

Abstract
To improve current cancer immunotherapies, strategies to modulate various immunosuppressive cells including myeloid derived suppressor cells (MDSC) which were shown to be negative factors in immune‐checkpoint blockade therapy, need to be developed. In the present study, we evaluated the role of the local renin‐angiotensin system (RAS) in the tumor immune‐microenvironment using murine models bearing tumor cell lines in which RAS was not involved in their proliferation and angiogenetic ability. Giving angiotensin II receptor blockers (ARB) to C57BL/6 mice bearing murine colon cancer cell line MC38 resulted in significant enhancement of tumor antigen gp70 specific T cells. ARB administration did not change the numbers of CD11b+ myeloid cells in tumors, but significantly reduced their T‐cell inhibitory ability along with decreased production of various immunosuppressive factors including interleukin (IL)‐6, IL‐10, vascular endothelial growth factor (VEGF), and arginase by CD11b+ cells in tumors. ARB also decreased expression of immunosuppressive factors such as chemokine ligand 12 and nitric oxide synthase 2 in cancer‐associated fibroblasts (CAF). Last, combination of ARB and anti‐programmed death‐ligand 1 (PD‐L1) antibodies resulted in significant augmentation of anti‐tumor effects in a CD8+ T cell‐dependent way. These results showed that RAS is involved in the generation of an immunosuppressive tumor microenvironment caused by myeloid cells and fibroblasts, other than the previously shown proliferative and angiogenetic properties of cancer cells and macrophages, and that ARB can transform the immunosuppressive properties of MDSC and CAF and could be used in combination with PD‐1/PD‐L1 immune‐checkpoint blockade therapy.
Keywords: angiotensin receptor antagonist, cancer‐associated fibroblast, myeloid cell, programmed cell death 1 receptor, renin‐angiotensin system
Abbreviations
- ACE
angiotensin‐converting enzyme
- Ang II
angiotensin II
- ARB
angiotensin II receptor blocker
- AT1R
type I angiotensin receptor
- ATCC
American Type Culture Collection
- CAF
cancer‐associated fibroblast
- CFSE
carboxyfluorescein succinimidyl ester
- CTLA‐4
cytotoxic T lymphocyte antigen‐4
- CXCL
chemokine (C‐X‐C motif) ligand
- DC
dendritic cell
- EGFP
enhanced green fluorescent protein
- IFN
interferon
- IL
interleukin
- MDSC
myeloid‐derived suppressor cell
- NF‐κB
nuclear factor‐κB
- nor‐NOHA
Nω‐hydroxy‐nor‐l‐arginine
- NOS
nitric oxide synthase
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand
- PGE2
prostaglandin E2
- RAS
renin‐angiotensin system
- RT‐PCR
real‐time polymerase chain reaction
- SLN
sentinel lymph node
- STAT3
signal transducer and activator of transcription 3
- TAM
tumor‐associated macrophage
- TGF‐β
transforming growth factor
- Treg
regulatory T cell
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Recent cancer immunotherapies including immune‐checkpoint blockade (ie blocking PD‐1, PD‐L1, or CTLA‐4) have produced durable clinical effects in some patients with various advanced cancers. However, only a subset of patients responds to this treatment, and not all responses continue indefinitely. Unresponsiveness to these immune‐checkpoint blockade therapies may be mediated by numerous immunosuppressive mechanisms that inhibit anti‐tumor T‐cell responses and T‐cell infiltration into tumor tissues.1, 2 Thus, combined therapies that can reverse such immunosuppression in non‐responders are urgently needed.
Hematopoietic cells and mesenchymal cells, which comprise the stromal microenvironment of tumors, play important roles in suppressing immune control of tumor growth. Among hematopoietic cells of interest are M2 macrophages, MDSC, and Treg. Accumulation of these suppressor cells has been associated with a poor prognosis in many cancers and with unfavorable clinical response to anti‐PD‐1/PD‐L1 or anti‐CTLA‐4 therapies.1, 3 The dominant mesenchymal cell component in tumor tissues is fibroblasts, which are strongly involved in cancer progression and metastasis.4, 5 Generally, CAF are thought to induce an immunosuppressive tumor microenvironment by producing a variety of cytokines and chemokines with an impact on tumor angiogenesis and remodeling of the extracellular matrix.6
One strategy to reduce the effects of these immunosuppressive cancer stromal cells is to use neutralizing antibodies or inhibitors of immunosuppressive effector molecules produced by the stromal cells. We and other groups previously reported that targeting signaling pathways such as STAT3 or NF‐κB in cancer‐associated immunosuppressive cells could augment anti‐tumor T‐cell immune responses by reducing cancer‐associated immunosuppression.7, 8 Several studies have shown that currently used molecular target drugs for these pathways have synergistic anti‐tumor effects when combined with immunotherapies through immune‐related mechanisms.9, 10, 11
Renin‐angiotensin system is generally thought of as an endocrine system that regulates hydromineral balance and blood pressure systemically. However, recent data have demonstrated that renin and angiotensinogen genes and their products, known as local RAS, are also expressed locally at many tissue sites, where they serve as fundamental regulators of many additional physiological and pathophysiological processes.12
In cancer microenvironments, the major components of RAS are expressed in cancer cells as well as in stromal cells, such as macrophages and CAF.12 This local RAS in cancer tissues has been reported to be involved in cellular migration, proliferation, inflammation, and angiogenesis in the tumor and supporting stromal cells.13, 14, 15 RAS antagonists suppress tumor progression in a variety of experimental cancer models, and retrospective studies in humans show evidence that long‐term use of RAS inhibitors, such as ACE inhibitors or ARB may protect against cancer.12 In tumor tissues, Ang II, the main effector molecule of RAS, acting through AT1R on both tumor cells and stromal cells, regulates secretion of a variety of growth factors and cytokines, such as IL‐6, IL‐8, and VEGF, partly through activating NF‐κB and STAT family members.16, 17 Although these molecules and transcriptional factors are well known to induce cancer‐promoting inflammation and restrain anti‐tumor immune responses, few studies have evaluated the effect of local RAS on anti‐tumor immune responses. Additionally, few studies have been concerned with in vivo interaction of cancer cells and stromal cells and separately evaluated roles of RAS in cancer cells or stromal cells.
In the present study, by using a mouse model bearing a tumor cell line which does not express RAS components, we have separately evaluated immunosuppressive roles of local RAS especially in cancer stromal cells such as macrophages, MDSC and CAF for the induction of anti‐tumor immune responses, and whether ARB could reverse RAS‐induced immunosuppression and enhance the effect of current PD‐1/PD‐L1 immune‐checkpoint blockade therapies.
2. MATERIALS AND METHODS
2.1. Animals, cell lines, culture supernatants
Mice were bred at the animal facilities of Keio University according to guidelines for animal experimentation. CT26 was purchased from ATCC. MC38 was a gift from Surgery Branch, National Cancer Institute, National Institutes of Health (Bethesda, MD, USA). See Appendix S1 for more information of cell lines. Cell culture supernatants were obtained by culturing the cells at 1 × 106 cells/1.5 mL per well for 24 hours. Transgenic mice that ubiquitously express EGFP under control of the CAG promoter18 were bred in our animal facility. All animal experiments in our study have been approved by the ethics committee on animal research of Keio University School of Medicine.
2.2. Real‐time RT‐PCR
Total RNA and cDNA synthesis and real‐time RT‐PCR were carried out using standard protocols. Primers for the target genes are listed in Appendix S1.
2.3. WST‐1 cell proliferation analysis
Cancer cell proliferation was evaluated using WST‐1 Solution Reagent (Roche, Basel, Switzerland) according to the manufacturer's instructions. See Appendix S1 for further details.
2.4. Tumor‐bearing mouse models
Six to eight‐week‐old C57B/6 and Balb/c mice were inoculated s.c. in the flank with 4 × 105 MC38 and with 4 × 105 CT26 cells on day 0, respectively. Mice were injected i.p. with vehicle (PBS), valsartan (15 mg/kg; LKT Laboratories, St Paul, MN, USA), or candesartan (6 mg/kg; Funakoshi, Tokyo, Japan) every day from day 6. In the combined therapy, anti‐PD‐L1 or isotype antibody (Ab) (200 μg/body; Bio X Cell, Lebanon, NH, USA) was given on day 7, 10, and 13. For cell depletion, Anti‐CD8 or isotype Ab (200 μg/body; Bio X Cell) was given i.p. on day 1, 4, 5, 6, 12, and 19. Tumor size was measured using the following formula: volume = 0.5 × (width)2 × (length). Tumor antigen (gp70)‐specific T‐cell responses were detected by modification of our previously reported method.19 See Appendix S1 for more information.
2.5. Flow cytometry and cell isolation
Twenty days after tumor inoculation, resected tumor tissues were digested in RPMI‐1640 medium containing collagenase (1 mg/mL; Wako Pure Chemical Industries Ltd, Osaka, Japan) and DNase type 4 (20 U/mL; Sigma‐Aldrich, St Louis, MO, USA) for 1 hour at 37°C. Digested tumor tissues were stained with fluorescein‐conjugated monoclonal Abs and analyzed using Gallios (Beckman Coulter, Carlsbad, CA, USA). For cell isolation, tumor‐infiltrating CD11b+ cells were positively selected using anti‐CD11b microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) from CD11c− cells which had been negatively selected from the digested tumor tissues using anti‐CD11c microbeads (Miltenyi Biotec). DC were positively selected using CD11c microbeads (Miltenyi Biotec) from draining lymph nodes 20 days after tumor inoculation. GFP+, DsRed, CD45−, CD31−, and Ter119− cells were isolated as murine CAF from DsRed‐labeled MC38 tumor tissues inoculated on EGFP mice using MoFlo XDP (Beckman Coulter). Purity of fresh CAF (ratio of GFP+ DsRed CD45− CD31− Ter119− cells in total sorted cells analyzed by FACS) was approximately 95% (data not shown). After 1‐day culture, approximately 95% (average value of positive cells counted in 10 randomly chosen images at 20× magnification) of the sorted cells were positive for both GFP and HSP47, a fibroblast marker (Fig. S1). For further details and antibodies, see Appendix S1.
2.6. Immunohistochemistry
Immunohistochemistry was done on frozen sections (for CD8) or formalin‐fixed paraffin‐embedded sections (for CD31) of MC38 tumor tissues using standard protocols as previously described.20 For further details and antibodies, see Appendix S1.
2.7. T‐cell proliferation assay
Activity of CD11b+ cells, DC or CAF to suppress or stimulate T cells was assessed by BrdU incorporation using a BrdU ELISA colorimetric assay (Roche) and the intracellular dye carboxyfluorescein succinimidyl ester (CFSE) (Dojindo, Tokyo, Japan). See Appendix S1 for more information.
2.8. Nuclear extract preparation and NF‐κB transcription activation assay
Nuclear extracts of the cells were obtained using an NE‐PER Nuclear and Cytoplasmic Extraction Reagent kit (Thermo Fisher Scientific, Rockford, IL, USA) and their NF‐κB transcription activity was measured using NF‐κB Transcription Factor Microplate Assay (Thermo Fisher Scientific), according to the manufacturer's instructions.
2.9. NOS activity assay
NOS activity was measured using Ultrasensitive Colorimetric NOS Assay Kits (Oxford Biomedical Research, Rochester Hills, MI, USA), according to the manufacturer's instructions.
2.10. Statistical analysis
All results are expressed as mean ± SD. Data were subjected to statistical analysis (unpaired t test and Bonferroni/Dunn's test) to determine differences among the means of the experimental, treated, and control groups. Differences were considered to be statistically significant at P < .05.
3. RESULTS
3.1. Angiotensin II receptor blockade enhanced induction of tumor antigen‐specific CD8+ cytotoxic T lymphocytes in vivo
To investigate the role of RAS in stromal cells such as immune cells and CAF in tumor microenvironments, we chose a tumor‐bearing mouse model in which a murine colorectal carcinoma cell line, MC38, which expresses little of the RAS component gene by itself (Figure 1A), was s.c. inoculated. Because there was little expression of AT1R in MC38 cells, it was expected that RAS would have little direct effect on MC38 cell characteristics, such as cell growth (Fig. S2). Using this model, we first evaluated the effect of ARB on induction of tumor antigen‐specific T cells in vivo. Six days after MC38 implantation, ARB were given i.p. every day and, 14 days later, cells from draining lymph nodes or spleens of the tumor‐bearing mice were stimulated in vitro with an immunodominant MC38 T‐cell epitope peptide, gp70, for 5 days. Induction of gp70‐specific T cells was evaluated by IFN‐γ release assay. Although in vivo growth of MC38 tumors was not affected (Figure 1B), induction of gp70‐specific T cells in the draining lymph nodes or in the spleens was significantly enhanced by the treatment of both ARB tested, valsartan and candesartan (lymph node: Figure 1C, spleen: data not shown). We also analyzed the character of tumor‐infiltrating T cells and found that gp70‐specific CTL (Figure 1D) was enhanced by valsartan treatment although the number of tumor‐infiltrating T cells was not increased (Fig. S3). In addition, similar enhancement of tumor antigen‐specific CTL induction in draining lymph nodes (Figure 1E), accompanied by no change in tumor growth (Figure 1F), was also observed after valsartan treatment of CT26‐bearing mice (a murine colorectal cancer cell line expressing AT1R [Figure 1A] in which proliferation was not affected by valsartan or angiotensin II [Fig. S2]). These results suggest that local RAS in tumor microenvironments might suppress tumor antigen‐specific T‐cell induction, an effect which can be reversed by giving systemic ARB.
Figure 1.
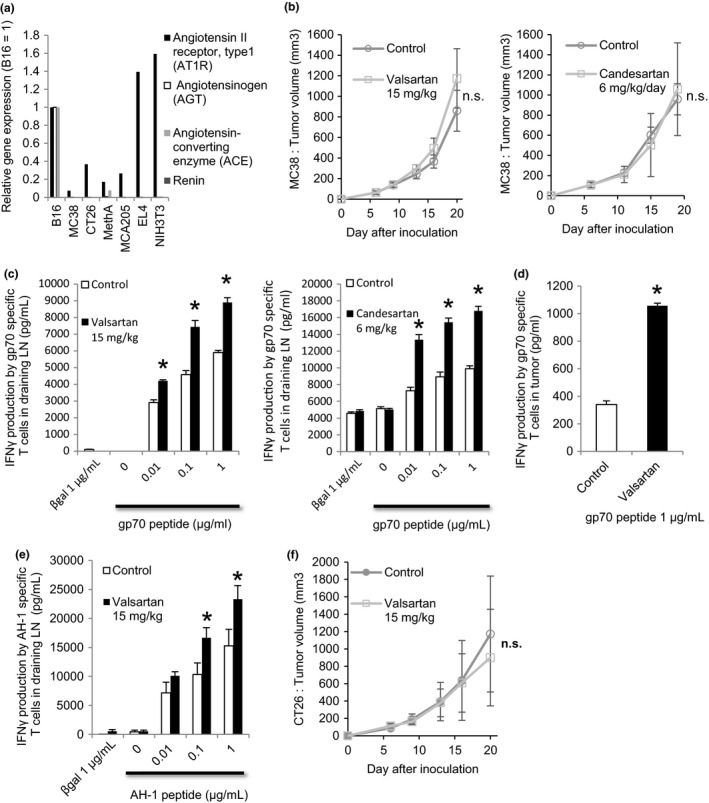
Angiotensin‐renin blockade enhanced induction of tumor antigen‐specific CTL. A, Expression of type I angiotensin receptor (AT1R), angiotensinogen, angiotensin‐converting enzyme (ACE), and renin mRNA relative to gene GAPDH RNA in murine cell lines measured by quantitative PCR (qPCR). Vertical axis shows the expression ratio of mRNAs. Expression level in B16 was defined as 1. B, C57BL/6 mice bearing MC38 tumors were treated with valsartan, candesartan, or DMSO (Control). Mean tumor size ± SD (n = 5). All data are from 3 independent experiments. C and D, At day 20, whole cells from draining lymph nodes were cultured and restimulated with gp70 peptide. Tumor‐infiltrating CD8+ T cells and irradiated syngeneic splenocytes were cocultured and restimulated with gp70 peptide. In vivo tumor antigen‐specific T‐cell induction from (C) draining lymph nodes and (D) tumor was evaluated by an interferon (IFN)‐γ releasing assay. All data are from 3 independent experiments. Error bars indicate SD. *P < .05 using a t test. E, In vivo induction of tumor antigen (AH‐1)‐specific T cells from draining lymph nodes was evaluated by an IFN‐γ releasing assay in Balb/c‐CT26 mouse models. All data are from 2 independent experiments. Error bars indicate SD. *P < .05 using a t test. F, Balb/c mice bearing CT26 tumors were treated with valsartan or DMSO (Control). Mean tumor size ± SD (n = 5). All data are from 2 independent experiments. Error bars indicate SD
3.2. ARB treatment reduced the T‐cell suppressive activity of tumor‐infiltrating CD11b+ cells
We further evaluated the effect of ARB treatment on tumor‐infiltrating immune cells and found that treatment led to phenotypic changes in CD11b+ cells, including MDSC and macrophages. Protein expression of certain immune suppressive molecules, such as PGE2, IL‐6,8, 21 VEGF, and arginase, in tumor infiltrating CD11b+ CD11c− cells containing macrophages and MDSC was significantly decreased by valsartan treatment (Figure 2A), whereas their expression in tumor tissues was not changed (Fig. S4a). mRNA expression of these molecules was also decreased in each CD11b+ myeloid cell fraction, including macrophages, monocytic‐MDSC, and granulocytic‐MDSC (Figure 2B). IL‐6 is preferentially decreased in TAM. Only protein level in VEGF was decreased, suggesting possible post‐transcriptional regulation22 by valsartan in vivo. Additionally, the proportion of each cell fraction of CD11b+ cells (Fig. S4a) and the expression of MHC class II molecules and PD‐L1 on macrophages and MDSC (Fig. S4b) were not affected by valsartan treatment. Although VEGF production from CD11b+ CD11c− cells was decreased in valsartan‐treated mice, angiogenesis in tumor tissues evaluated by immunohistochemical staining of CD31 was unaltered (Fig. S4c).
Figure 2.
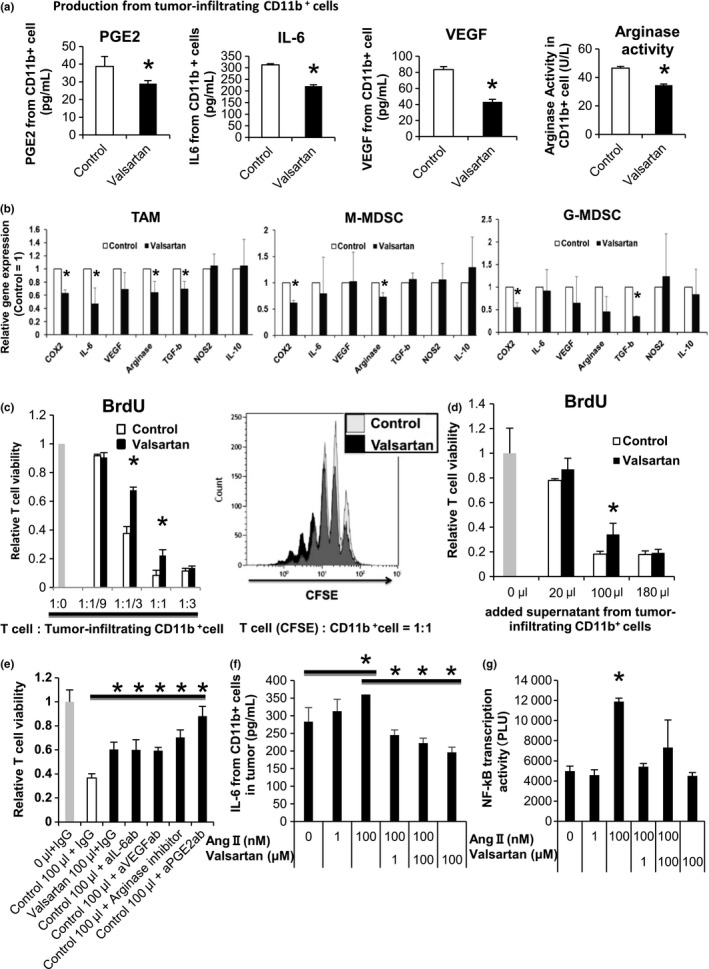
Angiotensin‐renin blockade reduced T‐cell suppressive activity of tumor‐infiltrating CD11b+ cells along with decreased production of immunosuppressive molecules. A, Prostaglandin E2 (PGE2), interleukin (IL)‐6 and vascular endothelial growth factor (VEGF) production and arginase activity of CD11b+ cells in tumors of MC38‐implanted mice were measured by ELISA and colorimetric method. B, COX2, interleukin (IL)‐6, VEGF, arginase, transforming growth factor β (TGF‐β), nitric oxide synthase (NOS)2 and IL‐10 mRNA expression relative to GAPDH mRNA in tumor‐infiltrating CD11b+ cells measured by quantitative PCR (qPCR). Expression level in the control group was defined as 1. C, Syngeneic T cells from C57BL/6 mice were cocultured with tumor‐infiltrating CD11b+ cells in the presence of anti‐CD3‐Ab for 3 days. T‐cell proliferation was measured by BrdU incorporation (left). T cells incubated without tumor‐infiltrating CD11b+ cells (1:0) served as a positive control and BrdU incorporation level in this group was defined as 1. T‐cell proliferation was also measured by flow cytometry using carboxyfluorescein succinimidyl ester (CFSE). CFSE intensity in CD3+ T cells is shown. Ratio of proliferating cells was increased by valsartan (right). D and E, Syngeneic T cells from C57BL/6 mice were cultured with culture supernatant from tumor‐infiltrating CD11b+ cells in the presence of anti‐CD3‐Ab (E) with or (D) without neutralizing antibodies (anti‐IL‐6 antibody, anti‐VEGF antibody, and anti‐PGE2 antibody) and an arginase inhibitor for 3 days. T‐cell proliferation was measured by BrdU incorporation. BrdU incorporation level in the group without supernatant was defined as 1. F and G, Tumor‐infiltrating CD11b+ cells were cultured with various concentrations of angiotensin II and valsartan in vitro. F, IL‐6 production from the tumor‐infiltrating CD11b+ cells was measured by ELISA. G, DNA‐binding activity of NF‐κB (p65) in nuclear lysates of tumor‐infiltrating CD11b+ cells was measured by NF‐κB Transcription Factor Microplate Assay (Thermo Fisher Scientific, Rockford, IL, USA). All data in Figure 2 are from 3 independent experiments. Error bars indicate SD. *P < .05 using a t test
Next, we evaluated the suppressive activity of CD11b+ CD11c− myeloid cells on T‐cell proliferation. The CD11b+ CD11c− cells isolated from the MC38 tumor tissues were cocultured in vitro with activated syngeneic splenic T cells stimulated by anti‐CD3 Abs, and suppression of T‐cell proliferation was evaluated by BrdU incorporation. The CD11b+ CD11c− cells from valsartan‐treated mice had less suppressive activity on T‐cell proliferation compared with those from control mice (Figure 2C). Similarly, culture supernatants of the isolated CD11b+ CD11c− myeloid cells also suppressed T‐cell proliferation. This suppressive activity was reduced by in vivo valsartan treatment (Figure 2D) or by neutralizing Abs or an inhibitor targeting each suppressive molecule, including PGE2, IL‐6, VEGF, and arginase (Figure 2E). Production of these molecules was shown to be inhibited by in vivo valsartan treatment (Figure 2A), indicating that ARB weaken the suppressive activity on T‐cell proliferation by macrophages and MDSC through reducing their production of PGE2, IL‐6, VEGF, and arginase.
Next, we evaluated whether RAS might directly affect IL‐6 production from CD11b+ cells. mRNA of AT1R was expressed on the tumor‐infiltrating CD11b+ cells (Fig. S4d). Increased IL‐6 production accompanied by activation of NF‐κB, a representative transcriptional factor regulating IL‐6 mRNA expression, was observed in in vitro Ang II‐treated tumor‐infiltrating CD11b+ CD11c− cells isolated from naive C57BL/6 mice, an effect that was inhibited by valsartan (Figure 2F,G). This demonstrates a direct effect of RAS on macrophages and MDSC.
Because inhibiting RAS has been reported to have suppressive effects on DC in vitro or in AT1R knockout mice23 and to induce Treg in autoimmunity models,24 we analyzed in vivo phenotypic changes of them in valsartan‐treated MC38‐bearing mice and found that the treatment affected neither T‐cell stimulatory activity of DC (Fig. S4e) nor the number of tumor‐infiltrating Treg (Fig. S4f).
These results suggest that local RAS activity in the tumor microenvironment affects mainly CD11b+ cells, such as macrophages and MDSC, to enhance suppression of T‐cell proliferation, partly by inducing expression of immunosuppressive molecules, such as IL‐6. This particular effect is inhibited by appropriate doses of ARB without affecting DC and Treg.
3.3. ARB treatment augmented the T‐cell stimulatory activity of CAF
Fibroblasts from various organs have been reported to express AT1R. CAF from the MC38 tumor tissue in this experiment also express AT1R (Fig. S4d). Ang II stimulates their proliferation and induces production of various cytokines, such as TGF‐β,25, 26 some of which have immunosuppressive functions. CAF, which are considered to be immunosuppressive cells in the cancer microenvironment,4 also express AT1R.27 Thus, we evaluated the effect of ARB treatment on CAF phenotypes in our mouse models. For sorting of pure CAF, we used specially devised tumor‐bearing mouse models in which MC38 cells labeled by a fluorescent protein, DsRed, were inoculated into CAG‐EGFP transgenic mice that expressed EGFP ubiquitously. We sorted out the stromal non‐immune cells (EGFP+ DsRed CD45− CD31− Ter119−) as CAF from the tumor tissues of this mouse model. CAF sorted from valsartan‐treated mice had significantly lowered expression of CXCL12 and NOS2 (Figure 3A,B), both of which are related to the inhibition of anti‐tumor immune responses.28, 29, 30, 31 When evaluating the effect of CAF on T‐cell proliferation in vitro, CAF from valsartan‐treated mice showed T‐cell stimulatory activities, whereas CAF from control mice had no effect on T‐cell proliferation (Figure 3C). These results suggest that local RAS in the tumor microenvironment might contribute to the immunosuppressive character of CAF, an effect that can be altered by ARB treatment.
Figure 3.
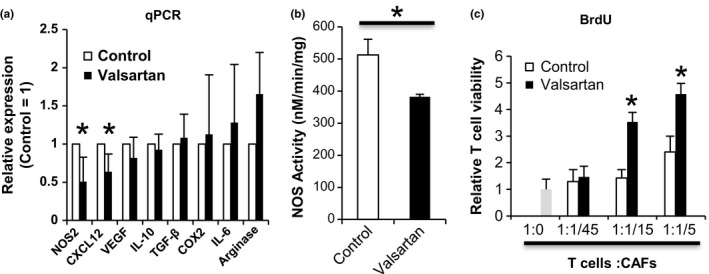
Angiotensin‐renin blockade increased T‐cell stimulatory function of cancer‐associated fibroblasts and decreased their expression of nitric oxide synthase (NOS)2 and chemokine (C‐X‐C motif) ligand (CXCL12). A, Genes expressed in cancer‐associated fibroblasts (CAF) were measured by quantitative PCR (qPCR). Levels are normalized to the control. B, NOS activity of CAF was measured using ultrasensitive colorimetric NOS assay kits. C, Murine CAF were isolated from dsRed‐labeled MC38 tumor‐bearing enhanced green fluorescent protein (EGFP) mice (n = 3). CAF were irradiated and cocultured with T cells from C57BL/6 mice in the presence of anti‐CD3‐Ab for 3 days. T‐cell proliferation was measured by BrdU incorporation. T cells incubated without CAF (1:0) served as a negative control and BrdU incorporation level in this group was defined as 1. All data in Figure 3 are from 3 independent experiments. Error bars indicate SD. *P < .05 using a t test.
3.4. Combined therapy with ARB and anti‐PD‐L1 antibody showed synergistic anti‐tumor effects
Although the induction of tumor antigen‐specific T cells was significantly augmented by ARB treatment (Figure 1C,D), no significant anti‐tumor effects were observed (Figure 1B). Therefore, we tried combined therapy with ARB and anti‐PD‐L1 Ab. Compared with the control group, tumor proliferation was significantly suppressed in the combined therapy group, whereas no significant suppression was observed with ARB or anti‐PD‐L1 antibody monotherapy (Figure 4A). Abrogation of the anti‐tumor effect of the combined therapy by CD8+ T‐cell depletion indicates an essential role of T‐cell‐mediated immune responses for the synergistic effect (Figure 4A). Additionally, Ang II did not affect PD‐L1 expression in MC38 cells in vitro, and there was no change in PD‐L1 expression in MC38 cells, TAM and CAF following ARB treatment in vivo. Figure 4B,C, Figs S4b and S5, suggesting the synergistic effect of this combined therapy was not a result of the direct effect of ARB on their PD‐L1 expression.
Figure 4.
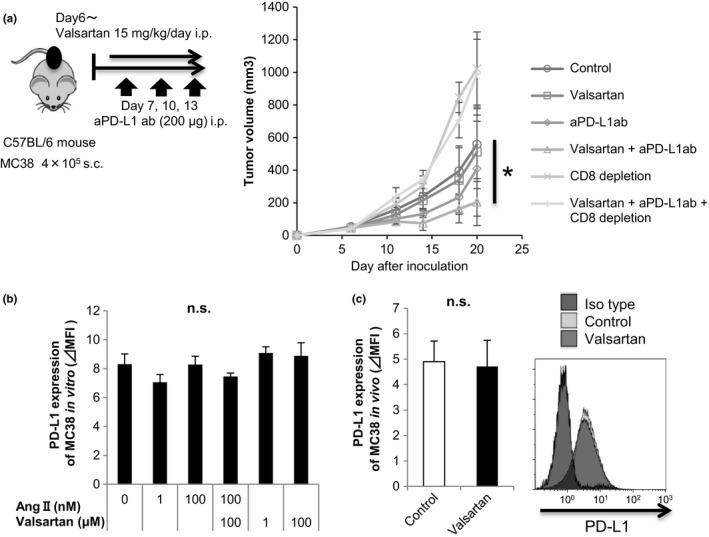
Combination of angiotensin‐receptor blockade and anti‐programmed death‐ligand (anti‐PD‐L1) antibody treatment inhibited tumor growth in MC38‐bearing mice. A, MC38 cells (4 × 105) were s.c. injected into C57BL/6 mice on day 0. Anti‐PD‐L1 antibody or isotype antibody (200 μg) was given on days 7, 10, and 13. Rat anti‐mouse CD8 or isotype antibody (200 μg) was given i.p. 1 day before tumor implantation and on days 4, 5, 6, 12, and 19 after tumor implantation. Tumor volumes were measured every 3‐4 days. Mean tumor size ± SD (n = 5). All data are from 3 independent experiments. *P < .05 comparing untreated mice (control) with valsartan + anti‐PD‐L1 antibody‐treated mice. B, PD‐L1 expression levels of MC38 cultured with various concentrations of angiotensin II and valsartan in vitro were measured by flow cytometry. All data are from 3 independent experiments. Error bars indicate SD. C, PD‐L1 expression levels of MC38 tumor from the untreated or valsartan‐administered mouse were measured by flow cytometry. All data are from 2 independent experiments. Error bars indicate SD. *P < .05 using a t test. n.s., not significant
Collectively, the results of the present study demonstrated that local RAS in cancer microenvironments induces immune suppression by affecting macrophages as well as MDSC and CAF, effects that could be counteracted by ARB treatment, supporting the combined use of ARB with PD‐1/PD‐L1 blockade therapies.
4. DISCUSSION
There are various in vitro studies evaluating the role of RAS in the induction of immune responses. For example, activation of RAS enhances antigen‐specific T‐cell responses, which can be blocked by ARB.32 ARB also inhibit the differentiation of human DC from monocytes and their maturation by LPS stimulation.23 In non‐tumor‐bearing mouse models, RAS has been reported to be involved in the induction of T‐cell responses.33 However, few studies have reported the role of RAS in the induction of anti‐cancer T‐cell responses. In the present study, we identified local RAS activity in cancer stromal cells, including macrophages, MDSC, and fibroblasts, as a potent inducer of immunosuppression in the cancer microenvironment. Inhibition of this local RAS using ARB increased the induction and infiltration of tumor antigen‐specific T cells, leading to enhanced anti‐tumor efficacy of anti PD‐L1 Ab therapies.
In animal models, accumulating evidence indicates that inhibition of RAS suppresses tumor growth mainly by inhibiting angiogenesis.13, 34 In our mouse model using MC38, although VEGF production from CD11b+ cells was reduced, angiogenesis evaluated by CD31 staining and tumor growth were not affected by ARB treatment. There are multiple factors regulating angiogenesis, which are specific for each model. Dose of ARB and characteristics of MC38 cells, which express few of the RAS component genes, may explain why we saw no change in angiogenesis in our models. Lack of change in tumor size and in angiogenesis enables precise evaluation of the role of RAS in suppression of the immune response to tumors in our study because tumor size itself influences tumor immune responses to some extent. Although it has been reported, in the past, that ARB inhibits tumor growth through angiogenesis, our results indicated that tumor growth can be inhibited through the immune response with no involvement of angiogenesis.
Physiologically, hematopoietic stem cells express ACE,35 and ACE promotes the generation of myeloid progenitors36 in an AT1R‐dependent way. Recently, Cortez‐Retamozo et al37 reported that overproduction of Ang II by tumor tissues in a tumor‐bearing mouse model amplified macrophage progenitors in the spleen, leading to increased accumulation of TAM. They also showed that enalapril, an ACE inhibitor, inhibited tumor progression, an effect accompanied by a reduced number of TAM. Their study suggests a significant role for systemic RAS in TAM differentiation. However, the role of RAS in myeloid‐mediated immune responses against cancers has not been well addressed. Furthermore, although Ang peptide levels have been reported to be elevated in some patients with cancer, this is not always the case.38, 39 In our study, plasma Ang II was not elevated in tumor‐bearing mice (data not shown), possibly because MC38 cells do not express RAS‐component genes. The number of CD11b+ cells, including macrophages and MDSC, was not affected by ARB treatment. Our observation suggests that even if Ang II was not elevated systemically, RAS might locally affect the suppressive activity of tumor‐infiltrating CD11b+ cells, leading to suppressed anti‐tumor T‐cell responses.
Accumulation of MDSC as a result of dysfunctional myelopoiesis and the polarization of macrophages toward the M2 phenotype in tumor tissues promote tumor growth by suppressing the immune response as well as by supporting angiogenesis, tumor cell survival, and metastasis. Although it is well known that AT1R is expressed on these cells, the role of local RAS in their suppressive activities remains largely unknown. In the transgenic ACE 10/10 mouse generated by the promoter‐swapping technique where ACE are overexpressed only in macrophages and myeloid‐lineages, tumor growth has been reported to be suppressed with an increased number of tumor‐specific T cells and M1‐like macrophages characterized by high IL‐12.40 As for MDSC, the number of CD11b+ Gr‐1+ cells were decreased in the spleens of ACE 10/10 mice.41 Unlike our findings, these observations suggest that ACE in macrophages and MDSC has anti‐tumor properties. However, the expression of ACE in that model was much higher than physiological levels, and these phenotypes of tumor‐bearing ACE 10/10 mice were not critically dependent on the AT1R signal, because ARB could not reverse these phenotypes in that study.40 Neither infiltration of MDSC into tumors nor their suppressive activity has been evaluated. Our study shows that physiological levels of Ang II (data not shown) in cancer microenvironments are involved in the suppressive activities of tumor‐infiltrating macrophages and MDSC.
Cancer‐associated fibroblasts are generally considered to be immunosuppressive cells in cancer microenvironments because of their secretion and expression of immunosuppressive molecules, such as nitric oxide, TGF‐β1, indoleamine‐pyrrole 2,3‐dioxygenase, PGE2, PD‐L1 and PD‐L2.6, 42 Although most reports have been based on in vitro experiments using cultured and expanded CAF, a recent study using genetically engineered mouse models where CAF were depleted, showed that CAF were directly involved in decreased anti‐tumor CD8+ T‐cell responses.43, 44 CAF also produce CXCL12, which has been shown to inhibit T‐cell infiltration into tumor tissues, leading to reduced anti‐tumor effects of anti‐CTLA‐4 Ab and anti‐PD‐L1 Ab in a pancreatic cancer model.28 These studies have shown in vivo immunosuppressive roles for CAF. Although CAF expressing AT1R27 and Ang II were reported to be involved in fibroblast proliferation and secretion of some of the above immunosuppressive factors, the role of RAS in CAF‐mediated anti‐tumor immune responses has not been well evaluated. Our study suggests that local RAS is involved in generating CAF immunosuppressive properties by enhancing its NOS2 and CXCL12 expression, effects that could be modified by ARB treatment.
Several retrospective evaluations of the effects of ACE inhibitors or ARB treatment on cancer incidence have produced conflicting conclusions. Decreases,45 increases,46 and no change47 in cancer incidence have been reported. However, the concurrent use of ARB with chemotherapy has shown synergistic clinical effects in many studies. Recently, it has been reported that the composition, localization, and function of tumor‐infiltrating lymphoid and myeloid cells, called the immune contexture, has major prognostic value in cancer patients treated with chemotherapy.48 One of the mechanisms of these synergistic effects might be improvement of the cancer immunological microenvironment by ACE inhibitors or ARB as shown in our study.
As it has been reported that large amounts of MDSC or VEGF are related to resistance to immunotherapy such as anti‐PD‐1 therapy, it is important to suppress MDSC and VEGF. Various drugs have been developed to control MDSC and VEGF in order to augment the treatment effects of immunotherapy. However, few have come into practical use because of the side‐effects. As ARB is already used in general clinical settings, it can be safely used without any serious side‐effects such as drug repositioning. ARB is useful when used in combination with immunotherapy because it can decrease the immunosuppressive ability such as VEGF production by CD11b+ cells including MDSC.
In conclusion, we have shown that local RAS in cancer microenvironments has a profound impact, inducing immunosuppression by enhancing the immunosuppressive activities of macrophages, MDSC, and CAF. This effect is reversed by ARB treatment. ARB is therefore a candidate for combined use with immune‐checkpoint blockade therapies, such as anti‐PD‐1 Ab. Our results suggest that ARB, a drug that is already in use, can transform the immunosuppressive properties of stromal cells shared by a diverse range of tumors, meaning that it could be used in a new treatment strategy in combination with immunotherapy.
CONFLICT OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Grants‐in‐aid for Scientific Research (26221005) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) and the Project for Cancer Research And Therapeutic Evolution (P‐CREATE) from Japan Agency for Medical Research and Development (AMED). We would like to thank Miyuki Saito and Kenji Morii for technical assistance and Misako Horikawa and Ryoko Suzuki for preparation of the manuscript.
Nakamura K, Yaguchi T, Ohmura G, et al. Involvement of local renin‐angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2018;109:54–64. https://doi.org/10.1111/cas.13423
Funding information
Japan Agency for Medical Research and Development (Grant/Award Number: ‘P‐CREATE/16cm0106305h0001’, ‘P‐DIRECT/15cm0106084h0005’) Ministry of Education, Culture, Sports, Science and Technology (Grant/Award Number: ‘Grants‐in‐aid for Scientific Research/26221005’).
Contributor Information
Tomonori Yaguchi, Email: beatless@rr.iij4u.or.jp.
Yutaka Kawakami, Email: yutakawa@keio.jp.
REFERENCES
- 1. Kim JM, Chen DS. Immune escape to PD‐L1/PD‐1 blockade: seven steps to success (or failure). Ann Oncol 2016;27:1492–1504. [DOI] [PubMed] [Google Scholar]
- 2. Yaguchi T, Kawakami Y. Cancer‐induced heterogeneous immunosuppressive tumor microenvironments and their personalized modulation. Int Immunol 2016;28:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meyer C, Cagnon L, Costa‐Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 2014;63:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 5. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med 2014;211:1503–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer 2016;16:582–598. [DOI] [PubMed] [Google Scholar]
- 7. Iwata‐Kajihara T, Sumimoto H, Kawamura N, et al. Enhanced cancer immunotherapy using STAT3‐depleted dendritic cells with high Th1‐inducing ability and resistance to cancer cell‐derived inhibitory factors. J Immunol 2011;187:27–36. [DOI] [PubMed] [Google Scholar]
- 8. Nishio H, Yaguchi T, Sugiyama J, et al. Immunosuppression through constitutively activated NF‐kappaB signalling in human ovarian cancer and its reversal by an NF‐kappaB inhibitor. Br J Cancer 2014;110:2965–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawakami Y, Yaguchi T, Sumimoto H, et al. Improvement of cancer immunotherapy by combining molecular targeted therapy. Front Oncol 2013;3:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007;7:41–51. [DOI] [PubMed] [Google Scholar]
- 11. Mineharu Y, Muhammad AK, Yagiz K, et al. Gene therapy‐mediated reprogramming tumor infiltrating T cells using IL‐2 and inhibiting NF‐kappaB signaling improves the efficacy of immunotherapy in a brain cancer model. Neurotherapeutics 2012;9:827–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. George AJ, Thomas WG, Hannan RD. The renin‐angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 2010;10:745–759. [DOI] [PubMed] [Google Scholar]
- 13. Egami K, Murohara T, Shimada T, et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 2003;112:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo F, Chen XL, Wang F, Liang X, Sun YX, Wang YJ. Role of angiotensin II type 1 receptor in angiotensin II‐induced cytokine production in macrophages. J Interferon Cytokine Res 2011;31:351–361. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi M, Suzuki E, Takeda R, et al. Angiotensin II and tumor necrosis factor‐alpha synergistically promote monocyte chemoattractant protein‐1 expression: roles of NF‐kappaB, p38, and reactive oxygen species. Am J Physiol Heart Circ Physiol 2008;294:H2879–H2888. [DOI] [PubMed] [Google Scholar]
- 16. Ruiz‐Ortega M, Lorenzo O, Ruperez M, Konig S, Wittig B, Egido J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: molecular mechanisms. Circ Res 2000;86:1266–1272. [DOI] [PubMed] [Google Scholar]
- 17. Marrero MB, Schieffer B, Paxton WG, et al. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995;375:247–250. [DOI] [PubMed] [Google Scholar]
- 18. Kawamoto S, Niwa H, Tashiro F, et al. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre‐mediated recombination. FEBS Lett 2000;470:263–268. [DOI] [PubMed] [Google Scholar]
- 19. Nakamura S, Yaguchi T, Kawamura N, et al. TGF‐beta1 in tumor microenvironments induces immunosuppression in the tumors and sentinel lymph nodes and promotes tumor progression. J Immunother 2014;37:63–72. [DOI] [PubMed] [Google Scholar]
- 20. Deng GM, Liu L, Kyttaris VC, Tsokos GC. Lupus serum IgG induces skin inflammation through the TNFR1 signaling pathway. J Immunol 2010;184:7154–7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bharadwaj U, Li M, Zhang R, Chen C, Yao Q. Elevated interleukin‐6 and G‐CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 2007;67:5479–5488. [DOI] [PubMed] [Google Scholar]
- 22. Arcondeguy T, Lacazette E, Millevoi S, Prats H, Touriol C. VEGF‐A mRNA processing, stability and translation: a paradigm for intricate regulation of gene expression at the post‐transcriptional level. Nucleic Acids Res 2013;41:7997–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nahmod K, Gentilini C, Vermeulen M, et al. Impaired function of dendritic cells deficient in angiotensin II type 1 receptors. J Pharmacol Exp Ther 2010;334:854–862. [DOI] [PubMed] [Google Scholar]
- 24. Platten M, Youssef S, Hur EM, et al. Blocking angiotensin‐converting enzyme induces potent regulatory T cells and modulates TH1‐ and TH17‐mediated autoimmunity. Proc Natl Acad Sci USA 2009;106:14948–14953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor‐beta expression in rat glomerular mesangial cells. J Clin Invest 1994;93:2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawano H, Do YS, Kawano Y, et al. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000;101:1130–1137. [DOI] [PubMed] [Google Scholar]
- 27. Dougherty U, Mustafi R, Sadiq F, et al. The renin‐angiotensin system mediates EGF receptor‐vitamin D receptor cross‐talk in colitis‐associated colon cancer. Clin Cancer Res 2014;20:5848–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA 2013;110:20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu W, Liu LZ, Loizidou M, Ahmed M, Charles IG. The role of nitric oxide in cancer. Cell Res 2002;12:311–320. [DOI] [PubMed] [Google Scholar]
- 30. Ito H, Ando T, Seishima M. Inhibition of iNOS activity enhances the anti‐tumor effects of alpha‐galactosylceramide in established murine cancer model. Oncotarget 2015;6:41863–41874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bronte V, Zanovello P. Regulation of immune responses by L‐arginine metabolism. Nat Rev Immunol 2005;5:641–654. [DOI] [PubMed] [Google Scholar]
- 32. Maeda A, Okazaki T, Inoue M, et al. Immunosuppressive effect of angiotensin receptor blocker on stimulation of mice CTLs by angiotensin II. Int Immunopharmacol 2009;9:1183–1188. [DOI] [PubMed] [Google Scholar]
- 33. Wysocki PJ, Kwiatkowska EP, Kazimierczak U, Suchorska W, Kowalczyk DW, Mackiewicz A. Captopril, an angiotensin‐converting enzyme inhibitor, promotes growth of immunogenic tumors in mice. Clin Cancer Res 2006;12:4095–4102. [DOI] [PubMed] [Google Scholar]
- 34. Fujita M, Hayashi I, Yamashina S, Fukamizu A, Itoman M, Majima M. Angiotensin type 1a receptor signaling‐dependent induction of vascular endothelial growth factor in stroma is relevant to tumor‐associated angiogenesis and tumor growth. Carcinogenesis 2005;26:271–279. [DOI] [PubMed] [Google Scholar]
- 35. Jokubaitis VJ, Sinka L, Driessen R, et al. Angiotensin‐converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood 2008;111:4055–4063. [DOI] [PubMed] [Google Scholar]
- 36. Lin C, Datta V, Okwan‐Duodu D, et al. Angiotensin‐converting enzyme is required for normal myelopoiesis. FASEB J 2011;25:1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cortez‐Retamozo V, Etzrodt M, Newton A, et al. Angiotensin II drives the production of tumor‐promoting macrophages. Immunity 2013;38:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loibner H, Poglitsch M, Schwager C, et al. Cancer and the renin angiotensin system (RAS): substantial activation as evidenced by quantification of various RAS peptides and their metabolites in plasma by LC‐MS. J Clin Oncol 2011;29:e21171. [Google Scholar]
- 39. Lieberman J. Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis. Am J Med 1975;59:365–372. [DOI] [PubMed] [Google Scholar]
- 40. Shen XZ, Li P, Weiss D, et al. Mice with enhanced macrophage angiotensin‐converting enzyme are resistant to melanoma. Am J Pathol 2007;170:2122–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen XZ, Okwan‐Duodu D, Blackwell WL, et al. Myeloid expression of angiotensin‐converting enzyme facilitates myeloid maturation and inhibits the development of myeloid‐derived suppressor cells. Lab Invest 2014;94:536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barnas JL, Simpson‐Abelson MR, Yokota SJ, Kelleher RJ, Bankert RB. T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron 2010;3:29–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kraman M, Bambrough PJ, Arnold JN, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein‐alpha. Science 2010;330:827–830. [DOI] [PubMed] [Google Scholar]
- 44. Ozdemir BC, Pentcheva‐Hoang T, Carstens JL, et al. Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang CC, Chan WL, Chen YC, et al. Angiotensin II receptor blockers and risk of cancer in patients with systemic hypertension. Am J Cardiol 2011;107:1028–1033. [DOI] [PubMed] [Google Scholar]
- 46. Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. Angiotensin‐receptor blockade and risk of cancer: meta‐analysis of randomised controlled trials. Lancet Oncol 2010;11:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bangalore S, Kumar S, Kjeldsen SE, et al. Antihypertensive drugs and risk of cancer: network meta‐analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet Oncol 2011;12:65–82. [DOI] [PubMed] [Google Scholar]
- 48. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 2015;28:690–714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials