

Abstract
Recent sequencing studies demonstrated the MYD88 L265P mutation in more than 70% of primary central nervous system lymphomas (PCNSL), and the clinical significance of this mutation has been proposed as diagnostic and prognostic markers in PCNSL. In contrast, mutational analyses using cell‐free DNAs have been reported in a variety of systemic lymphomas. To investigate how sensitively the MYD88 L265P mutation can be identified in cell‐free DNA from PCNSL patients, we carried out droplet digital PCR (ddPCR) and targeted deep sequencing (TDS) in 14 consecutive PCNSL patients from whom paired tumor‐derived DNA and cell‐free DNA was available at diagnosis. The MYD88 L265P mutation was found in tumor‐derived DNA from all 14 patients (14/14, 100%). In contrast, among 14 cell‐free DNAs evaluated by ddPCR (14/14) and TDS (13/14), the MYD88 L265P mutation was detected in eight out of 14 (ddPCR) and in 0 out of 13 (TDS) samples, implying dependence on the detection method. After chemotherapy, the MYD88 L265P mutation in cell‐free DNAs was traced in five patients; unexpectedly, the mutations disappeared after chemotherapy was given, and they remained undetectable in all patients. These observations suggest that ddPCR can sensitively detect the MYD88 L265P mutation in cell‐free DNA and could be used as non‐invasive diagnostics, but may not be applicable for monitoring minimal residual diseases in PCNSL.
Keywords: cell‐free DNA, droplet digital PCR, MYD88 L265P, non‐invasive diagnosis, primary central nervous system lymphoma
1. INTRODUCTION
Primary central nervous system lymphoma (PCNSL) is a rare subtype of diffuse large B‐cell lymphoma (DLBCL) that arises within the brain or eyes. PCNSL accounts for up to 2%‐3% of primary malignant brain tumors and less than 1% of non‐Hodgkin lymphomas (NHL) in adults.1 Almost all PCNSL patients undergo invasive surgical procedures for appropriate diagnosis. However, histological diagnosis of PNCSL is sometimes difficult because of deep brain structure involvement. Therefore, there is a need for a more reliable and minimally invasive biomarker detection method aiding the diagnosis of PCNSL.
Recently, circulating tumor DNAs, which are fragmented DNAs released through the apoptotic process of tumor cells, in plasma and serum have received attention as materials for non‐invasive diagnosis.2 Many genetic alterations identified in tumors have been used to detect circulating tumor DNAs in patients with both solid cancers and hematological malignancies.2, 3, 4
Whole exome and targeted sequencing studies have determined the genetic profiles of PCNSL. Notably, mutations in genes associated with the nuclear factor kappa B (NF‐κB) and B‐cell receptor signaling pathways are highly frequent in PCNSL.5, 6, 7, 8, 9, 10, 11, 12 In particular, the MYD88 L265P mutation was found in 38%‐85.4% of PCNSL patients but never in those with non‐hematological brain tumors, suggesting that this mutation is useful for differential diagnosis of PCSNL among central nervous system (CNS) tumors.5, 6, 7, 8, 9, 10, 11, 12
Herein, we examined how sensitively the MYD88 L265P mutation is detected in cell‐free DNAs in PCNSL patients at diagnosis and during the disease course.
2. MATERIALS AND METHODS
2.1. Patient characteristics
From October 2014 to June 2017, a total of 30 consecutive patients diagnosed with PCNSL were identified in the clinical records at the University of Tsukuba Hospital. Sixteen patients were excluded from this study because CNS tumor or serum samples before treatment were not available. The remaining 14 patients were analyzed in a retrospective way (Table S1).
This retrospective study was approved by the institutional review board of University of Tsukuba Hospital. Samples were provided in accordance with the “Guidelines for clinical measurement and diagnosis technology improvement project”. The guidelines are promoted by Tsukuba Medical Laboratory of Education and Research Center and University of Tsukuba Hospital, Japan.
2.2. Serum and plasma collection
Serum samples were collected before first‐line therapy in all 14 patients. A plasma sample before treatment was available in one patient only (TP103). Serial serum samples were preserved during the clinical courses from five patients (TP87, TP89, TP90, TP92, and TP99). Cell‐free DNAs were extracted from 700 to 3000 μL serum or plasma using the QIAmp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). Genomic DNAs were also extracted from 14 formalin‐fixed, paraffin‐embedded (FFPE) CNS tumor samples using the FFPE tissue Kit (Qiagen). Final DNAs were dissolved in 30‐50 μL AVE buffer (Qiagen, Hilden, Germany) and were stored at −20°C until use.
2.3. ddPCR assay
Droplet digital polymerase chain reaction (ddPCR) reagents and Primer/probe mix for MYD88 L265P were purchased from Bio‐Rad (Hercules, CA, USA). The 20 μL PCR mix, composed of 10 μL of 2× ddPCR Supermix for Probes (No dUTP) (Bio‐Rad), 1 μL ddPCR Mutation Assay (Bio‐Rad) that used an amplicon of 65 nt, 5 μL ultra pure distilled water and 4 μL cell‐free DNA was loaded into sample wells of an 8‐channel disposable droplet generator cartridge (Bio‐Rad). Additional 70 μL droplet generation oil (Bio‐Rad) was loaded into the oil well for each channel. After droplet generation, droplets were transferred carefully into a 96‐well PCR plate. The plate was heat‐sealed with the PX1 PCR Plate Sealer (Bio‐Rad) and proceeded to thermal cycling. Amplification was carried out on the 20 μL reaction mixture on the QX200 Droplet digital PCR system (Bio‐Rad).
After PCR, the 96‐well PCR plate was subjected to the QX‐200 droplet reader (Bio‐Rad). ddPCR data were analyzed by QuantaSoft analysis software (Bio‐Rad) that accompanied the droplet reader. In our analyses, MYD88 L265P mutation‐specific signals were generated in the FAM channel, whereas MYD88 wild‐type signals were in the HEX channel.
2.4. Targeted deep sequencing
Targeted deep sequencing (TDS) was carried out on Ion Torrent PGM platform (Thermo Fisher Scientific, Waltham, MA, USA) for MYD88 L265P mutation as previously described with minor modifications.13 The Ion Plus Fragment Library Kit (Thermo Fisher Scientific) was used to prepare the libraries according to the recommended protocol for libraries of short amplicons.
Primers were designed to amplify the genomic regions encoding codon 265 in MYD88 (Table S2). PCR was carried out in a final volume of 10 μL with tumor‐derived DNAs and cell‐free DNAs, 5 nmol/L primers, and KOD Neo reagents (Toyobo, Osaka, Japan). The PCR protocol was as follows: 10 minutes at 95°C, followed by 39 cycles of 15 seconds at 95°C and 60 seconds at 60°C. Then, PCR amplicons were ligated to barcode adapters and P1 adapters, and amplified to generate libraries. Quantification of the amplified libraries was done by quantitative PCR with the Ion Library Quantification kit according to the manufacturer's instructions (Thermo Fisher Scientific). The libraries were then subjected to deep sequencing on Ion Torrent PGM with the Ion 318 chip according to the standard protocol for 300 base pair single‐end reads (Thermo Fisher Scientific). The data were analyzed by Variant caller 5.2 (Thermo Fisher Scientific). The sequencing variations were adopted as mutations when frequencies were higher than 2% for tumor‐derived DNA and 0.2% for cell‐free DNA.
2.5. Statistical analysis
Statistical analysis was conducted using the EZR software.14 We used two‐sided tests in all calculations. P‐value ≤ .05 was considered statistically significant.
3. RESULTS
3.1. Validation of the ddPCR assay and TDS for detecting the MYD88 L265P mutation
The signal emitted by the MYD88 L265P mutation was clearly distinguished from that by wild‐type DNA in a 2D fluorescent amplitude plot (Figure S1). Lower limits of detection (LOD) and quantification (LOQ) in ddPCR were determined using serial dilutions of DNA with the MYD88 L265P mutation. We chose a frozen tissue‐derived DNA in which AmpliSeq measured 24.2% variant allele frequency (VAF) at the depth of 1920. This sample was serially diluted at 1/4 each down to (1/4)7, by mixing with the copy number‐adjusted DNA without MYD88 L265P mutation. All dilutions were then applied to ddPCR. Mutation ratio was calculated for each sample by QuantaSoft and plotted (Figure 1).
Figure 1.
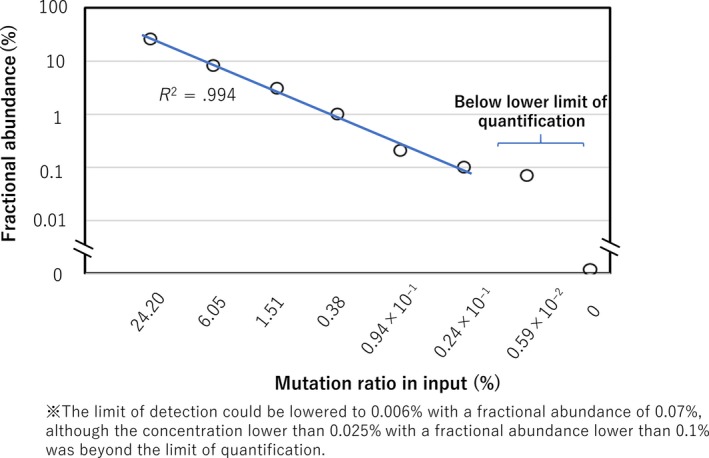
Sensitivity of our droplet digital PCR (ddPCR) assay. DNA with MYD88 L265P mutation was mixed with DNA without the mutation at the concentrations indicated on the horizontal axis. All diluted samples were individually subjected to ddPCR. Fractional abundance (mutation ratio calculated by QuantaSoft) for each sample is plotted
In the 1/4 (corresponding to 6.05% by calculation), (1/4)2 (1.5125%), (1/4)3 (0.378125%), (1/4)4 (0.09453125%), (1/4)5 (0.0236328125%), dilutions, fractional abundance (referring to the proportion of mutated DNA by QuantaSoft) measured by ddPCR was 8.3%, 3.1%, 1.0%, 0.2% and 0.1%. After being converted to log‐log scale, data showed precision and linearity down to a dilution of 0.024%; this material corresponded to a fractional abundance of 0.1% (coefficient of determination [R 2] was .994). The positive control diluted to (1/4)6 (0.005908203125%) was judged as positive by ddPCR, whereas the fractional abundance was 0.07%, implying that the quantification was inaccurate (Figure 1). All dilutions less than (1/4)6 showed 0%.
Based on this result, the LOQ of ddPCR was defined as 0.024% with a fractional abundance of 0.1%. The LOD could be lowered to 0.0059% with a fractional abundance of 0.07%.
The same dilution series was also applied to amplicon‐based sequencing at the approximate depth of 20 000 reads and the mutation ratio was plotted (Figure 2). In the 1/4 (corresponding to 6.05% by calculation), (1/4)2 (1.5125%), and (1/4)3 (0.378125%) dilutions, VAF measured by amplicon‐based sequencing was 7.2%, 2.7%, and 0%. All dilutions less than (1/4)3 showed 0%. Based on this, the cut‐off value was determined at 1.51%. After being converted to log‐log scale, data showed precision and linearity down to a dilution of 1.51% (R 2 was 1; Figure 2).
Figure 2.
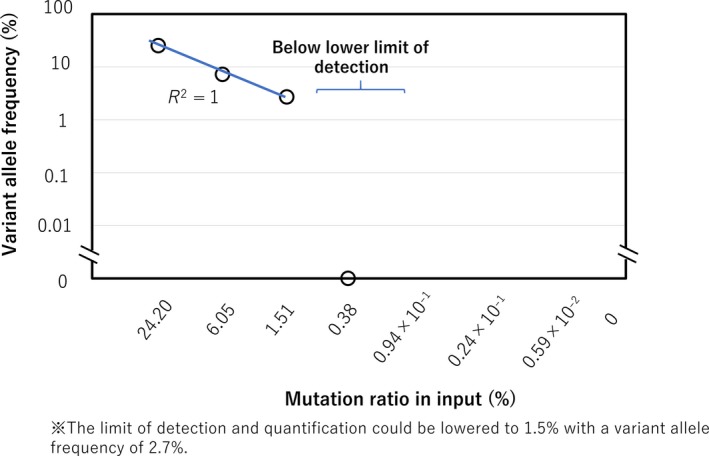
Sensitivity of our targeted deep sequencing (TDS) assay. DNA with MYD88 L265P mutation was mixed with DNA without the mutation at the concentrations indicated on the horizontal axis. All diluted samples were individually subjected to TDS assay. Variant allele frequency for each sample is plotted
Therefore, LOQ and LOD of TDS were defined as 1.51% with a VAF of 2.7%.
3.2. MYD88 L265P mutation in tumor‐derived DNA
We examined the MYD88 L265P mutation in tumor‐derived DNA prepared from 14 PCNSL patients by ddPCR and TDS (Table 1). In this cohort, MYD88 L265P mutation was found in all patients (14/14, 100%; range of VAF, 10.5%‐87.0%) and mean depths for the mutation in tumor‐derived DNAs were 16868x to 19993x (Table 1).
Table 1.
Targeted sequencing of MYD88 L265P for 14 PCNSL samples
| Mutation ratio of MYD88 L265P (%) | ||||||
|---|---|---|---|---|---|---|
| Tumors | Cell‐free DNAs in serum | |||||
| NGS | ddPCR | NGS | ddPCR | |||
| VAF (%) | Reads showing the mutation/total reads | FA (%) | VAF | Reads showing the mutation/total reads | FA (%) | |
| TP73 | 43.2 | 8584/19870 | ND | 0 | 0/19996 | 0 |
| TP87 | 65.5 | 13070/19954 | ND | 0 | 0/19930 | 0.40 |
| TP89 | 35.1 | 6979/19882 | ND | 0 | 0/19981 | 0.19 |
| TP90 | 40.9 | 8137/19895 | ND | 0 | 0/19980 | 0.09 |
| TP92 | 53.3 | 10534/19763 | ND | 0 | 0/19985 | 0.10 |
| TP94 | 61.3 | 12233/19956 | ND | 0 | 0/19948 | 0.38 |
| TP95 | 36.4 | 7230/19862 | ND | 0 | 0/19963 | 0.14 |
| TP96 | 56.1 | 11122/19825 | ND | 0 | 0/19972 | 0.47 |
| TP98 | 88.7 | 17736/19993 | 87 | 0 | 0/19987 | 0 |
| TP99 | 56.6 | 11299/19974 | 55.9 | 0 | 0/19994 | 0.69 |
| TP100 | 75.2 | 15025/19980 | 79 | ND | ND | 0 |
| TP101 | 9.2 | 1844/19988 | 10.5 | 0 | 0/12560 | 0 |
| TP102 | 43 | 7260/16868 | 38.3 | 0 | 0/19991 | 0 |
| TP103 | 19.7 | 3928/19973 | 17.1 | 0 | 0/19989 | 0 |
ddPCR, droplet digital PCR; FA, fractional abundance; ND, not done; NGS, next‐generation sequencing; PCNSL, primary central nervous system lymphoma; VAF, variant allele frequency.
3.3. Detection of the MYD88 L265P mutation in cell‐free DNAs at diagnosis
Amounts of cell‐free DNA, prepared from 1 mL serum or plasma that was collected before treatment, ranged from 107 ng to 2410 ng (Figure 3).
Figure 3.
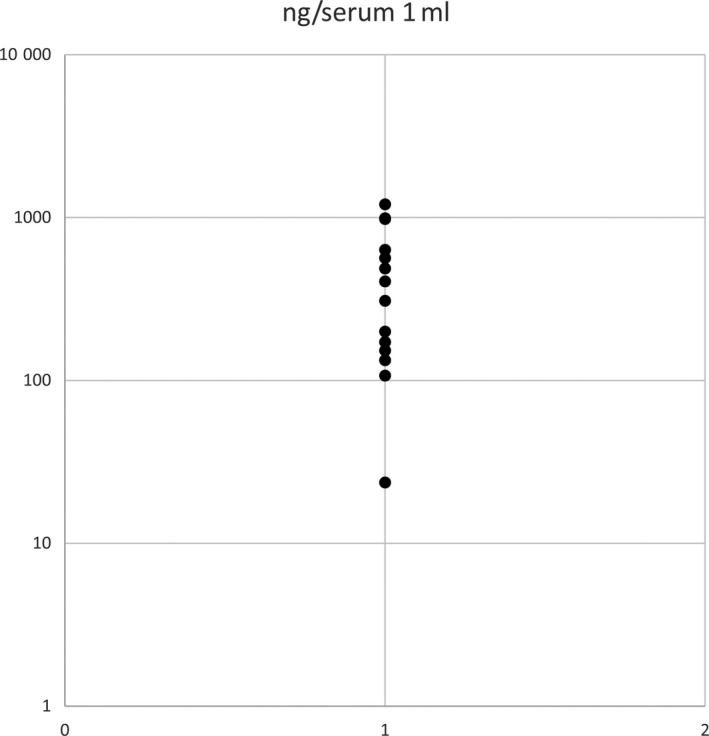
Concentrations of cell‐free DNAs of primary central nervous system lymphoma (PCNSL). Distribution of the cell‐free DNA concentrations in serum in patients with PCNSL is shown by plots. The y‐axis has a log scale
The MYD88 L265P mutation was detected by ddPCR in eight out of 14 serum‐derived cell‐free DNAs at diagnosis (Figure 1; Figure S1). Among six negative subjects, we prepared plasma‐derived cell‐free DNA from only 1; the mutation was also undetectable in this preparation (TP103, Table S3). VAF of the MYD88 L265P mutation in cell‐free DNAs was significantly lower than those of the tumors (mean, 0.34% [range, 0.1%‐0.69%] vs 48.5% [10.5%‐87%], P < .001). Thirteen samples, except for one sample of TP100 because the amount of DNA in this sample was not sufficient, were also evaluated by TDS. Mean depths for MYD88 L265P in cell‐free DNA were 12 560x to 19 996x. In this method, the mutations were not detected at all irrespective of whether or not they were detectable by ddPCR.
Presence of mutations in cell‐free DNAs was not significantly associated with laboratory data, such as lactate dehydrogenase (LDH) (P = .360) and soluble interleukin‐2 receptor (sIL2R) (P = .354) levels, concentrations of cell‐free DNAs (P = .105), tumor sizes (P = .611) and clinical outcomes (OS, P = .386; PFS, P = .629).
3.4. MYD88 L265P mutation in cell‐free DNAs during the disease course
To investigate whether the MYD88 L265P mutation in cell‐free DNAs is a useful marker for estimating disease course, such as remission and/or progression of disease (PD), we analyzed the MYD88 L265P mutation in cell‐free DNAs during and after chemotherapies in five patients who showed positivity of the MYD88 L265P mutation in cell‐free DNAs at diagnosis.
Patients TP92 and TP99 received the modified EORTC regimen15 and the R‐MPV regimen,16 respectively. They obtained complete remission (CR) during the observation period. However, the mutations were not detected at all throughout the treatment courses (Figure 4).
Figure 4.
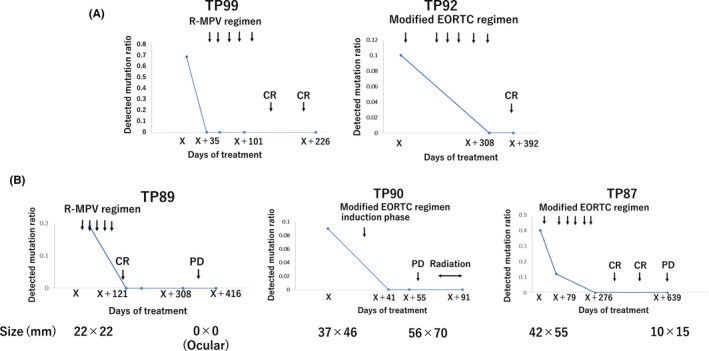
Mutation ratio in cell‐free DNAs and disease course. A, Patients who maintained complete remission (CR). B, Patients who progressed during or after the therapy
In two patients (TP87 and TP89), once achieving CR, lymphomas relapsed at 3 months (TP89) and 12 months (TP87) after completion of the therapies. In TP89, relapse occurred in intraocular tissues without reappearance of a tumor in the CNS. In TP87, tumor size at the time of progression was smaller than that at diagnosis (10 × 15 mm vs 42 × 55 mm). In contrast, TP90 was treated by the modified EORTC regimen, but it was abandoned after induction therapy because of PD. Tumor size at the time of progression was bigger than that at diagnosis (56 × 70 mm vs 37 × 46 mm). In these patients, the MYD88 L265P mutation was not detected in cell‐free DNAs despite disease progress after the relapse (Figure 4).
4. DISCUSSION
The present study demonstrated that the MYD88 L265P mutation was detected by the ddPCR assay in 57.1% of cell‐free DNAs of PCNSL patients at diagnosis. Further analysis is required to determine the actual detection rate of this mutation in cell‐free DNAs of PCNSL. Consistent with the previous report on liquid biopsies, our results confirmed that the ddPCR assay is a reliable method to detect mutations in cell‐free DNAs and has an advantage over TDS in detecting very small numbers of mutation copies, avoiding biases during the PCR procedures that happen in TDS.
Nakamura et al reported that almost all PCNSL had MYD88 and CD79B mutations, whereas they were not detected in glioblastomas (GBM).12, 17 These mutations may serve as a genetic hallmark that help a differential diagnosis among these two most common malignant brain tumors.12
In our analysis, concentration of cell‐free DNAs in PCNSL was 107‐2410 ng/mL. This estimation implies a much higher level than that of cell‐free DNA prepared from healthy volunteers, which were reported to be 1‐10 ng/mL.18 Also, the estimated range appears to be higher than that of cell‐free DNA amounts in patients with non‐hematological neoplasms, such as colorectal, lung, and breast cancer (range 0.5‐1980 ng/mL).19, 20, 21 However, this range of concentration was lower than that of cell‐free DNAs in patients with systemic lymphomas (range 100‐14 180 ng/mL).22, 23
In our analysis, the MYD88 L265P mutation was not detected in cell‐free DNAs by TDS. However, Fontanilles et al recently reported that a variety of somatic mutations specific to CNS tumors, including MYD88 L265P mutation, were detected in cell‐free DNAs of PCNSL patients by TDS.24 In their study, VAF of the MYD88 L265P mutation in cell‐free DNAs was magnitudinally higher than those in our study (mean, 4.7% vs 0.34%).24 The higher VAFs in their study may be a major reason why Fontanilles et al succeeded in the detection of mutations by TDS. In their study, materials used for extraction of cell‐free DNAs were plasma, whereas ours were serum. This difference might explain the discordant VAF, despite the fact that our analysis of one paired cell‐free DNA prepared from serum and plasma failed to demonstrate it. Another potential cause of the discordance could be the bias among the cohorts. Several previous studies have shown that cell‐free DNA in several types of lymphomas could also be a surrogate marker for tumor burden.25, 26, 27 If the tumor burden in our cohort was less than in others because of the early setting of biopsies, the discordance might be explained.
Diagnosis and identifying minimal residual diseases (MRD) by using cell‐free DNA have also been examined in DLBCL: cancer personalized profiling by deep sequencing (CAP‐seq) and high‐throughput sequencing identifying clonotypic Ig rearrangement using cell‐free DNA were shown to be useful for early detection of relapse.21, 25, 26 Recently, we showed that gene mutations specific to angioimmunoblastic T cell lymphoma in cell‐free DNAs could be useful as sensitive markers for circulating tumor DNAs and might be applicable for non‐invasive monitoring of MRD.27 However, in the current study, the MYD88 L265P mutation was not detected in cell‐free DNAs even in the PD state. Our results suggest that circulating tumor DNAs in patients with PCNSL may not be applicable to monitoring the disease course of PCNSL, although those in patients with systemic lymphomas were shown to be useful for monitoring of MRD.26, 27 It remains to be elucidated why the MYD88 L265P mutation was undetectable in the PD state. Tumor size may not explain the reason, considering that tumor size in the CNS at the time of disease progression was bigger than that at diagnosis in a case (TP 90). Moreover, there was a possibility that the MYD88 L265P mutation‐negative clone may expand in relapsed tumors or that cell‐free DNA may not circulate in peripheral blood after chemotherapy. Unfortunately, we analyzed the course of MYD88 L265P mutation in cell‐free DNAs in five cases only. Further analysis will enable us to determine whether MYD88 mutations are applicable for detecting MRD.
To our knowledge, this is the first study for the detection of gene mutations in cell‐free DNAs in PCNSL patients using the ddPCR assay. Ratio of detection of the MYD88 L265P mutation in cell‐free DNA among the mutation‐positive patients was 57%. The ddPCR‐based detection of the MYD88 L265P mutation in cell‐free DNA may serve as a useful method for non‐invasive diagnosis of PCNSL.
CONFLICTS OF INTEREST
Authors declare no financial conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
All authors have contributed significantly to the content of the manuscript.
Hattori K, Sakata‐Yanagimoto M, Suehara Y, et al. Clinical significance of disease‐specific MYD88 mutations in circulating DNA in primary central nervous system lymphoma. Cancer Sci. 2018;109:225–230. https://doi.org/10.1111/cas.13450
Funding information
Leukemia Research Fund and Takeda Science Foundation for Cancer Research to S.C. Grant/Award Number: ‘n/a’, Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, and Science of Japan (Grant/Award Numbers: ‘15H01504’, ‘16H02660’, ‘25112703’).
Contributor Information
Mamiko Sakata‐Yanagimoto, Email: sakatama-tky@umin.net.
Shigeru Chiba, Email: schiba-tky@umin.net.
REFERENCES
- 1. Kluin P, Deckert M, Ferry J. Primary diffuse large B‐cell lymphoma of the CNS In: Swerdlow SH, Camp E, Harris NL, et al., eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edn. Lyon, France: IARC Press; 2008:240‐241. [Google Scholar]
- 2. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199‐1209. [DOI] [PubMed] [Google Scholar]
- 4. Wang S, An T, Wang J, et al. Potential clinical significance of a plasma‐based KRAS mutation analysis in patients with advanced non‐small cell lung cancer. Clin Cancer Res. 2010;16:1324‐1330. [DOI] [PubMed] [Google Scholar]
- 5. Gonzalez‐Aguilar A, Idbaih A, Boisselier B, et al. Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res. 2012;18:5203‐5211. [DOI] [PubMed] [Google Scholar]
- 6. Bruno A, Boisselier B, Labreche K, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. 2014;5:5065‐5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braggio E, Van Wier S, Ojha J, et al. Genome‐wide analysis uncovers novel recurrent alterations in primary central nervous system lymphomas. Clin Cancer Res. 2015;21:3986‐3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vater I, Montesinos‐Rongen M, Schlesner M, et al. The mutational pattern of primary lymphoma of the central nervous system determined by whole‐exome sequencing. Leukemia. 2015;29:677‐685. [DOI] [PubMed] [Google Scholar]
- 9. Yamada S, Ishida Y, Matsuno A, Yamazaki K. Primary diffuse large B‐cell lymphomas of central nervous system exhibit remarkably high prevalence of oncogenic MYD88 and CD79B mutations. Leuk Lymphoma. 2015;56:2141‐2145. [DOI] [PubMed] [Google Scholar]
- 10. Fukumura K, Kawazu M, Kojima S, et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016;131:865‐875. [DOI] [PubMed] [Google Scholar]
- 11. Hattori K, Sakata‐Yanagimoto M, Okoshi Y, et al. MYD88 (L265P) mutation is associated with an unfavourable outcome of primary central nervous system lymphoma. Br J Haematol. 2017;177:492‐494. [DOI] [PubMed] [Google Scholar]
- 12. Nakamura T, Tateishi K, Niwa T, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42:279‐290. [DOI] [PubMed] [Google Scholar]
- 13. Nguyen TB, Sakata‐Yanagimoto M, Nakamoto‐Matsubara R, et al. Double somatic mosaic mutations in TET2 and DNMT3A—origin of peripheral T cell lymphoma in a case. Ann Hematol. 2015;94:1221‐1223. [DOI] [PubMed] [Google Scholar]
- 14. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee SY, Okoshi Y, Kurita N, et al. Prognosis factors in Japanese elderly patients with primary central nervous system lymphoma treated with a nonradiation, intermediate‐dose methotrexate‐containing regimen. Oncol Res Treat. 2014;37:378‐383. [DOI] [PubMed] [Google Scholar]
- 16. Hattori K, Sakata‐Yanagimoto M, Okoshi Y, et al. A single institutional retrospective evaluation for younger patients with primary central nervous lymphomas on a modified R‐MPV regimen followed by radiotherapy and high dose cytarabine. J Clin Exp Hematop. 2017; 57: 41‐46. https://doi.org/10.3960/jslrt.17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wan JC, Massie C, Garcia‐Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223‐238. [DOI] [PubMed] [Google Scholar]
- 19. Mouliere F, El Messaoudi S, Gongora C, et al. Circulating cell‐free DNA from colorectal cancer patients may reveal high KRAS or BRAF mutation load. Transl Oncol. 2013;6:319‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoon KA, Park S, Lee SH, Kim JH, Lee JS. Comparison of circulating plasma DNA levels between lung cancer patients and healthy controls. J Mol Diagn. 2009;11:182‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarzenbach H, Pantel K. Circulating DNA as biomarker in breast cancer. Breast Cancer Res. 2015;17:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scherer F, Kurtz DM, Newman AM, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med 2016;8:364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M, Jia Y, Xu J, Cheng X, Xu C. Assessment of the circulating cell‐free DNA marker association with diagnosis and prognostic prediction in patients with lymphoma: a single‐center experience. Ann Hematol. 2017;96:1343‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fontanilles M, Marguet F, Bohers E, et al. Non‐invasive detection of somatic mutations using next‐generation sequencing in primary central nervous system lymphoma. Oncotarget. 2017;8:48157‐48168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roschewski M, Dunleavy K, Pittaluga S, et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B‐cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16:541‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kurtz DM, Green MR, Bratman SV, et al. Noninvasive monitoring of diffuse large B‐cell lymphoma by immunoglobulin high‐throughput sequencing. Blood. 2015;125:3679‐3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sakata‐Yanagimoto M, Nakamoto‐Matsubara R, Komori D, et al. Detection of the circulating tumor DNAs in angioimmunoblastic T‐ cell lymphoma. Ann Hematol. 2017;96:1471‐1475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials