted to be resistant. MIC thresholds for susceptibility and resistance for clinical data and model predictions are based on current CLSI breakpoints36.
Table 1.
Accuracies for the entire XGBoost model and for the individual antibiotics.
| Antibiotic | Samples | Accuracya | 95% C.I.b |
|---|---|---|---|
| All | 32705 | 0.92 | 0.92, 0.92 |
| Amikacin | 1667 | 0.97 | 0.96, 0.98 |
| Ampicillin | 1666 | 1.00 | 0.99, 1.00 |
| Ampicillin/Sulbactam | 1664 | 0.99 | 0.99, 1.00 |
| Aztreonam | 1644 | 0.89 | 0.89, 0.90 |
| Cefazolin | 1667 | 0.96 | 0.95, 0.96 |
| Cefepime | 1571 | 0.61 | 0.58, 0.64 |
| Cefoxitin | 1645 | 0.90 | 0.89, 0.91 |
| Ceftazidime | 1667 | 0.92 | 0.91, 0.93 |
| Ceftriaxone | 1667 | 0.89 | 0.87, 0.90 |
| Cefuroxime sodium | 1575 | 0.99 | 0.99, 1.00 |
| Ciprofloxacin | 1664 | 0.98 | 0.97, 0.98 |
| Gentamicin | 1667 | 0.95 | 0.93, 0.96 |
| Imipenem | 1666 | 0.94 | 0.93, 0.95 |
| Levofloxacin | 1666 | 0.97 | 0.96, 0.97 |
| Meropenem | 1660 | 0.93 | 0.91, 0.95 |
| Nitrofurantoin | 895 | 0.96 | 0.95, 0.97 |
| Piperacillin/Tazobactam | 1662 | 0.78 | 0.77, 0.79 |
| Tetracycline | 1667 | 0.89 | 0.87, 0.90 |
| Tobramycin | 1666 | 0.95 | 0.94, 0.96 |
| Trimethoprim/Sulfamethoxazole | 1667 | 0.95 | 0.94, 0.96 |
aAverage accuracy within ±1 two-fold dilution factor, based on a 10-fold cross validation. b95% confidence interval.
The average raw accuracy of the entire model, testing on all available MICs for 20 antibiotics was 69% with a 95% confidence interval of [68%; 69%]. The within 1-tier accuracy was much higher, at 92% with a 95% confidence interval of [92%; 92%] (Table 1, Supplemental Table S1). The large difference between the raw accuracy and the within 1-tier accuracy is probably the result of a variety of factors including the inherent error in the laboratory MIC testing procedure37, variations in growth and testing conditions, MICs with > and < values (which may actually represent a range of values), and a possible lack of discriminating genetic features between adjacent MIC dilutions. The raw accuracies and within 1-tier accuracies for the individual antibiotics track similarly, with the raw accuracies being lower and the within 1-tier accuracies being markedly higher. Overall, 15 of the 20 antibiotics have within 1-tier accuracies ≥90%. Three antibiotics have within 1-tier accuracies = 89%, while piperacillin/tazobactam and cefepime had lower within 1-tier accuracies of 78% and 61%, respectively (Table 1).
The accuracy of the model varies across the MICs for each antibiotic, in part, due to nonuniform representation of genomes for every possible value; however, the included genomes are representative of K. pneumoniae strains causing human infections. Figure 2 depicts the number of genomes and the within 1-tier accuracy for each MIC and antibiotic. Overall, MICs represented by many genomes tend to have high accuracies and MICs represented by fewer genomes tend to have lower accuracies. For example, the model has higher accuracies for β-lactam resistant MICs than susceptible MICs because there are fewer genomes for susceptible strains in our data set. In some cases, the model appears to be able to successfully interpolate over bins with a small number of samples. For example tobramycin MIC = 4 μg/ml, which is in between bins with deep sampling and high accuracy had 23 samples and an accuracy equal to 90%. Accuracies and confidence intervals for all bins are shown in Supplemental Table S2. The actual and predicted MICs for each genome are displayed in Supplemental Table S3.
Figure 2.
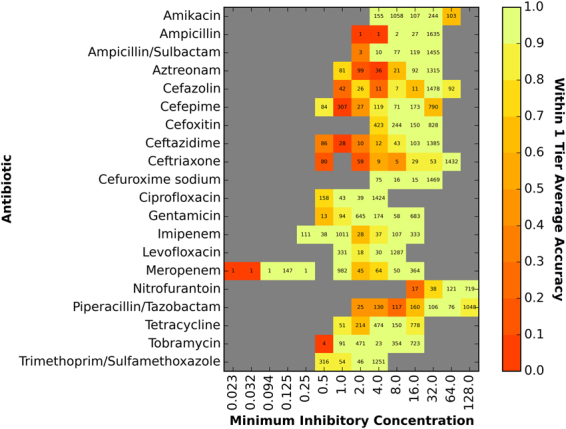
The accuracy of the XGBoost model for individual MICs. The X-axis of the heatmap shows the actual MIC (μ g/ml) for a bin and the Y-axis lists the antibiotics. The within ±1-tier accuracy of a particular antibiotic-MIC bin is denoted by color, with red and orange being least accurate and bright yellow and green being most accurate. The number within each cell represents the number of samples (genomes with the MIC) within the bin.
The average VME rate for the entire model was 3.1% with a 95% confidence interval of [2.8%;3.4%](Table 2). The average ME rate was 3.7% with a confidence interval of [3.3%;4.1%] (Table 2). The VME ranged from 0 for ampicillin and ceftriaxone to 30% for amikacin. Poorer prediction of amikacin MICs is likely due to the lower representation of amikacin resistant genomes in the dataset. Eleven of the antibiotics have VME rates with confidence intervals between 1.5 and 7.5% for the lower and upper bounds respectively. Likewise, 12 of the antibiotics have ME rates ≤3%. These results align with FDA diagnostic device standards34, suggesting that our model may be suitable for clinical use. However, it must be noted that susceptible and resistant MICs are not balanced in the data set, so the lowest VME rates tend to track with antibiotics that have the largest number of resistant genomes. We plan to test additional K. pneumoniae strains with these AMR phenotypes to fill this gap.
Table 2.
Error rates for the entire XGBoost model for the individual antibiotics.
| Antibiotic | Resistant | Susceptible | VMEa | VME 95% C. I.b | MEc | ME 95% C. I.b |
|---|---|---|---|---|---|---|
| All | 21404 | 9410 | 0.031 | 0:028, 0:034 | 0.037 | 0:033, 0:041 |
| Amikacin | 103 | 1320 | 0.298 | 0:239, 0:358 | 0.000 | 0:000, 0:000 |
| Ampicillin | 1635 | 4 | 0.000 | 0:000, 0:000 | 0.000 | 0:000, 0:000 |
| Ampicillin/Sulbactam | 1455 | 90 | 0.003 | 0:000, 0:007 | 0.032 | −0:021, 0:085 |
| Aztreonam | 1407 | 216 | 0.001 | −0:001, 0:002 | 0.398 | 0:333, 0:462 |
| Cefazolin | 1570 | 97 | 0.060 | 0:047; 0:072 | 0.018 | −0:009, 0:046 |
| Cefepime | 963 | 418 | 0.007 | 0:002, 0:012 | 0.137 | 0:077, 0:197 |
| Cefoxitin | 828 | 667 | 0.077 | 0:060, 0:095 | 0.009 | −0:001, 0:019 |
| Ceftazidime | 1488 | 136 | 0.005 | 0:001, 0:008 | 0.123 | 0:069, 0:177 |
| Ceftriaxone | 1528 | 80 | 0.000 | 0:000, 0:000 | 0.188 | 0:101, 0:274 |
| Cefuroxime sodium | 1469 | 91 | 0.002 | 0:000, 0:004 | 0.010 | −0:013, 0:033 |
| Ciprofloxacin | 1424 | 201 | 0.005 | 0:000, 0:010 | 0.025 | 0:000, 0:050 |
| Gentamicin | 683 | 926 | 0.072 | 0:061, 0:082 | 0.009 | 0:001, 0:017 |
| Imipenem | 478 | 1160 | 0.040 | 0:012, 0:067 | 0.032 | 0:021, 0:043 |
| Levofloxacin | 1287 | 349 | 0.016 | 0:006, 0:025 | 0.020 | 0:006, 0:034 |
| Meropenem | 481 | 1134 | 0.048 | 0:034, 0:062 | 0.027 | 0:017, 0:038 |
| Nitrofurantoin | 719 | 55 | 0.018 | 0:009, 0:027 | 0.227 | 0:098, 0:356 |
| Piperacillin/Tazobactam | 1048 | 432 | 0.025 | 0:011, 0:038 | 0.012 | 0:000, 0:023 |
| Tetracycline | 778 | 739 | 0.114 | 0:095, 0:134 | 0.008 | 0:001, 0:015 |
| Tobramycin | 723 | 589 | 0.040 | 0:023, 0:057 | 0.012 | 0:002, 0:022 |
| Trimethoprim/Sulfamethoxazole | 1251 | 416 | 0.119 | 0:098, 0:140 | 0.108 | 0:082, 0:134 |
aVME, Average very major error rate, which is defined as the percentage of resistant samples that are incorrectly predicted to be susceptible by the model.
b95% confidence interval for the average VME and ME rates, respectively.
cME, Average major error rate, which is defined as the percentage of susceptible samples that are incorrectly predicted to be resistant by the model.
Correlation with AMR Genes
The XGBoost algorithm generates a regression model that is based on an ensemble of decision trees. In our model, these decision trees split the data based on k-mers that distinguish the different antibiotics and MICs. Interpreting the features that are being selected by the model and subsequently understanding their relationship to a given antibiotic and resistance mechanism can be challenging for several reasons. Primarily, understanding the feature importance is an open graph theory problem. Furthermore, if an important k-mer maps to an uncharacterized gene or genomic region, it becomes difficult to determine if it is a hallmark of resistance or susceptibility. Finally, unambiguously associating a k-mer to a given antibiotic may inappropriately confer resistance to several related antibiotics, and we know that this dataset contains multiple resistance gene types such as the Bla-KPC and TEM β-lactamases3.
In general, the AMR that is detected in K. pneumoniae strains isolated from patients is usually conferred by the acquisition of antibiotic resistance genes rather than an accumulation of SNPs3,38. We reasoned that in many cases, the MICs for an antibiotic should correlate with the occurrence of genes known to confer resistance to that antibiotic. By comparing the correlation of MICs and functions with the correlation that is based on the predicted MICs from the model, we gain an understanding of the relationship between MICs and AMR genes. We can also observe how closely the model is replicating these relationships. To do this, we computed the Pearson correlation coefficient (PCC) between the MICs for each genome and the presence or absence of each unique function in each genome (Table 3, Supplemental Table S4).
Table 3.
The function that is most highly correlated with the MICs for each antibiotic.
| Antibiotic | PATRIC Function | PCC Actual MICa | PCC Predicted MICb | Top 10 Coveragec |
|---|---|---|---|---|
| Meropenem | Class A beta-lactamase (EC 3.5.2.6) => KPC family, carbapenem-hydrolyzing | 0.923 | 0.814 | 0.7 |
| Trimethoprim Sulfamethoxazole | Dihydropteroate synthase type-2 (EC 2.5.1.15) @ Sulfonamide resistance protein | 0.919 | 0.758 | 0.9 |
| Imipenem | Class A beta-lactamase (EC 3.5.2.6) => KPC family, carbapenem-hydrolyzing | 0.891 | 0.905 | 0.8 |
| Cefepime | Class A beta-lactamase (EC 3.5.2.6) => CTX-M family, extended-spectrum | 0.848 | 0.648 | 0.9 |
| Tobramycin | Aminoglycoside N(6′)-acetyltransferase (EC 2.3.1.82) => AAC(6′)-Ib/AAC(6′)-II | 0.837 | 0.853 | 0.8 |
| Tetracycline | Tetracycline resistance regulatory protein TetR | 0.829 | 0.717 | 0.8 |
| Ceftriaxone | Class A beta-lactamase (EC 3.5.2.6) => CTX-M family, extended-spectrum | 0.823 | 0.700 | 0.7 |
| Gentamicin | Aminoglycoside N(3)-acetyltransferase (EC 2.3.1.81) => AAC(3)-II,III,IV,VI,VIII,IX,X | 0.818 | 0.862 | 0.6 |
| Ampicillin Sulbactam | Class A beta-lactamase (EC 3.5.2.6) => TEM family | 0.780 | 0.787 | 0.8 |
| Ciprofloxacin | Integron integrase IntI1 | 0.715 | 0.713 | 0.8 |
| Aztreonam | Integron integrase IntI1 | 0.678 | 0.614 | 0.7 |
| Cefazolin | Class A beta-lactamase (EC 3.5.2.6) => CTX-M family, extended-spectrum | 0.676 | 0.667 | 0.9 |
| Cefuroxime sodium | Aminoglycoside N(3)-acetyltransferase (EC 2.3.1.81) => AAC(3)-II,III,IV,VI,VIII,IX,X | 0.668 | 0.616 | 0.3 |
| Ceftazidime | Integron integrase IntI1 | 0.657 | 0.623 | 0.6 |
| Levofloxacin | probable bacteriophage protein STY1063 | 0.588 | 0.584 | 0.7 |
| Piperacillin Tazobactam | plasmid stabilization system | 0.583 | 0.501 | 0.1 |
| Amikacin | IncI1 plasmid conjugative transfer prepilin PilS | 0.577 | 0.478 | 0.2 |
| Cefoxitin | Class A beta-lactamase (EC 3.5.2.6) => KPC family, carbapenem-hydrolyzing | 0.550 | 0.571 | 0.6 |
| Nitrofurantoin | Integron integrase IntI1 | 0.433 | 0.507 | 0.6 |
| Ampicillin | Class A beta-lactamase (EC 3.5.2.6) => TEM family | 0.357 | 0.327 | 0.0 |
aPearson correlation coefficient between the occurrences of the given function and the actual MICs.
bPearson correlation coefficient between the occurrences of the given function and the predicted MICs.
cThe fraction of the top 10 functions (by PCC) for the predicted MICs that occur in the top 10 for the actual MICs.
In the case of 12 antibiotics, we observe high PCCs between the MICs and the functions for well-known AMR genes. For example, the β-lactam antibiotics correlate with the CTX, KPC and TEM type β-lactamase genes and the aminoglycoside antibiotics correlate with aminoglycoside acetyltransferase genes. For 8 of the 20 antibiotics, the association between the functions and AMR is not obvious. In some of these cases, the functions appear to be related to horizontal gene transfer, and this may have resulted in their high PCCs. In other cases, the genes may not be sufficiently characterized by the raw 10-mer counts. When the analysis is repeated using MICs that are predicted by the model, the PCCs track very closely with those generated from the actual MICs, indicating that the model is not only learning that these genes exist (the model is only fed 10-mer counts, not whole genes), but it is also placing importance on these genes. Similarly, there is considerable overlap between the top ten most highly correlated functions based on the actual and predicted MICs (Table 3, last column).
In previous work, we built 16 AdaBoost-based classifiers for predicting susceptibility and resistance and installed them in the RAST annotation system3. Thirteen of the antibiotics that were used to build these classifiers are represented in our MIC prediction model. The top genomic regions predicted by the K. pneumoniae AdaBoost classifiers correspond to the most most highly correlated functions from the MIC prediction model in for four of the thirteen antibiotics. These include gentamicin, imipenem, meropenem and tetracycline. The top AdaBoost feature can be found found among the top five most highly correlated functions in three more cases including aztreonam, cefoxitin, and trimethoprim/sulfamethoxazole. Since the AdaBoost classifiers match SNPs in gyrase and topoisomerase genes conferring resistance to ciprofloxacin and levofloxacin, assessing the correlation to the presence of functions will not work for these two antibiotics. It is not immediately clear why the top features for the other four antibiotics (amikacin, cefepime, piperacillin/tazobactam and tobramicin) are not highly correlated, but no characterized AMR functions appear in the top 10 most highly correlated features.
Building a Model Based on AMR Genes
Although we chose to build a model that was based on data from the entire genome, previous studies have built MIC prediction models using the known AMR genes28–30. We wanted to know whether a model based on whole genome data, or a model based on only AMR genes would be more accurate. On one hand, the extra k-mers used by the whole genome model could be a source of noise, but on the other hand, they may represent useful data that could making the model more accurate. To make this comparison, we built a model that used only the AMR genes as the source of k-mers, keeping all other parameters identical. Both the PATRIC annotation service18,39 and the CARD database15 were used as sources of potential AMR genes. The overall accuracies for the whole genome and AMR gene-based models are both 92%. Likewise, the individual accuracies for each antibiotic are also nearly identical, differing by no more than 2% for any antibiotic (Supplemental Table S5, Supplemental Figure S2). This indicates that the extra k-mers used by the whole genome are not a source of noise for the XGBoost model. Since the AMR genes model is much smaller than the whole-genome model (20GB vs. 148GB), it is more efficient to compute, and it is therefore tempting to conclude that a model built from AMR genes is sufficient for performing MIC prediction for K. pneumoniae. However, due to the smaller number of susceptible and intermediate samples in this data set, more genome sequencing is necessary to determine if a model built from AMR genes is sufficient for accurately predicting these lower MIC values.
When we repeat this analysis building an identical model for the leftover non-AMR genes, we also observe an overall accuracy of 92%, and accuracies for individual antibiotics that track closely with the models built from full contigs and AMR genes (Supplemental Table S5, Supplemental Figure S2). This indicates that there is enough residual data in the non-AMR genes to accurately predict MIC values as well. This could be due to the presence of uncharacterized AMR genes in the data set, the existence of relevant information such as non-AMR genes co-occurring with AMR genes, or variants in non-AMR genes that have an impact on MICs. In the case of all three models, it is unlikely that the accuracy is due to the model mapping to a phylogenetic pattern between strains, since nearly clonal strains of the same MLST type can have a variety of different MIC values per antibiotic, and closely related strains can be quite diverse in their AMR gene content (Supplemental Table S6, Supplemental Figure S3).
Discussion
Using the clinical laboratory determined MICs and whole genome sequence data for 1668 K. pneumoniae strain recovered from infected patients, we built an XGBoost regression model that can predict the MICs for 20 antibiotics with an average within ±1-two fold dilution factor accuracy of 92%. Individually, 15 of the 20 antibiotics have within 1-tier accuracies >90%. These results demonstrate that accurate genome-based MIC prediction is possible for K. pneumoniae isolates. Herein, we provide the necessary groundwork for building a complete in silico panel.
To date, our whole genome sequencing efforts have focused on the most highly antibiotic resistant pathogens, including extended spectrum β-lactamase-(ESBL) producing K. pneumoniae 3, so the model currently lacks sufficient inclusion of organisms with MICs in the lower range. We also currently lack sufficient data to predict MICs for some less-commonly tested antibiotics, including amoxicillin/clavulanate, ertapenem, fosfomycin, moxifloxacin ticarcillin/clavulanate and tigecycline. Ideally, selecting a balanced number of organisms at each possible MIC could improve the overall accuracy of the model. Our future efforts will seek to close these gaps. Inclusion of additional strains will also improve the ME and VME rates. The K. pneumoniae strains used in our model were collected as part of a large, comprehensive, population-based study in Houston. Although our sampling capacity is extensive and we treat a diverse population of patients from Houston and around the world, the model may be further improved by including samples from other geographic locations.
In previous work, we built binary classifiers that can predict susceptibility or resistance for a given species and antibiotic18,20. Although somewhat simplistic in approach, the method provides a straightforward way to extract the genomic features relating to resistance. In this study, in order to achieve high accuracies for predicting each MIC, we combined the antibiotics and k-mers into a large matrix and used XGBoost as the ML method. While the approach is clearly advantageous because it provides a more refined phenotype prediction, feature extraction from this kind of model remains quite challenging. Although we have shown strong correlations between the actual MICs and predicted MICs with known AMR genes, we will continue to explore ways to extract the key gene data that contributes to each MIC. Importantly, these studies may provide new insight to the molecular mechanisms conferring intermediate phenotypes.
We chose to present a model that was built using k-mers from the entire genome because it requires no underlying information about the gene content of the organisms. Furthermore, since it is likely that genomic features that are not annotated as being directly involved in conferring AMR may be causing the distinction between lower MICs, a whole-genome approach may eventually lead to higher accuracies as we acquire more data. Using a whole-genome-based approach may also lead to an improved understanding the genomic features that are causing the various MIC phenotypes. However at present, the current data set and MIC data for K. pneumoniae published by other groups40–43 lacks sufficient sampling of genomes with susceptible and intermediate phenotypes to determine if either approach will ultimately differ in accuracy. If the XGBoost model remains robust on the smaller datasets like the AMR and non-AMR gene sets, this implies that it may be possible to build models that can predict AMR phenotypes from incomplete genomic data, which may help to enable the characterization of metagenomic samples.
In this study, we found that deeper trees, with a depths of 3–4, were optimal for the XGBoost model. A logical next step will be to train a deeper model, like a neural network, on this data set to determine if the accuracy can be further improved. A deep learning approach may also provide more efficient memory usage and reduced computational times. However, this strategy would not improve the extraction and interpretation of AMR-related genomic features, since feature extraction from deep learning methods is more challenging compared to ensemble methods such as XGBoost.
The genomes used in this study were sequenced using Illumina sequencing technology. In order to generate genome sequence data cost effectively, we collected samples and performed highly multiplexed runs in batches. However, this is not a clinically time efficient strategy. Newer devices such as the PacBio Sequel and Oxford MinION could potentially be used for point of care sequencing, and thus, may become a model for whole genome sequence-based diagnostic strategies44. However, at present, the cost of sequencing individual pathogens using these technologies is higher, and their error rates may be too high for effective MIC prediction with our ML method45. In order to couple the MIC prediction model outlined in this study with these sequencing platforms, we may need to either incorporate an error correction model for processing MinION or PacBio reads, or regenerate the model using genomes sequenced with these technologies. Further analyses to sequence based off of blood enrichment cultures, or from the actual wound source, rather than the pure culture, would also provide more rapid results, but require algorithms for identifying pathogens and eliminating host DNA and other contaminants. Regardless, sequence-based MIC prediction appears to be feasible as a diagnostic strategy.
Methods
Strain collection
Klebsiella pneumoniae isolates were cultured from patient specimens in the Houston Methodist Hospital System between September of 2011 and March of 2017. Strains were tested for minimum inhibitory concentrations to 20 antibiotics using the BD-Phoenix system (BD Diagnostics, Sparks, MD, USA). All of the strains collected before 2015 were originally part of two studies by Long and colleagues designed to track extended spectrum β-lactamase- (ESBL) producing strains3,46. A total of 1497 strains from the Long et al. study with BD-Phoenix MIC data were used in this analysis (Supplemental Table S3). An additional 171 isolates, 93 ESBL-producing and 78 non-ESBL-producing (Phoenix ESBL test; BD), were also panel tested, whole genome sequenced, and used in this study. In total, we analyzed 1668 Klebsiella pneumoniae genomes. Strains were collected with informed consent under human subjects protocol IRB1010-0199 as approved by the Institutional Review Board, Houston Methodist Research Institute.
DNA extraction and whole-genome sequencing
Genomic DNA was extracted using the manufacturer’s Gram-negative protocol for the DNeasy Blood and Tissue kit (Qiagen) and quantified using a Qubit 3.0 instrument (Invitrogen). Whole-genome sequencing libraries were prepared using NexteraXT reagents (Illumina) and sequenced on a MiSeq or NextSeq. 500 instrument (Illumina).
Data preparation
When the MICs produced from the BD-Phoenix tests exceeded the testing thresholds, they were cleaned to remove the >, <, ≥, and ≤ symbols. If the MIC was >x, the MIC label was changed to 2x, if it was <x, the MIC was changed to , and if the MIC was ≥x or ≤x, the symbol was removed and the MIC remained unchanged. The Log 2 value of these cleaned MICs were used for all ML tasks. For dual antibiotics with two MICs, such as trimethoprim/sulfamethoxazole, the first value was used in all cases, since the second value is either constant or dependent on the first.
Experimental design
Genomes were assembled with SPAdes47 using the PATRIC assembly service39. Contigs with ≤500 bp and ≤5-fold coverage were removed. The contigs were divided into overlapping 10-mer oligonucleotide k-mers, sorted in lexicographical order and counted using the software package KMC248. K-mer counts and antibiotic names were used as input features into the model. Antibiotic names were one-hot encoded. In this study, we chose to use 10-mers as features, rather than a longer k-mer length, in order to reduce the size of the matrix. This enabled us to load the matrix into memory and perform cross validation on a machine with 1.5TB RAM. Shorter k-mers of size 4 and 8 were also evaluated, but were not chosen because they are more likely to be redundant within a genome and because they had lower initial accuracies. The dataset was then split into equal training, validation and testing sets. Each subset was created so that it contained the same number of MICs for a given antibiotic. The final distribution antibiotics used in the model is uniform, but the number of samples for a given MIC per antibiotic was not uniformly distributed.
The prediction of MICs can be cast as a regression problem or a multi-class classification problem with each MIC representing an individual class. We explored several popular ML algorithms including AdaBoost (Adaptive Boosting)49, bagging50, random forest51, extremely randomized trees52, and support vector machines53 from the scikit-learn python package54, as well as XGBoost (Extreme Gradient Boosting)33. Using default parameter choices, we attempted to cast the problem as both a classification and regression problem depending on the capabilities of the algorithm. Algorithms were compared based on accuracy and computational resources required. An XGBoost regression model was ultimately chosen for this study because it produced the highest default accuracies and had reasonable computation costs for the current data set.
To assess the sensitivity of the model with regard to the training data, we performed a ten-fold cross validation by taking all samples and randomly splitting them into 10 mutually exclusive sets. Each set was split such that all sets had an equal (or nearly equal) number of antibiotic-MIC combinations. Ten models were then generated using 8 of the sets for training, one for validation, and one for testing. In total, 10 models were created. The accuracy within ±1 two-fold dilution factor was computed along with the 95% confidence intervals for each model. This aligns with clinical practice and FDA device guidelines34.
Hyperparameter tuning
Important hyperparameters from XGBoost were then selected and tuned using a 2k factorial design55 and a grid search, respectively. The model was tested for stability using a 10-fold cross validation. The 2k factorial design was used to tune 3 XGBoost parameters: maximum tree depth, column subsampling and row subsampling. The maximum tree depth parameter limits the maximum height of a decision tree while creating the ensemble. A value that is too high tends to overfit data whereas a value that is too low tends to underfit data33. A low value of 1 and a high value of 6 (6 is the default) were used to evaluate the impact of tree depth on the accuracy of the model. The column subsampling parameter limits the number of features that are chosen for training each tree in the ensemble. For example, if 0.5 is chosen, 50% of the features (k-mers) will be randomly chosen to train the first tree, then a different 50% for the second, and so on. A low value of 0.25 and a high value of 1 (1 is the default and maximum value allowed for XGBoost) were chosen. The row subsampling parameter limits the number of samples that are chosen for training for each tree in the ensemble. For example, if 0.5 is chosen, 50% of the samples (MIC tests) will be randomly chosen to train the first tree, then a different 50% for the second, etc. A low value of 0.25 and a high value of 1 (1 is the default and maximum value allowed for XGBoost) were chosen for evaluation.
The accuracy of the XGBoost model was evaluated in two ways. First, the accuracy of each individual MIC prediction over the test set was assessed within ±1 two-fold dilution factor. Secondly the coefficient of determination, or R2, was also used as a metric during the hyperparameter tuning.
A 2k factorial design assumes that as a parameter is increased or decreased, the metric that is being tested will either increase or decrease. Additionally, it only gives an idea of the parameters that are important and not the optimal value for a given parameter. Since the accuracy is not known to always increase or decrease as the maximum tree depth value goes up33, this parameter needed to be tested systematically. A grid search56 was designed based on the results of the 2k factorial design to deal with both issues. Since learning rate is known to have a relationship with maximum tree depth, we systematically varied the maximum tree depth and the learning rate, in tandem, in a grid search experiment with respect to accuracy33.
The results of the 2k factorial design showed that when tuning the model, a higher maximum tree depth was preferable. Different values of row and column subsampling did not cause variance in the accuracy of the model, though a larger value was deemed to produce a more accurate model. Both factors had already been maximized at 1 during the 2k factorial. Supplemental Figure S1a shows this in better detail.
The applied grid search showed that a lower value for the learning rate always returned a better solution. However, as the learning rate decreased, the training time increased. The gain in accuracy was deemed too small for the price (time) with a learning rate of 2−4. During testing we also found that a maximum depth between 3 and 4 was optimal for the Klebsiella data used to train the algorithm. Supplemental Figure S1b shows this in better detail.
Genome Annotation and AMR gene analyses
All genomes were annotated using the PATRIC annotation service in August of 201739. MLST (Multi Locus Sequencing Typing) was performed by the PATRIC annotation service and was based on Diancourt et al.57. AMR genes are defined as those having AMR-related functions (annotations) defined by PATRIC18 as well as all genes encoding proteins matching the CARD database15 with BLASTP identity scores >= 80%58. Non-AMR genes are defined as those that do not match the AMR set. A MIC prediction model built only from AMR genes was computed the same as the final whole genome-based model described above.
Correlation analyses were performed by first gathering the set of uniquely occurring functions from every K. pneumoniae genome. For each genome, the presence (+1) or absence (−1) of a function was compared to the MIC for an antibiotic. The Pearson correlation coefficient was computed for every function and antibiotic combination.
The phylogenetic tree in Supplemental Figure S3 was built by generating alignments for the seven MLST housekeeping genes, rpoB, gapA, mdh, pgi, phoE, infB, and tonB57 using MAFFT59, and then concatenating the them. The tree was generated using FastTree60. Genomes with identical sets of AMR genes are depicted by a single genome. The tree was rendered using iTOL61.
Data Availability
The model and software for predicting MICs in for K. pneumoniae genomes can be found at the PATRIC github page: https://github.com/PATRIC3/mic_prediction. All genomes were submitted to the SRA under bioprojects (PRJNA376414, PRJNA386693 and PRJNA396774). SRA run accession numbers for individual genomes can be found in Supplemental Table S3.
Accession codes
All genomes were submitted to the SRA under bioprojects (PRJNA376414, PRJNA386693 and PRJNA396774).
Electronic supplementary material
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Service [Contract No. HHSN272201400027C] and the Fondren Foundation. We thank Concepcion Cantu, Matthew Ojeda Saavedra and Sarah Linson for technical assistance. We thank Emily Dietrich for her help preparing the manuscript.
Author Contributions
M.N., designed study, conducted experiments, performed data analysis, wrote paper; T.B., designed study; S.W.L., designed study, obtained strains, generated MIC data, performed data analysis, revised manuscript; J.M.M., designed study; R.O. conducted software engineering and distribution; R.J.O., designed study, obtained strains, generated MIC data, performed data analysis, revised manuscript; M.S., integration of data into PATRIC; R.L.S., designed study; F.X., designed study; H.Y. integration of data into PATRIC; J.J.D., designed study, performed data analysis, wrote paper. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-18972-w.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chen, L., Todd, R., Kiehlbauch, J., Walters, M. & Kallen, A. Notes from the Field: Pan-Resistant New Delhi Metallo-Beta-Lactamase-Producing Klebsiella pneumoniae –” Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep 66 (2017). [DOI] [PMC free article] [PubMed]
- 2.Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiology Reviews. 2017;41:252. doi: 10.1093/femsre/fux013. [DOI] [PubMed] [Google Scholar]
- 3.Long SW, et al. Population Genomic Analysis of 1,777 Extended-Spectrum Beta-Lactamase-Producing Klebsiella pneumoniae Isolates, Houston, Texas: Unexpected Abundance of Clonal Group 307. mBio. 2017;8:e00489–17. doi: 10.1128/mBio.00489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snitkin ES, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Science translational medicine. 2012;4:148ra116–148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Endimiani A, et al. Emergence of blakpc-containing Klebsiella pneumoniae in a long-term acute care hospital: a new challenge to our healthcare system. Journal of Antimicrobial Chemotherapy. 2009;64:1102–1110. doi: 10.1093/jac/dkp327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tacconelli, E. & Magrini, N. Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organization (2017).
- 7.Llor C, Bjerrum L. Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem. Therapeutic Advances in Drug Safety. 2014;5:229–241. doi: 10.1177/2042098614554919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer HR, Palavecino EL, Johnson JW, Ohl CA, Williamson JC. Clinical and microbiological implications of time-to-positivity of blood cultures in patients with Gram-negative bacilli bacteremia. European Journal of Clinical Microbiology & Infectious Diseases. 2013;32:955–959. doi: 10.1007/s10096-013-1833-9. [DOI] [PubMed] [Google Scholar]
- 9.Kumar, A. et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Critical Care Medicine34 (2006). [DOI] [PubMed]
- 10.Reller LB, Weinstein M, Jorgensen JH, Ferraro MJ. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clinical Infectious Diseases. 2009;49:1749. doi: 10.1086/647952. [DOI] [PubMed] [Google Scholar]
- 11.Opota O, Croxatto A, Prod’hom G, Greub G. Blood culture-based diagnosis of bacteraemia: state of the art. Clinical Microbiology and Infection. 2015;21:313–322. doi: 10.1016/j.cmi.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg B, Sichtig H, Geyer C, Ledeboer N, Weinstock GM. Making the Leap from Research Laboratory to Clinic: Challenges and Opportunities for Next-Generation Sequencing in Infectious Disease Diagnostics. mBio. 2015;6:e01888–15. doi: 10.1128/mBio.01888-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leski TA, et al. Molecular Characterization of Multidrug Resistant Hospital Isolates Using the Antimicrobial Resistance Determinant Microarray. PLOS ONE. 2013;8:1–12. doi: 10.1371/journal.pone.0069507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pulido MR, García-Quintanilla M, Martín-Peña R, Cisneros JM, McConnell MJ. Progress on the development of rapid methods for antimicrobial susceptibility testing. Journal of Antimicrobial Chemotherapy. 2013;68:2710. doi: 10.1093/jac/dkt253. [DOI] [PubMed] [Google Scholar]
- 15.McArthur AG, et al. The Comprehensive Antibiotic Resistance Database. Antimicrobial Agents and Chemotherapy. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. The ISME Journal. 2015;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.NCBI. Bacterial Antimicrobial Resistance Reference Gene Database. https://www.ncbi.nlm.nih.gov/bioproject/?term=3130472017 (2017).
- 18.Antonopoulos, D. A. et al. PATRIC as a unique resource for studying antimicrobial resistance. Briefings in Bioinformatics 1–9 (2017). [DOI] [PMC free article] [PubMed]
- 19.Bush, K., Pazkill, T. & Jacoby, J. ß-Lactamase Classification and Amino Acid Sequences for TEM, SHV and OXA Extended-Spectrum and Inhibitor Resistant Enzymes. http://www.lahey.org/Studies/.
- 20.Davis JJ, et al. Antimicrobial Resistance Prediction in PATRIC and RAST. Scientific Reports. 2016;6:27930. doi: 10.1038/srep27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drouin A, et al. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genomics. 2016;17:754. doi: 10.1186/s12864-016-2889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradley P, et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nature Communications. 2015;6:10063. doi: 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niehaus, K. E., Walker, T. M., Crook, D. W., Peto, T. E. A. & Clifton, D. A. Machine learning for the prediction of antibacterial susceptibility in Mycobacterium tuberculosis. In IEEE-EMBS International Conference on Biomedical and Health Informatics (BHI), 618–621, 10.1109/BHI.2014.6864440 (2014).
- 24.Pesesky MW, et al. Evaluation of Machine Learning and Rules-Based Approaches for Predicting Antimicrobial Resistance Profiles in Gram-negative Bacilli from Whole Genome Sequence Data. Frontiers in Microbiology. 2016;7:1887. doi: 10.3389/fmicb.2016.01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeukens, J. et al. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa. Annals of the New York Academy of Sciences n/a–n/a, 10.1111/nyas.13358 (2016). [DOI] [PMC free article] [PubMed]
- 26.Coelho JR, et al. The Use of Machine Learning Methodologies to Analyse Antibiotic and Biocide Susceptibility in Staphylococcus aureus. PLoS ONE. 2013;8:e55582. doi: 10.1371/journal.pone.0055582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoesser N, et al. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. Journal of Antimicrobial Chemotherapy. 2013;68:2234–2244. doi: 10.1093/jac/dkt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, et al. Penicillin-Binding Protein Transpeptidase Signatures for Tracking and Predicting β-Lactam Resistance Levels in Streptococcus pneumoniae. mBio. 2016;7:e00756–16. doi: 10.1128/mBio.00756-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eyre, D. W. et al. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. Journal of Antimicrobial Chemotherapy (2017). [DOI] [PMC free article] [PubMed]
- 30.Metcalf B, et al. Using whole genome sequencing to identify resistance determinants and predict antimicrobial resistance phenotypes for year 2015 invasive pneumococcal disease isolates recovered in the United States. Clinical Microbiology and Infection. 2016;22:1002.e1–1002.e8. doi: 10.1016/j.cmi.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 31.Long SW, et al. A genomic day in the life of a clinical microbiology laboratory. Journal of Clinical Microbiology. 2013;51:1272–1277. doi: 10.1128/JCM.03237-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olsen RJ, et al. Clinical laboratory response to a mock outbreak of invasive bacterial infections: a preparedness study. Journal of Clinical Microbiology. 2014;52:4210–4216. doi: 10.1128/JCM.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen, T. & Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22Nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD’16, 785–794, 10.1145/2939672.2939785 (ACM, New York, NY, USA, 2016).
- 34.Food and Drug Administration. Guidance for industry and FDA. Class II special controls guidance document: antimicrobial susceptibility test (AST) systems (Center for Devices and Radiological Health, Food and Drug Administration, US Department of Health and Human Services, Silver Spring, MD, 2009).
- 35.Jorgensen JH. Selection criteria for an antimicrobial susceptibility testing system. Journal of Clinical Microbiology. 1993;31:2841–2844. doi: 10.1128/jcm.31.11.2841-2844.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: 27th Informational Supplement (Clinical and Laboratory Standards Institute, Wayne, PA, 2017).
- 37.Carroll KC, et al. Evaluation of the bd phoenix automated microbiology system for identification and antimicrobial susceptibility testing of Enterobacteriaceae. Journal of Clinical Microbiology. 2006;44:3506–3509. doi: 10.1128/JCM.00994-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holt KE, et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E3574–E3581. doi: 10.1073/pnas.1501049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wattam AR, et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Research. 2017;45:D535–D542. doi: 10.1093/nar/gkw1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Runcharoen C, et al. Whole genome sequencing reveals high-resolution epidemiological links between clinical and environmental Klebsiella pneumoniae. Genome. Medicine. 2017;9:6. doi: 10.1186/s13073-017-0397-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moradigaravand D, Martin V, Peacock SJ, Parkhill J. Evolution and epidemiology of multidrug-resistant Klebsiella pneumoniae in the united kingdom and ireland. mBio. 2017;8:e01976–16. doi: 10.1128/mBio.01976-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.FDA - CDC. FDA-CDC Antimicrobial Resistance Isolate Bank | Antimicrobial Resistance Isolate Bank | Antibiotic/Antimicrobial Resistance | CDC. https://www.cdc.gov/drugresistance/resistance-bank/ (2017).
- 43.DeLeo FR, et al. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae. Proceedings of the National Academy of Sciences. 2014;111:4988–4993. doi: 10.1073/pnas.1321364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain M, Olsen HE, Paten B, Akeson M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biology. 2016;17:239. doi: 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laver T, et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomolecular Detection and Quantification. 2015;3:1–8. doi: 10.1016/j.bdq.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long, W. et al. Whole-Genome Sequencing of Human Clinical Klebsiella pneumoniae Isolates Reveals Misidentification of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. American Society forMicrobiology (2017). [DOI] [PMC free article] [PubMed]
- 47.Bankevich A, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deorowicz S, Kokot M, Grabowski S, Debudaj-Grabysz A. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics. 2015;31:1569. doi: 10.1093/bioinformatics/btv022. [DOI] [PubMed] [Google Scholar]
- 49.Freund Y, Schapire RE. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. Journal of Computer and System Sciences. 1997;55:119–139. doi: 10.1006/jcss.1997.1504. [DOI] [Google Scholar]
- 50.Breiman L. Bagging predictors. Machine Learning. 1996;24:123–140. [Google Scholar]
- 51.Ho, T. K. Random Decision Forests. In Proceedings of the Third International Conference on Document Analysis and Recognition (Volume 1) - Volume 1, ICDAR’95, 278– http://dl.acm.org/citation.cfm?id=844379.844681 (IEEE Computer Society, Washington, DC, USA, 1995).
- 52.Geurts P, Ernst D, Wehenkel L. Extremely randomized trees. Machine Learning. 2006;63:3–42. doi: 10.1007/s10994-006-6226-1. [DOI] [Google Scholar]
- 53.Vapnik, V. N. The Nature of Statistical Learning Theory (Springer-Verlag New York, Inc., New York, NY, USA, 1995).
- 54.Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning ResearchURL http://hal.inria.fr/hal-00650905 (2011).
- 55.Winer, B. J., Brown, D. R. & Michels, K. M. Statistical principles in experimental design, vol. 2 (McGraw-Hill New York, 1971).
- 56.Lerman PM. Fitting Segmented Regression Models by Grid Search. Journal of the Royal Statistical Society. Series C (Applied Statistics) 1980;29:77–84. [Google Scholar]
- 57.Diancourt L, Passet V, Verhoef J, Grimont PAD, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. Journal of Clinical Microbiology. 2005;43:4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 59.Katoh K, Misawa K, Kuma K-i, Miyata T. Mafft: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Research. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Price MN, Dehal PS, Arkin AP. Fasttree 2 - approximately maximum-likelihood trees for large alignments. PLOS ONE. 2010;5:1–10. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Letunic I, Bork P. Interactive tree of life (itol) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The model and software for predicting MICs in for K. pneumoniae genomes can be found at the PATRIC github page: https://github.com/PATRIC3/mic_prediction. All genomes were submitted to the SRA under bioprojects (PRJNA376414, PRJNA386693 and PRJNA396774). SRA run accession numbers for individual genomes can be found in Supplemental Table S3.