

Abstract
It is widely accepted that aberrant activation of the Wnt signaling pathway is responsible for the development of precursor lesions of colorectal cancer (CRC). However, the molecular mechanisms involved in the process of progression from these precursor lesions to invasive lesions of CRC are not fully understood. Recently, we reported that constitutive activation of MAPK accompanied by downregulation of dual‐specificity phosphatase 4 (DUSP4), a MAPK phosphatase, contributes to the progression of precursor lesions in the pancreas. In this study, we found that downregulation of DUSP4 was related to constitutive activation of ERKs in CRC cells. Restoration of DUSP4 resulted in inactivation of ERKs, leading to suppression of both proliferation and invasiveness, as shown by treatment with an MEK inhibitor. Furthermore, immunohistochemistry revealed that DUSP4 expression was upregulated in the superficial region of CRC tissue, whereas it was significantly downregulated in the deep region. In contrast, ERKs in the deep region were markedly hyperactivated compared to those in the superficial region. These results suggest that activation of the MAPK signaling pathway caused by downregulation of DUSP4 is responsible for progression of CRCs and would be a promising therapeutic target.
Keywords: colorectal carcinoma, DUSP4, invasion, MAPK signalling, proliferation
Abbreviations
- 5‐aza‐dC
5‐aza‐2′‐deoxycytidine
- CRC
colorectal cancer
- DUSP4
dual‐specificity phosphatase 4
- EGFR
epidermal growth factor receptor
- RT
room temperature
- TSA
trichostatin A
- UNC5C
unc‐5 netrin receptor C
1. INTRODUCTION
Aberrant activation of the Wnt signaling pathway due to somatic or germ‐line mutation in the APC or CTNNB1 gene has been shown to be responsible for the development of precursor lesions of CRCs.1, 2, 3, 4 Recently, it has been reported that, in addition to mutations of these driver genes, EGFR, a member of the receptor tyrosine kinase family, is overexpressed in 20%‐80% of CRCs, conferring a growth and survival advantage on the cancer cells.5 In the light of those findings, it was suggested that EGFR might be a molecular target for therapeutic interventions. Therefore, targeted therapy using an antibody specific to EGFR was developed on the basis of a clinical study, and is now clinically available for treatment of CRCs showing EGFR overexpression but without mutated KRAS.6, 7 However, as such cases account for only a limited percentage of total CRC cases, other therapeutic molecular targets need to be sought for the majority of advanced CRCs.
In our previous study, we found that ERKs are constitutively activated by downregulation of DUSP4 through genomic loss, and that this activation is important for cell growth and invasiveness in pancreatic ductal adenocarcinoma.8 Based on these findings, we hypothesized that a similar mechanism might also play an important role in the growth and invasion of other cancers, including CRC. In the present study, we analyzed CRCs and found evidence of DUSP4 downregulation and hyperphosphorylation of ERKs in the deep region of CRC tissues. In contrast, DUSP4 was strongly expressed and ERK phosphorylation was reduced in the superficial region. Furthermore, in vitro studies suggested that constitutive phosphorylation of ERKs caused by DUSP4 downregulation is important for the growth and invasiveness of CRC and might be a promising therapeutic target.
2. MATERIALS AND METHODS
2.1. Cell lines
The CRC cell lines used in this study were selected on the basis of DUSP4 expression data from the Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle). Five CRC cell lines, COLO‐320, SNU‐283, CW‐2, SNU‐503, and SNU‐1033, express low levels of DUSP4, and three cell lines, SNU‐C2A, SNU‐C5, and HCT 116, express high levels of DUSP4. The CRC cell lines COLO‐320, CW‐2, and HCT 116 were obtained from RIKEN BioResource Center (Ibaraki, Japan), and SNU‐283, SNU‐503, SNU‐1033, SNU‐C2A, and SNU‐C5 were obtained from the Korean Cell Line Bank (Seoul, Korea). The human colonic epithelial cells (HCoEpiC) were purchased from ScienCell (Carlsbad, CA, USA). The human glioblastoma cell line A‐172, in which the promoter region of DUSP4 is known to be hypermethylated,9 was obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Miyagi, Japan). All of the cell lines were cultured in accordance with the suppliers' instructions. The genotypes of EGFR, KRAS, HRAS, NRAS, BRAF, APC, CTNNB1, TP53 and CDKN2A genes in all eight CRC cell lines were obtained from the Cancer Cell Line Encyclopedia. All of them except for HRAS, NRAS, and CDKN2A are summarized in Table 1, because these three genes are not mutated in any of the cell lines.
Table 1.
Genotype data of colorectal cancer cell lines used in this study
| Cell line | EGFR | KRAS | BRAF | APC | CTNNB1 | TP53 |
|---|---|---|---|---|---|---|
| COLO‐320 | WT | WT | WT | p.S811a | WT | p.R248W |
| SNU‐283 | WT | WT | WT | p.N1815K | WT | WT |
| CW‐2 | p.E543fs, p.E928G | p.F566L, p.P140H | WT | p.A528V, p.G470R, p.K1462fs, p.R302a | p.R582Q | WT |
| SNU‐503 | WT | WT | WT | p.K1543a | WT | p.R273L |
| SNU‐1033 | WT | p.G12D | WT | p.K1030a | WT | p.P190L |
| SNU‐C5 | WT | WT | p.V600E | p.R1790fs | WT | p.R248W, p.V218L |
| HCT 116 | WT | p.G13D | WT | WT | p.S45del | WT |
| SNU‐C2A | p.R165Q | p.G12D | WT | p.P2048fs | WT | p.R273Y |
*Nonsense mutation.
fs, frame shift.
2.2. Patients and tissues
Tissue specimens were obtained from 59 Japanese patients who underwent surgical resection for primary CRC at Oita University Hospital (Yufu City, Japan) between 2007 and 2008. None of the patients had received preoperative treatments such as irradiation or chemotherapy. Written informed consent was obtained from all patients and the study was approved by the Oita University ethics committee (approval no. 540). The surgical specimens were fixed in 10% formalin and embedded in paraffin for histopathology and immunohistochemistry. Histopathologic diagnosis was carried out in accordance with the TNM Classification of Malignant Tumours 7th edition.10 Clinicopathological data of all patients are summarized in Table 2.
Table 2.
Characteristics of Japanese patients with primary colorectal cancer (CRC) from clinical and histological analyses (n = 59)
| Factor | Category | Case (n = 59) |
|---|---|---|
| Age, y, mean ± SD | 70 ± 13 | |
| Gender | Female/male | 27/32 |
| Tumor size, mm, mean ± SD | 44 ± 19 | |
| Location | Colon/rectum | 48/11 |
| Histopathological type | Tub1/Tub2/Pap/Por1/Muc | 11/39/3/1/5 |
| pT | T1/T2/T3/T4 | 5/10/28/16 |
| pN | Presence/absence | 34/25 |
| Lymphatic invasion | Presence/absence | 39/20 |
| Venous invasion | Presence/absence | 19/40 |
| pStage | I/II/III | 14/19/26 |
Pathological classification of invasion (pT), lymph node metastasis (pN), and pStage of individual CRC cases according to the UICC TNM Classification of Malignant Tumours 7th Edition. Muc, Mucinous carcinoma; Pap, papillary carcinoma; Por, poorly differentiated carcinoma; Tub, tubular carcinoma.
2.3. Treatment of cells with 5‐aza‐dC and TSA
SNU‐503 and SNU‐1033 cells were seeded into 12‐well plates on day 0 and exposed to 10 μmol L−1 5‐aza‐dC (Sigma‐Aldrich, St. Louis, MO, USA) from day 1 to day 4. Trichostatin A (Sigma‐Aldrich) was added to cells on day 3 at a concentration of 500 nmol L−1. On day 4, total RNA was recovered from the cells. The efficacy of these epigenetic modifiers in the CRC cells was confirmed by demonstrating the robust increase of UNC5C expression (Figure S1), which has been reported to be downregulated epigenetically in CRCs.11, 12
2.4. Combined bisulfite restriction analysis and bisulfite sequencing
Genomic DNAs of HCoEpiC, SNU‐503, SNU‐1033, and A‐172 were extracted using NucleoSpin Tissue (MACHEREY‐NAGEL, Düren, Germany). Bisulfite treatment of genomic DNA was carried out using the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). Methylation‐specific PCR for DUSP4 promoter, combined bisulfite restriction analysis, and bisulfite sequencing were carried out as described previously.9
2.5. Total RNA extraction and first‐strand cDNA synthesis
Total RNA was extracted from HCoEpiC and CRC cell lines with an RNeasy mini kit (Qiagen). Five hundred nanograms of total RNA was reverse‐transcribed to cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany).
2.6. Quantitative real‐time PCR
Quantitative real‐time PCR analysis was carried out using a LightCycler 480 real‐time PCR System with a universal probe library and a LightCycler 480 probe master (Roche Diagnostics, Indianapolis, IN, USA), in accordance with the manufacturer's instructions. The primer sets for DUSP4, UNC5C, and GAPDH were as follows: DUSP4‐F, 5′‐TGCATCCCAGTGGAAGATAA‐3′ and DUSP4‐R, 5′‐GCAGTCCTTCACGGCATC‐3′; UNC5C‐F, 5′‐CCAGACGAGAGGCCATGA‐3′ and UNC5C‐R, 5′‐TGGATTTGGTGGCAAAGTAAT‐3′; and GAPDH‐F, 5′‐AGCCACATCGCTCAGACAC‐3′ and GAPDH‐R, 5′‐GCCCAATACGACCAAATCC‐3′. All assays were normalized against GAPDH as an internal control.
2.7. Western blot analysis
Western blot analysis was carried out as described previously,8 with some modifications. Briefly, cultured cells were lysed in SDS‐modified RIPA buffer (0.1% w/v SDS, 40 mmol L−1 HEPES‐NaOH [pH 7.4], 1% v/v Nonidet P‐40, 0.5% w/v sodium deoxycholate, 150 mmol L−1 NaCl, and 4 mmol L−1 EDTA) with Complete Mini and PhosSTOP (both Roche Diagnostics). For preparation of tissue lysates, cryosections of frozen tissues were transferred to a microtube and lysed in lysis buffer as described above. Protein concentrations of the lysates were measured using the DC Protein Assay (Bio‐Rad, Hercules, CA, USA). The lysates containing 10 μg protein were suspended in Laemmli sample buffer and then subjected to SDS (10% w/v)‐PAGE. The samples were transferred to NitroBind nitrocellulose membranes (0.45 μm; Osmonic, Gloucester, MA, USA), which were blocked for 1 hour in BlockAce (DS Pharma Biomedical, Osaka, Japan) at RT, then incubated for 16 hours at 4°C with the primary antibody. The primary antibodies used for Western blot analysis were anti‐DUSP4 antibody (1:1000; #5149, Cell Signaling Technology, Danvers, MA, USA), anti‐phosphorylated ERK antibody (Thr202/Tyr204; 1:3000; #4370, Cell Signaling Technology), and anti‐ERK antibody (1:3000; #9102, Cell Signaling Technology). The filters were washed thoroughly with 1× PBS containing 0.1% v/v Tween‐20, then incubated for 1 hour at RT with a goat anti‐rabbit IgG HRP‐linked whole antibody (BioSource, Camarillo, CA, USA) diluted 1:1000 in 1× PBS containing 10% v/v BlockAce, and rewashed with 1× PBS containing 0.1% v/v Tween‐20. Finally, the signals were visualized on Hyperfilm (Amersham Biosciences, Little Chalfont, UK) using an ECL Western blotting detection kit (Amersham Biosciences, Piscataway, NJ, USA) in accordance with the manufacturer's instructions. The filters were re‐incubated with anti‐GAPDH (Ambion, Austin, TX, USA) diluted 1:10 000 in 1× PBS containing 10% v/v BlockAce as an internal loading control, followed by the procedure described above, except for use of a rabbit anti‐mouse IgG HRP‐linked whole antibody (Cappel, Aurora, OH, USA) as the detection antibody.
2.8. Lentivirus
Lentivirus encoding DUSP4 cDNA or encoding no cDNAs for transient transduction were generated as described previously.8 Lentivirus vector expressing constitutively active rat ERK2 mutant was prepared by subcloning of Myc‐tagged rat ERK2 (L73P, S151D)13 cDNA fragment into pLenti7.3/V5‐DEST (Thermo Fisher Scientific, Carlsbad, CA, USA). Transduction of CRC cell lines was carried out at an optimized MOI of 5 with Polybrene (Sigma‐Aldrich) at a final concentration of 6.0 μg/mL. Forty‐eight hours after transduction, the cells were used for the following experiments.
2.9. Cell proliferation assay
Two thousand cells transduced with the lentiviruses or treated with 10 μmol L−1 PD0325901 (LC Laboratories, Woburn, MA, USA) were cultured in 100 μL medium in a 96‐well tissue culture plate (Corning, Corning, NY, USA) at 37°C in 5% CO2 for the indicated duration. Proliferation was determined using the CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega, Madison, WI, USA) in accordance with the manufacturer's protocols.
2.10. Invasion assay
Invasiveness of CRC cell line SNU‐1033 was determined by the assay using 24‐well Transwell culture chambers (Corning, Tewksbury, MA, USA) as described previously,8 with some modifications. Briefly, the lower and upper surfaces of filters with an 8.0 μm pore size were coated with 1 μg fibronectin (Roche Diagnostics) and 5 μg Matrigel (BD Biosciences, San Jose, CA, USA), respectively. A total of 1 × 105 cells in 100 μL culture medium were added to the upper compartment of the chamber, and 600 μL culture medium was added to the lower chamber. To exclude any influence of cell proliferation on invasiveness, 50 ng/mL proliferation inhibitor mitomycin C (Nacalai Tesque, Kyoto, Japan) was applied to the culture medium in both upper and lower chambers. After incubation for 24 hours at 37°C in 5% CO2, the filter was fixed with 30% methanol and stained with 0.5% crystal violet in 20% ethanol. The non‐invading cells on the upper surface of the filter were then removed using a cotton swab. Subsequently, the filter was excised from the chamber using a scalpel and the number of invaded cells was counted using a microscope in three predetermined fields at a magnification of ×200.
2.11. Immunohistochemistry
For immunohistochemistry, rabbit polyclonal antisera was raised against synthetic peptides corresponding to the amino terminal sequences of human DUSP4 (NH2‐VHSAPSSLPYLHSP‐COOH), and purified by affinity chromatography (Unitech, Chiba, Japan). The specificity of antisera against DUSP4 was confirmed by both immunocytochemistry and Western blot analysis using pancreatic cancer cell lines, PANC‐1 stably re‐expressing DUSP4 (PANC‐1/DUSP4‐1) and PANC‐1 expressing only the blasticidin resistance gene (PANC‐1/cont).8
Immunohistochemistry was performed as described previously8 with some modifications. Briefly, 3‐μm‐thick sections were deparaffinized, rehydrated, and autoclaved in 10 mmol L−1 citrate buffer (pH 6.0) for 10 minutes at 120°C for antigen retrieval. After blocking with 3% hydrogen peroxide for 15 minutes at RT, the sections were incubated in 10% normal goat serum for 30 minutes at RT to block any non‐specific binding of the secondary goat anti‐rabbit antibody. The sections were then incubated with antisera against DUSP4 (1:1000) in diluting solution (Dako North America, Carpinteria, CA, USA) overnight at 4°C. Next, the sections were washed with 1× PBS and incubated with biotinylated goat anti‐rabbit IgG (Nichirei, Tokyo, Japan) for 30 minutes at RT. After a further wash with 1× PBS, the sections were incubated with a solution of avidin‐conjugated HRP using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) for 20 minutes at RT in accordance with the manufacturer's recommendations. Finally, after washing with 1× PBS, the signals were visualized by incubation with H2O2/diaminobenzidine substrate solution. The sections were counterstained with hematoxylin before dehydration and mounting.
Evaluation of the immunohistochemical data was undertaken by two independent pathologists (N.H. and T.U.) based on the following criteria. The staining intensity was scored as: 0, no staining; 1, weak (less than that in fibroblasts in the positive internal control); 2, moderate (as intense as in positive internal control cells); or 3, strong (more than that in positive internal control). The staining population was scored according to the ratio of positive cells as follows: 0, 0% of cells positive; 1, 1%‐9% of cells positive; 2, 10%‐50% of cells positive; and 3, >50% of cells positive. Scoring was carried out in three distinct fields per case, and the three scores were averaged and rounded off to the nearest whole number. The sum of the scores for intensity and population was defined as the DUSP4 score in this study. Because the DUSP4 score in normal epithelium adjacent to cancer tissues was 2.58 ± 1.45, DUSP4 scores ≥4 and ≤3 were considered to indicate upregulation and downregulation of DUSP4, respectively.
2.12. Statistical analysis
Statistical analyses in this study were carried out with the JMP statistical software package (SAS Institute, Cary, NC, USA). The effects of re‐expression of DUSP4 on cell proliferation and invasiveness was examined using anova and Student's t‐test. Comparisons of DUSP4 scores in normal epithelium, superficial region, and deep region were evaluated using Fisher's exact probability test. Differences at P ≤ .05 were considered statistically significant.
3. RESULTS
3.1. Downregulation of DUSP4 in SNU‐503 and SNU‐1003 cells with constitutive activation of ERKs
In our previous study, we found that, under physiologic conditions, DUSP4 is induced by phosphorylated ERKs. It then dephosphorylates ERKs, and is finally downregulated after the ERKs have been inactivated.8 Therefore, regulation of DUSP4 expression is controlled by a negative feedback loop mechanism mediated by the phosphorylation status of ERKs. Based on these previous data, we first examined the relationship between the expression level of DUSP4 and the phosphorylation level of ERKs in eight CRC cell lines and a normal colonic epithelial cell line, HCoEpiC. As shown in Figure 1A,B, two of these cell lines, SNU‐503 and SNU‐1033, showed constitutive activation of ERKs and downregulation of DUSP4. In addition, DUSP4 expression in these two cell lines was increased 2‐3.5‐fold by treatment with the epigenetic modifiers 5‐aza‐dC and TSA (Figure 1C). However, the promoter region of DUSP4 was not hypermethylated in either cell line (Figure 1D,E), suggesting that the hypermethylation of the promoter region may not be responsible for this small recovery of epigenetic modifiers. These results suggest that, in CRC cells, DUSP4 downregulation may contribute to ERK activation, similar to the process occurring in pancreatic cancer cells.
Figure 1.
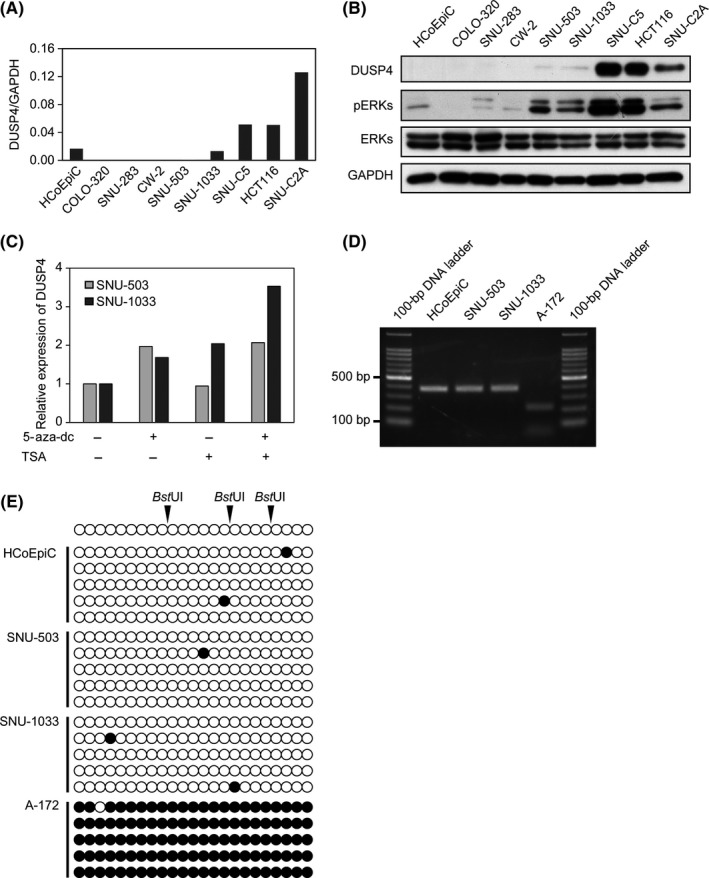
Downregulation of dual‐specificity phosphatase 4 (DUSP4) in colorectal cancer (CRC) cells is related to activation of ERKs. A, Expression of DUSP4 mRNA in HCoEpiC and CRC cell lines was detected by quantitative RT‐PCR. B, Expression of DUSP4 protein and phosphorylation of ERKs (pERKs) in HCoEpiC and CRC cell lines were analyzed by Western blotting. C, DUSP4 expression in SNU‐503 and SNU‐1033 was increased by treatment with 5‐aza‐2′‐deoxycytidine (5‐aza‐dc) and/or trichostatin A (TSA). D, The 363‐bp products of methylation‐specific PCR for the DUSP4 promoter were digested with BstUI for combined bisulfite restriction analysis. E, PCR products containing 23 putative CpG methylation sites were subcloned, and five single clones of each cell were sequenced. Black and white circles indicate the methylated and unmethylated CpG site, respectively
3.2. Downregulation of DUSP4 may enhance both proliferation and invasiveness of CRC cells
Next, to confirm whether downregulated DUSP4 in the SNU‐503 and SNU‐1033 cell lines may cause hyperactivation of ERKs, resulting in a growth advantage and invasiveness, they were transduced with lentivirus encoding DUSP4 and then analyzed for ERK phosphorylation status, cell proliferation, and invasiveness. As shown in Figure 2A, the phosphorylation level of ERKs was clearly reduced in both cell lines following re‐expression of DUSP4. Furthermore, the invasiveness of SNU‐1033 cells and the proliferation of SNU‐503 and SNU‐1033 were evidently suppressed by re‐expression of DUSP4 (Figure 2B‐D). However, these suppressive effects of DUSP4 on ERK phosphorylation, invasiveness, and proliferation were significantly overridden by introduction of a constitutively active ERK2 mutant into CRC cells (Figure 2A‐D).
Figure 2.
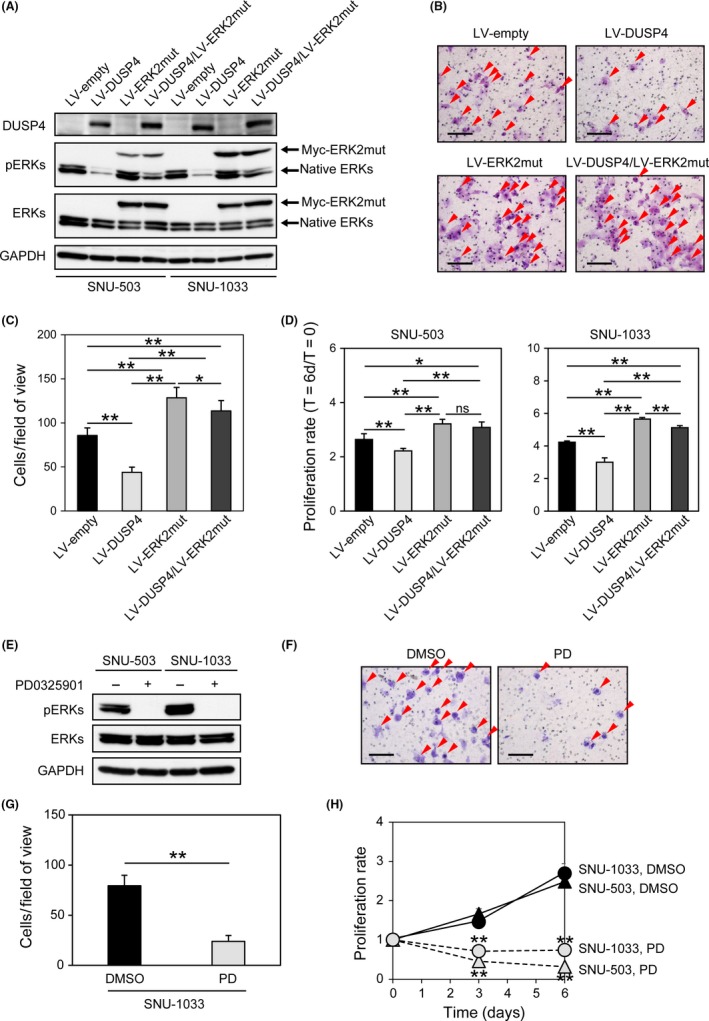
Activation of ERKs caused by downregulation of dual‐specificity phosphatase 4 (DUSP4) enhances the proliferation and invasiveness of colorectal cancer cells. A, Re‐expression of DUSP4 using lentiviruses (LV) suppressed the phosphorylation of ERKs (pERKs), and additional introduction of a constitutively active ERK2 mutant (ERK2mut) abolished the suppressive effect of DUSP4 on ERK phosphorylation in SNU‐503 and SNU‐1033 cell lines. B, Representative images of invasive SNU‐1033 cells with and without re‐expression of DUSP4 and introduction of constitutively active ERK2 mutant. Red arrowheads indicate invaded cells. Scale bar = 100 μm. C, Effects of DUSP4 re‐expression and constitutively active ERK2 introduction on SNU‐1033 invasiveness. The number of invaded cells on the filters was counted using microscopy (n = 9). D, Effects of DUSP4 re‐expression and constitutively active ERK2 introduction on proliferation of the SNU‐503 and SNU‐1033 cell lines. The proliferation was measured at T = 0 and T = 6 days (D). The proliferative activities are presented as fold‐change values, T = 6d/T = 0. E, MEK inhibition using PD0325901 suppressed the phosphorylation of ERKs in SNU‐503 and SNU‐1033 cells. F, Representative images of invasive SNU‐1033 cells with or without PD0325901 (PD) treatment. Red arrowheads indicate invaded cells. Scale bar = 100 μm. G, Inhibitory effect of PD0325901 on SNU‐1033 invasiveness. The number of invaded cells on the filters was counted using microscopy (n = 9). H, Treatment with PD0325901 markedly reduced the proliferation of SNU‐503 and SNU‐1033 cells. Data in C, D, G, and H are shown as mean ± SD. Data in A‐D and E‐H are representative of two and three independent experiments, respectively. *P ≤ .05, **P ≤ .001. ns, not significant, P > .05 by anova and Student's t‐test
We then investigated whether ERK inhibition actually led to suppression of cell proliferation and invasiveness in SNU‐503 and SNU‐1033 cells. As shown in Figure 2E‐H, cell proliferation and invasiveness were both evidently suppressed by inhibition of ERKs using MEK inhibitor PD0325901. These findings suggested that constitutive activation of ERKs due to downregulation of DUSP4 may lead to enhanced proliferation and invasiveness in CRC.
3.3. Downregulation of DUSP4 may lead to activation of ERKs and invasiveness of CRC
To validate our findings from in vitro experiments, we analyzed the expression pattern of DUSP4 and the phosphorylation level of ERKs in CRC tissues. First, to detect DUSP4 expression immunohistochemically, we raised a novel rabbit anti‐DUSP4 polyclonal antibody and then verified its specificity by immunocytochemistry and Western blot analysis (Figure 3A,B). Immunohistochemistry using this antibody (Figure 3C) revealed that DUSP4 was weakly and focally expressed in the nuclei of normal epithelium adjacent to cancer tissues (Figure 3D), but was strongly and diffusely detected in nuclei of CRC cells in the superficial region in 54 of 59 cases (91.5%) (Figure 3D‐F). In contrast, nuclear expression of DUSP4 was significantly reduced in cells located in the deep region in 46 of 59 cases (78.0%) (Figure 3D‐F).
Figure 3.
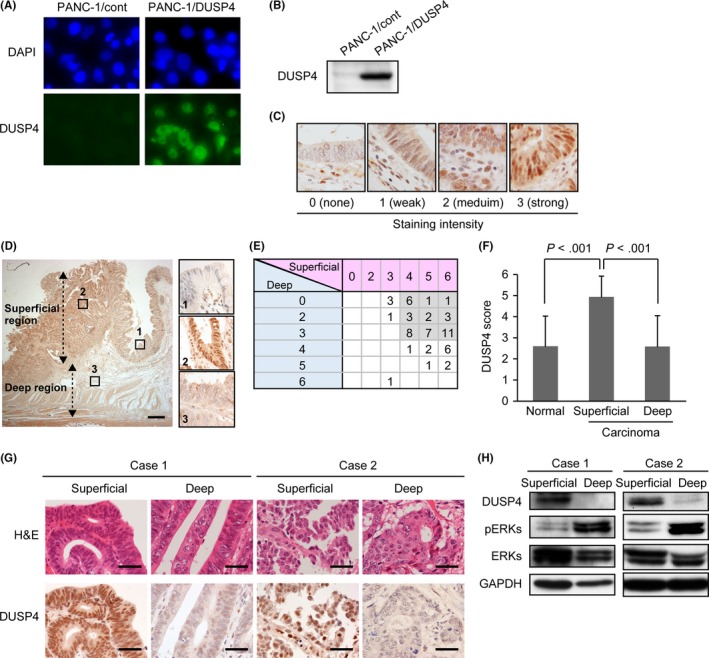
Expression of dual‐specificity phosphatase 4 (DUSP4) is upregulated in the superficial region of colorectal cancer (CRC) tissues, but downregulated in the deep region. A, Validation of a novel rabbit anti‐DUSP4 polyclonal antibody using PANC‐1 cells stably re‐expressing DUSP4 (PANC‐1/DUSP4‐1) and its control clones (PANC‐1/cont), which express only the blasticidin resistance gene. Representative images of immunocytochemistry using DAPI and the anti‐DUSP4 antibody. The antibody specifically recognized the nucleus of PANC‐1/DUSP4‐1 but not that of PANC‐1/cont. B, Specificity of the antibody was further determined by Western blot analysis using lysates of PANC‐1/cont and PANC‐1/DUSP4‐1. The antibody specifically detected DUSP4 protein in PANC‐1/DUSP4‐1. C, Representative images of immunohistochemistry for DUSP4 in CRC tissues. The staining intensity in the nucleus of cancer cells was scored as: 0, none; 1, weak; 2, medium; and 3, strong. D, Representative images of immunohistochemistry for DUSP4 in CRC tissues. The insets indicate normal epithelium (1), the superficial region of CRC (2), and the deep region of CRC (3). Scale bar = 1 mm. E, Immunohistochemical DUSP4 scores for 59 CRC cases. In 42 cases (71.2%, gray boxes), DUSP4 was upregulated and downregulated in the superficial region and deep region, respectively. F, DUSP4 score was significantly higher in the superficial region than in normal epithelium, and was significantly lower in the deep region than in the superficial region. Data shown as mean ± SD. G, H & E staining and immunohistochemistry for DUSP4 in frozen tissues from superficial and deep regions in representative CRC cases. In all sections, the proportion of tumor cells included exceeded 70% of the total. Scale bar = 40 μm. H, Expression levels of DUSP4 protein and phosphorylation levels of ERKs (pERKs) in CRC tissues analyzed by Western blotting. DUSP4 expression was downregulated in the deep region relative to the superficial region, whereas ERKs were hyperactivated in the deep region relative to the superficial region
Then, we further attempted to determine the phosphorylation levels of ERKs in CRC tissues using immunohistochemistry. However, commercially available antibodies failed to detect phosphorylated ERKs on formalin‐fixed, paraffin‐embedded sections, as had been reported previously.14 Therefore, we attempted to compare the phosphorylation status of ERK between the superficial and deep regions of fresh CRC tissues by Western blot analysis. To achieve this, we first excised fresh cancer tissues from the superficial region and deep region of surgically resected CRC tissues, and confirmed microscopically that a sufficient population of cancer cells for Western blot analysis was present in each tissue specimen (Figure 3G). Furthermore, using these fresh tissues, we confirmed immunohistochemically that DUSP4 was downregulated in the deep region relative to the superficial region (Figure 3G). As shown in Figure 3H, ERKs were relatively hyperphosphorylated in the deep region, whereas DUSP4 expression was obviously downregulated in the deep region. This contrasting pattern of DUSP4 expression and ERK phosphorylation is consistent with our proposal that activation of ERKs in the deep region of CRC may be caused by downregulation of DUSP4.
4. DISCUSSION
Somatic mutation of KRAS, NRAS, or BRAF, which is involved in the MAPK signaling pathway, has frequently been found in CRCs.15, 16 In addition to these mutations, EGFR overexpression detected in a smaller proportion of CRCs is also involved in MAPK signaling,17 suggesting its importance in the development of CRCs. However, it is still unclear how this pathway is constitutively activated in CRCs. In this study, we revealed that DUSP4 expression was apparently downregulated in the deep region of CRC tissues compared with the superficial region, and that ERK phosphorylation was conversely increased in the deep region relative to the superficial region. Furthermore, in CRC cell lines in vitro, we found that re‐expression of DUSP4 or administration of a MEK inhibitor reduced the degree of cell proliferation and invasiveness. These findings suggest that downregulation of DUSP4 in CRC might promote cell proliferation and invasiveness through activation of ERK.
The molecular mechanisms of DUSP4 downregulation in the deep region of CRC are still unclear. We have recently reported that DUSP4 is downregulated in pancreatic cancer due to genomic loss of 8p, on which the DUSP4 gene is located.8 In pancreatic cancer, DUSP4 was upregulated in carcinoma in situ without 8p loss through the positive feedback loop of ERK phosphorylation status, but downregulated due to 8p loss in the invasive region of pancreatic cancer. It has already been reported that 8p loss was detectable in 29% of CRCs.18 Therefore, it appears that 8p loss in a proportion of CRCs could contribute to downregulation of DUSP4. Another possible mechanism of DUSP4 downregulation could be epigenetic modification. It has been reported that hypermethylation in the promotor region of DUSP4 contributes to its reduced expression in glioblastoma,9 hepatocellular carcinoma,19 breast cancer,20 and lymphoma.21 In the present study, we were able to detect augmentation of DUSP4 expression in SNU‐503 and SNU‐1033 cells after treatment with the epigenetic modifiers 5‐aza‐dc and TSA, but the increase was limited to 2‐3.5‐fold relative to untreated cells. Furthermore, the promoter region of DUSP4 was not hypermethylated in either cell line. Thus, it appears possible that mechanisms other than epigenetic silencing might also be involved in the downregulation of DUSP4 in CRCs. Recently, Gaggianesi et al22 have reported that the pro‐inflammatory cytokine interleukin‐4, secreted by cells in the microenvironment, might play an important role in DUSP4 downregulation in breast cancer cells mediated by nuclear factor‐κB activation, leading to enhancement of MAPK signaling and promotion of cell proliferation and invasiveness. It is still unclear whether this attractive hypothesis proposed by Gaggianesi et al might hold true for CRCs. Further studies are required to confirm this possibility.
Monoclonal antibodies targeting EGFR are known to be effective for patients with advanced CRCs, but their indications are restricted to CRCs without mutation in both the RAS and RAF genes.6, 7, 23, 24 Therefore, a novel molecular targeting therapy is required to further improve the outcomes of patients with advanced CRCs, especially those harboring mutations in the KRAS or BRAF gene. Our findings suggest that abrogation of the MAPK signaling pathway using a MEK inhibitor may be therapeutically beneficial for patients with invasive CRC. However, a recent clinical trial of single MEK inhibitors failed to show any impact on the prognosis of patients with advanced CRC,25 largely because of compensatory upregulation of other signaling pathways, such as the PI3K pathway, through cross‐talk with MAPK.26, 27 Therefore, to enhance the therapeutic effects of MEK inhibitors in CRCs, identification of additional therapeutic targets responsible for resistance to MEK inhibitors would be required. Our present findings shed new light on how CRC cells acquire more malignant phenotypic characteristics, including augmented proliferation and invasiveness. Future studies to yield a more detailed understanding of DUSP4 downregulation in CRC cells would be useful for development of novel therapeutic strategies for patients with advanced CRC.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank Ms. Mami Kimoto (Department of Molecular Pathology, Faculty of Medicine, Oita University) and Ms. Mayumi Takeda (Department of Gastroenterological and Pediatric Surgery, Faculty of Medicine, Oita University) for technical assistance. This work was supported in part by the Japan Society for the Promotion of Science (KAKENHI Grant Nos. 16K08651 and 17K08696).
Ichimanda M, Hijiya N, Tsukamoto Y, et al. Downregulation of dual‐specificity phosphatase 4 enhances cell proliferation and invasiveness in colorectal carcinomas. Cancer Sci. 2018;109:250–258. https://doi.org/10.1111/cas.13444
Funding information
Japan Society for the Promotion of Science.
Ichimanda and Hijiya contributed equally to this work.
REFERENCES
- 1. Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665‐669. [DOI] [PubMed] [Google Scholar]
- 2. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159‐170. [DOI] [PubMed] [Google Scholar]
- 3. Morin PJ, Sparks AB, Korinek V, et al. Activation of beta‐catenin‐Tcf signaling in colon cancer by mutations in beta‐catenin or APC. Science. 1997;275:1787‐1790. [DOI] [PubMed] [Google Scholar]
- 4. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531‐7537. [DOI] [PubMed] [Google Scholar]
- 5. Di Fiore F, Sesboue R, Michel P, Sabourin JC, Frebourg T. Molecular determinants of anti‐EGFR sensitivity and resistance in metastatic colorectal cancer. Br J Cancer. 2010;103:1765‐1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Karapetis CS, Khambata‐Ford S, Jonker DJ, et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757‐1765. [DOI] [PubMed] [Google Scholar]
- 7. Amado RG, Wolf M, Peeters M, et al. Wild‐type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626‐1634. [DOI] [PubMed] [Google Scholar]
- 8. Hijiya N, Tsukamoto Y, Nakada C, et al. Genomic loss of DUSP4 contributes to the progression of intraepithelial neoplasm of pancreas to invasive carcinoma. Cancer Res. 2016;76:2612‐2625. [DOI] [PubMed] [Google Scholar]
- 9. Waha A, Felsberg J, Hartmann W, et al. Epigenetic downregulation of mitogen‐activated protein kinase phosphatase MKP‐2 relieves its growth suppressive activity in glioma cells. Cancer Res. 2010;70:1689‐1699. [DOI] [PubMed] [Google Scholar]
- 10. Sobin LH, Gospodarowicz MK, Wittekind C. International Union Against Cancer (UICC) TNM Classification of Malignant Tumors, 7th edn Oxford, UK: Wiley‐Blackwell; 2009. [Google Scholar]
- 11. Bernet A, Mazelin L, Coissieux MM, et al. Inactivation of the UNC5C Netrin‐1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology. 2007;133:1840‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shin SK, Nagasaka T, Jung BH, et al. Epigenetic and genetic alterations in Netrin‐1 receptors UNC5C and DCC in human colon cancer. Gastroenterology. 2007;133:1849‐1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emrick MA, Hoofnagle AN, Miller AS, Ten Eyck LF, Ahn NG. Constitutive activation of extracellular signal‐regulated kinase 2 by synergistic point mutations. J Biol Chem. 2001;276:46469‐46479. [DOI] [PubMed] [Google Scholar]
- 14. Holck S, Nielsen HJ, Pedersen N, Larsson LI. Phospho‐ERK1/2 levels in cancer cell nuclei predict responsiveness to radiochemotherapy of rectal adenocarcinoma. Oncotarget. 2015;6:34321‐34328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature. 2002;418:934. [DOI] [PubMed] [Google Scholar]
- 16. Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosom Cancer. 2011;50:307‐312. [DOI] [PubMed] [Google Scholar]
- 17. Ooi A, Takehana T, Li X, et al. Protein overexpression and gene amplification of HER‐2 and EGFR in colorectal cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol. 2004;17:895‐904. [DOI] [PubMed] [Google Scholar]
- 18. Douglas EJ, Fiegler H, Rowan A, et al. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817‐4825. [DOI] [PubMed] [Google Scholar]
- 19. Deng YB, Nagae G, Midorikawa Y, et al. Identification of genes preferentially methylated in hepatitis C virus‐related hepatocellular carcinoma. Cancer Sci. 2010;101:1501‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balko JM, Cook RS, Vaught DB, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. 2012;18:1052‐1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmid CA, Robinson MD, Scheifinger NA, et al. DUSP4 deficiency caused by promoter hypermethylation drives JNK signaling and tumor cell survival in diffuse large B cell lymphoma. J Exp Med. 2015;212:775‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaggianesi M, Turdo A, Chinnici A, et al. IL‐4 primes the dynamics of breast cancer progression via DUSP4 inhibition. Cancer Res. 2017;77:3268‐3279. [DOI] [PubMed] [Google Scholar]
- 23. Waring P, Tie J, Maru D, Karapetis CS. RAS mutations as predictive biomarkers in clinical management of metastatic colorectal cancer. Clin Colorectal Cancer. 2016;15:95‐103. [DOI] [PubMed] [Google Scholar]
- 24. Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386‐1422. [DOI] [PubMed] [Google Scholar]
- 25. Bennouna J, Lang I, Valladares‐Ayerbes M, et al. A Phase II, open‐label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY‐142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29:1021‐1028. [DOI] [PubMed] [Google Scholar]
- 26. Little AS, Balmanno K, Sale MJ, Smith PD, Cook SJ. Tumour cell responses to MEK1/2 inhibitors: acquired resistance and pathway remodelling. Biochem Soc Trans. 2012;40:73‐78. [DOI] [PubMed] [Google Scholar]
- 27. Migliardi G, Sassi F, Torti D, et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient‐derived xenografts of RAS‐mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515‐2525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials