

Abstract
Chronic infection with hepatitis B virus (HBV) increases the risk of developing fibrosis, cirrhosis or hepatocellular carcinoma. Current therapies are limited to type‐I interferons and/or nucleos(t)ide analogues; however, these are only partially effective. The development of novel anti‐HBV agents for new treatment strategies has been hampered by the lack of a suitable system that allows the in vitro replication of HBV. Studies of virus infection/replication at the molecular level using wild‐type HBV are labor‐intensive and time‐consuming. To overcome these problems, we previously constructed a recombinant reporter HBV bearing the NanoLuc gene and showed its usefulness in identifying factors that affect HBV proliferation. Because this system mimics the early stage of the HBV life cycle faithfully, we conducted a quantitative analysis of HBV infectivity to several human hepatocyte cell lines as well as the effect of dimethyl sulfoxide and HBV protein X on the early stage of HBV proliferation using this system. Furthermore, we developed a system to produce a reporter HBV expressing a pol gene. These reporter HBV may provide an opportunity to enhance our understanding of the HBV life cycle and aid strategies for the development of new anti‐HBV agents.
Keywords: anti‐ hepatitis B virus agents, hepatitis B virus, replication, reporter gene, reporter hepatitis B virus
1. INTRODUCTION
Chronic infection with hepatitis B virus (HBV) affects nearly 240 million people worldwide, and increases their risk of developing fibrosis, cirrhosis or hepatocellular carcinoma.1 Current therapies are limited to type‐I interferon and/or nucleos(t)ide analogues, but these are only partially effective. The development of anti‐HBV agents for new treatment strategies has been hampered by the lack of a suitable in vitro HBV replication system.
The study of virus infection/replication at the molecular level using wild‐type virus is time consuming and labor‐intensive. Moreover, exploring antivirals using high‐throughput screening demands rapid, sensitive and quantitative methods, which wild‐type virus, especially HBV, does not always tolerate. Although HBV replication mechanisms have been reported using genetic and biochemical approaches,2 simple in vitro infection systems were limited to human primary hepatocytes and their derivative cell line HepaRG3 until the recent discovery of the HBV receptor, sodium taurocholate co‐transporting polypeptide, NTCP.4 NTCP‐transduced human hepatocytes have been used to derive cell lines including HepG2 and HuH7, which are susceptible to HBV infection in vitro.5 However, despite the increased availability of HBV infectious cell lines, assays of wild‐type HBV infection and replication are still time consuming and laborious.
Compared with wild‐type HBV, reporter‐expressing viruses offer advantages such as reduced labor and the effective screening of anti‐HBV agents for the quantitative analysis of HBV infection/replication.
There have been numerous intensive attempts to engineer reporter HBV. However, the HBV genome is compact and contains indispensable cis‐elements, forming approximately one‐third of the genome, and which are clustered closely together between the C‐terminal part of polymerase and the N‐terminal of the core. These factors limit the size of pregenomic RNA (pgRNA) that can be packaged, the location of the insertion site, and the size of foreign genes inserted when engineering recombinant virus HBV.6
Previous studies have successfully inserted a foreign gene into specific regions of the HBV genome (small envelope [S] gene, core coding open reading frame [ORF] spanning from downstream of the poly‐A site and the spacer region of the pol gene of pgRNA), indicating that these regions tolerate the insertion of foreign genes.6, 7, 8, 9, 10, 11, 12
We previously developed a reporter HBV bearing the NanoLuc (NL) gene, which replaced 562 nucleotides of the core ORF, for the quantitative analysis of HBV infection/replication and for high throughput screening of anti‐HBV agents.5 The production of HBV/NL from HepG2/NTCP#22 cells bearing pHBV/NL and pHBV/D, a helper plasmid that provides missing virus proteins in HBV/NL, was approximately one‐fifth that of wild‐type HBV produced from the same cell line bearing a plasmid that produces wild‐type HBV. Because NL is approximately 100‐fold brighter than firefly (Photinus pyralis) or Renilla reniformis luciferase, HBV/NL allows the quantitative analysis of the HBV life cycle with high sensitivity and overcomes the low virus production for the quantitative analysis of HBV life cycle and high throughput screening.
NanoLuc activity of HBV/NL in virus‐infected cells represents the amount of cellular HBV RNA, especially RNA transcribed from the preCore promoter.5 Thus, this system is applicable for the analysis of various aspects of HBV entry/replication even to the level of HBV RNA. Indeed, using HBV/NL, we identified host factors that affected the entry of HBV and the transcription of its life cycle.5 We used this advantage of HBV/NL to study HBV replication. We evaluated the role of HBV X protein in the early events of HBV life cycle and the effect of dimethyl sulfoxide on HBV infection and its proliferation, as well as monitoring HBV susceptible cells. We also constructed a reporter HBV bearing the entire pol coding sequence and examined its use in the HBV life cycle.
2. MATERIALS AND METHODS
2.1. Cells
Cells used in this work are described previously.5 Briefly, human hepatocyte‐derived cell lines were cultured in DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg⁄mL streptomycin and 100 U/mL nonessential amino acids (Life Technologies). HepG2/NTCP#22 and HuH7/NTCP cells are HepG2‐derived and HuH7‐derived cell lines transduced by pcNTCP‐myc. Primary human hepatocytes (PHH), PXB cells, isolated from urokinase‐type plasminogen activator transgenic/SCID mice inoculated with PHH (PhoenixBio, Hiroshima, Japan), were cultured in the medium according to the manufacturer's protocol.
2.2. Plasmids
Hepatitis B virus genotype C cloned into pUC19, pUC1.2xHBV, was used (accession number AB246345). pHBV/NL was constructed as described previously5 with slight modification. The 540 nucleotide DNA fragment containing the NL gene from pNL2.1 N1061 NLuc (Promega, Madison, WI, USA) was substituted with a region of 587 nucleotides from 224 to 810 of the pregenome in pUC1.2xHBV. pHBV/NL(S+pol) was obtained by deleting 224 to 505 of the pgRNA coding region from the above plasmid. pHBV/NL(S+polS) has a deletion from 1192 to 1424 in the pgRNA of pHBV/NL(S+pol). pHBV/secNL was generated by substituting the NL gene of pHBV/NL with a DNA fragment of 588 bases carrying the NL gene flanked with a signal sequence fragment, GGAGTCAAAGTTCTGTTTGCCCTGATCTGCATCGCTGTGGCCGAGGCC, of GLuc. pHBV/GLuc was obtained by substitution of the NL gene from HBV/NL with a DNA fragment of 576 bases that carried the GLuc gene (pTK GLuc; New England Biolabs, Tokyo, Japan). A summary of plasmid structures is shown in Figure 1. pUC1.2xHBV/D was described previously5 and is renamed pHBV/D in this study. pHBV/D/MDH and pHBV/D/MHD are mutated forms of pHBV/D generated by introducing a mutation in the amino acid sequence of the catalytic domain of the pol gene: methionine‐asparagine‐asparagine (MDD) to methionine‐asparagine‐histidine (MDH) or methionine‐histidine‐asparagine (MHD), respectively. Premature termination of the X gene in pHBV/NL was generated by changing cytosine to thymine at 2827, and at both 2827 and 3117 in the pgRNA coding sequence to produce pHBV/NL‐X ter1 and pHBV/NL‐X ter2, respectively. These mutations do not alter pol coding amino acid residues. HBV/NL‐X ter1 and HBV/NL‐X ter2 have putative ORF for 7 amino acid residues from the initiator codon and for 76 or 25 amino acid residues from the second methionine in the wild‐type X coding frame, respectively. The HBV X expressing plasmid, pCAG X, was constructed by inserting the entire X coding sequence of HBV downstream of the CAG promoter.
Figure 1.
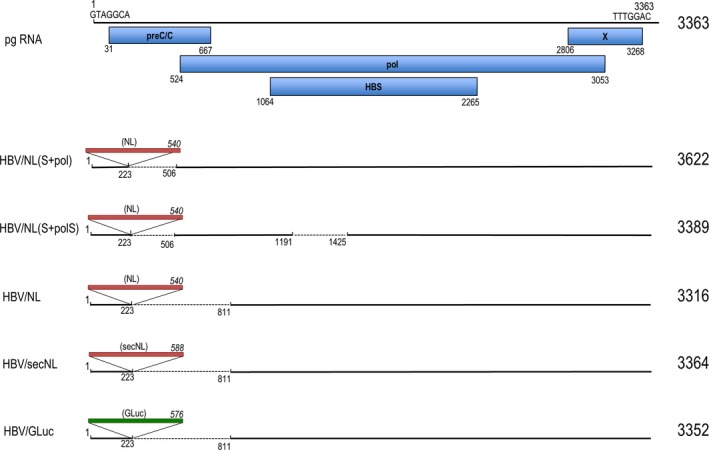
Schematic diagram of reporter hepatitis B virus generated in this study. pgRNA contains 3363 nucleotides. Nucleotide sequences of seven bases at both ends in pgRNA are shown. Open reading frames shown with blue rectangles indicate nucleotide numbers of the initiation and the C‐terminal in each protein. Red rectangles indicate the NanoLuc gene or secNL gene with several flanking nucleotide sequences. GLuc gene containing DNA is shown as a green rectangle. Dotted lines indicate deleted sequences in the pgRNA. Insertion site of the reporter genes is between 223 and 224 of pgRNA. Nucleotide numbers of the 5′ and 3′ end of the deleted sequences in pgRNA are also shown. Numbers in italics represent the DNA size of the inserted genes. Numbers on the right indicate the size of the recombinant pgRNA
2.3. Production of recombinant virus
Plasmids encoding the HBV genome carrying a reporter gene were co‐transfected with a helper virus expressing plasmid(s) to HepG2 or HuH7 cells using Lipofectamine 3000 (Life Technologies). Preparation of virus in medium was described previously.5
2.4. Infection and assay of NanoLuc activity
Cells were infected with HBV or with reporter HBV (10‐100 DNA copies quantitated by PCR per cell, unless otherwise described) in the presence of 4% PEG8000 (Sigma‐Aldrich, Tokyo, Japan) and 2% DMSO for one day and then the culture medium was changed to medium containing 2% DMSO. An aliquot of culture medium or cell lysate was used directly to analyze NL activity and cell viability using the NL Luciferase Assay Kit and CellTiter‐Glo Luminescent Cell Viability Assay Kit (Promega) according to the manufacturer's protocols. GLuc activity was analyzed by the BioLux Gaussia Luciferase Assay Kit (New England BioLabs) according to the manufacturer's protocols. Primer sequences for RT‐PCR to quantitate HBV RNA were 5′‐CCTCTGCCTAATCATCTCATGTTC‐3′ (forward) and 5′‐CGGTGTCGAGGAGATCTCGAATAG‐3′ (reverse).
Human hepatitis B immunoglobulin (Hebsublin) was purchased from Benesis, Japan. Interferon beta was from Mochida Pharmaceutical, Japan. Adefovir was from Toronto Research Chemicals, Canada. Tenofvir and lamivudine were from Tokyo Kasei, Japan. Entecavir and heparin were from Sigma‐Aldrich. Data represent the mean ± SD of three independent experiments.
3. RESULTS
3.1. Selection of hepatitis B virus susceptible cells using hepatitis B virus/NanoLuc
Hepatitis B virus/NanoLuc mimics the wild‐type HBV life cycle and was used to analyze early stages of HBV life cycle.5 We used this reporter virus to select HBV susceptible cell lines. Several human hepatocyte‐derived cell lines were chosen and HeLa cells were used as a negative control. Most cell lines were negative for HBV infection but after the ectopic expression of NTCP, some cell lines became susceptible to infection (Figure 2A). By analyzing HepG2/NTCP cell clones using end‐point dilution, we observed differences in HBV/NL infectivity for each clone (Figure 2B). However, infectivity did not strictly correlate with NTCP expression detected by western blot (Figure 2C), which suggests requirement of an additional factor(s) for HBV infection. HepG2 and HuH7 cells transduced by NTCP‐myc were examined for susceptibility to HBV infection. We obtained data consistent with that using HBV/NL (Figure 2D). The expression of NTCP‐myc was higher in HepG2 than HuH7 (Figure 2D), indicating that high infection to HuH7/NTCP‐myc is not a consequence of high NTCP‐myc expression. HepG2/NTCP#22 cell clone among NTCP‐myc expressing clones had the highest NL activity upon HBV/NL infection and, therefore, this cell clone was mainly used in this work.
Figure 2.
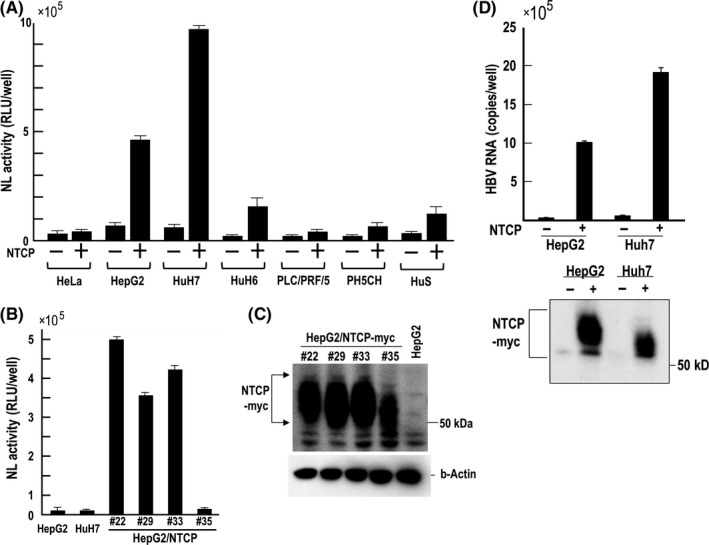
Analysis of human hepatocyte‐derived cell lines and HeLa cells for hepatitis B virus/NanoLuc (HBV/NL) infectivity. HeLa, HepG2, HuH7 and PLC/PRF/5 were obtained from ATCC. PH5CH and HuS cells are described elsewhere.23, 24 (A) Each cell line (−) and NTCP transduced cell line (+) was infected with HBV/NL, and NL activity was measured 6 days after infection. (B) A single cell clone of NTCP‐myc gene transduced HepG2 cells was obtained by end‐point dilution. After 6 days of HBV/NL infection to each cell clone, NL activity was measured. (C) Western blot of NTCP‐myc using the anti‐Myc antibody and of β‐actin. Plural bands reflect modification in NTCP. (D) Infection of HBV to NTCP‐myc transduced HepG2 and HuH7 as well as western blot to detect expression of NTCP‐myc. At 6 days after infection, HBV RNA in cell lysates was measured by RT‐PCR. Detection of NTCP‐myc was conducted using anti‐Myc antibody
3.2. Evaluation of the effect of DMSO on hepatitis B virus/NanoLuc entry and post‐entry events
The addition of DMSO and PEG in culture medium increases the infection of HBV.13, 14 DMSO increases HBV attachment to the cell surface8 and increases NTCP levels in NTCP‐transduced HepG2 cells.9 We analyzed the effect of DMSO by treating cells with DMSO during two steps of virus infection: entry and/or post‐entry steps (Figure 3). A control experiment demonstrated that HepG2 and HuH7 were barely infected with HBV/NL in the presence or absence of DMSO. A slight increase of NL activity was observed in PXB and HepG2/NTCP#22 in the absence of DMSO. However, the presence of DMSO during the entire periods of virus entry and post‐entry sharply increased NL activity in PXB and HepG2/NTCP#22. The absence of DMSO during the virus entry period significantly reduced NL activity in cells at post‐infection periods. The presence of DMSO in the virus entry period but not in the post‐entry period further reduced NL activity. Decreased HBV/NL entry in the absence of DMSO in PXB may be explained by a reduction of NTCP similar to that of HepG2/NTCP.9 Interestingly, we observed only a 20‐30% reduction of NL activity in HepG2/NTCP#22 when DMSO was depleted during the entry period, but nearly a 70% reduction of NL activity was observed in PXB cells under the same protocol of DMSO treatment. We analyzed whether transcription of the NTCP gene was altered by DMSO. PXB and HepG2/NTCP#22 were treated with 2% DMSO for 24 hours and the amount of NTCP RNA was evaluated by RT‐PCR. DMSO did not affect the level of NTCP (Figure S1). Because the amount of NTCP in HepG2/NTCP#22 was not affected by DMSO, this suggests that endogenous NTCP produced in PXB was also not affected by DMSO (data not shown). Cellular factors including genes for drug‐metabolizing enzymes as well as hepatocyte nuclear factors (HNF) and CCAAT‐enhancer‐binding proteins (C/EBP) that affect cell differentiation are also affected by DMSO,10, 11 and some of these factors are necessary for HBV gene transcription.12 Therefore, HBV/NL may be useful to clarify as yet unidentified functions of DMSO on HBV proliferation.
Figure 3.
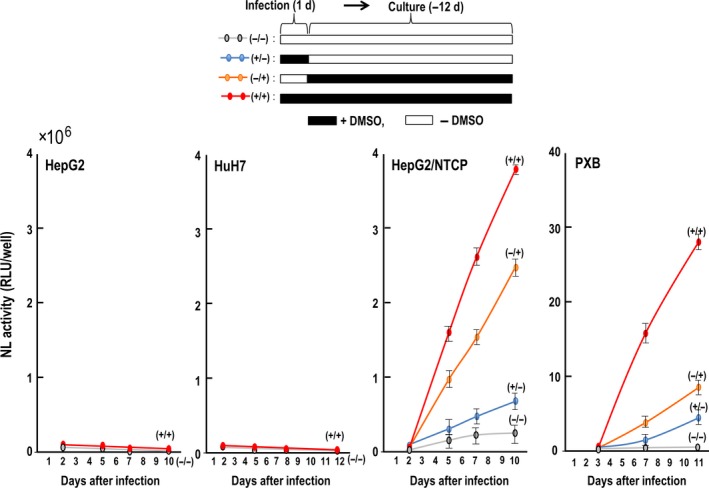
Effect of DMSO on hepatitis B virus/NanoLuc (HBV/NL) infection. The experimental protocol is shown at the top. HBV/NL was used to infect cells for 24 h in the presence of 4% PEG with or without 2% DMSO. Then, the medium was changed to new medium with or without 2% DMSO in the presence of PEG. HepG2 and HuH7 cells were used as a negative control
3.3. Hepatitis B virus X enhances hepatitis B virus/NanoLuc infection
It is well documented that HBV X protein, HBV X, exerts various effects on HBV infection/replication and the fate of HBV infected cells.12 We examined the effect of HBV X by taking advantage of the HBV/NL system. We prepared X‐mutated HBV/NL, HBV/NL‐X ter1 and HBV/NL‐X ter2, none of which produces HBV X. Viruses were rescued from pHBV/NL‐X ter1 and pHBV/NL‐X ter2 by transfecting them into HepG2 together with pHBV/D. HepG2/NTCP#22 was infected with each virus fraction and NL activity was measured after 5 days of infection. NL activity of HBV/NL‐X ter1 and HBV/NL‐X ter2 were 37 and 25% that of HBV/NL, respectively (Figure 4). However, by infecting HepG2/NTCP#22 transduced with viruses containing the HBV X producing plasmid, pCAG X, NL activity increased 1.5‐4‐fold. This observation indicates that HBV X enhances HBV/NL infection/replication. Because HBV X exerts its function by associating with the HBV minichromosome to trans‐activate transcription from HBV cccDNA,15 enhanced NL activity by ectopic HBV X production may reflect such an event. Therefore, this assay system might also allow the analysis of an as yet unidentified function of HBV X in the HBV life cycle.
Figure 4.
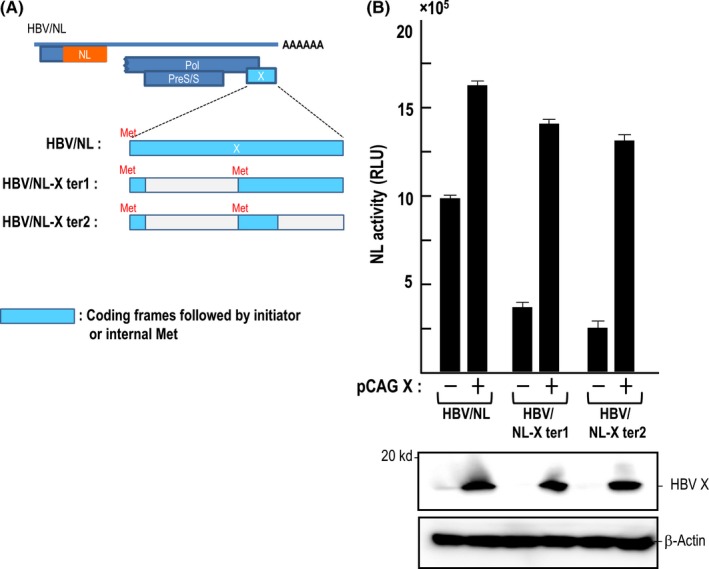
Effect of hepatitis B virus (HBV) X gene expression on the NanoLuc (NL) activity of cells infected with HBV/NL and its derivatives. (A) Amino acid in the protein X open reading frame (ORF) in the HBV/NL genome was mutated as described in the Materials and Methods. Putative ORF in the mutated X gene are shown in blue. (B) Suppressed NL activity of HBV/NL derivatives in infected cells was enhanced by ectopic expression of the HBV X gene. Viruses were used to infect HepG2/NTCP#22 cells (−) or HepG2/NTCP#22 cells transfected with pCAG X 1 day before infection (+). NL activity was measured at day 6 after infection. Western blot of HBV X and β‐actin is shown
3.4. Construction of a reporter hepatitis B virus that secretes a reporter protein into culture medium upon infection
Hepatitis B virus/NL infected cells produce NL in cells. Thus, preparation of cell lysate to measure NL activity is required. Recombinant HBV that produce a secreting reporter protein may contribute to kinetic analysis of virus replication without killing virus infected cells. We prepared reporter HBV that secrete reporter protein in culture medium as described in the Materials and Methods. The amounts of HBV RNA in cell lysates infected with GLuc and secNL expressing reporter HBV were nearly 80% of that in cells infected with HBV/NL (Figure 5A). We conducted time course analysis of reporter activity and of HBV RNA to determine any correlations (Figure 5B,C). We found that NL activity and GLuc activity produced from cells infected with HBV/secNL and HBV/GLuc, respectively, correlated with the level of reporter HBV RNA in cells infected with the relevant reporter viruses (Figure 5B,C).
Figure 5.
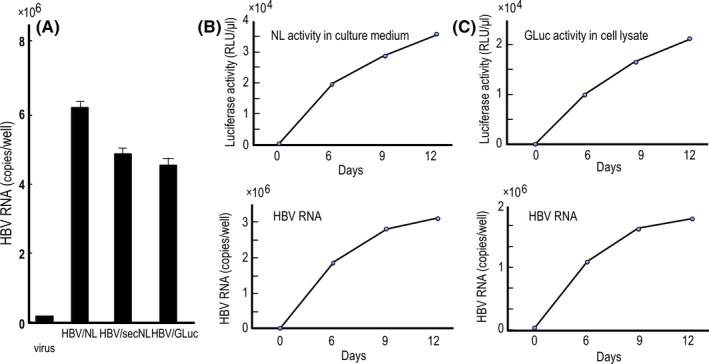
Comparative analysis of the RNA level of reporter virus in cells infected with hepatitis B virus/NanoLuc (HBV/NL), HBV/secNL and HBV/GLuc as well as analysis of the correlation of reporter activity with virus RNA. (A) Relative HBV RNA level in cells infected with HBV/NL, HBV/secNL and HBV/GLuc. Recombinant viruses equivalent to 100 DNA copies per cell were infected to 1 × 106 HepG2/NTCP#22 cells. RNA in cell lysates after 6 days of infection were analyzed for HBV RNA by quantitative RT‐PCR. (B,C) Time course‐dependent measurement of the relative amount of reporter activity and RNA of the reporter HBV in infected cells. One hundred copies of HBV/secNL (B) or HBV/GLuc (C) per cell were used to infect 1 × 106 HepG2/NTCP#22 cells. At the indicated days, culture medium of HBV/secNL infected cells or HBV/GLuc infected cells were measured for reporter activity. Simultaneously, virus RNA in a 1/10 volume of cell lysate was quantitated by RT‐PCR using the primer set described in the Materials and Methods
3.5. Production of infectious reporter virus bearing the expression of virus polymerase
Hepatitis B virus/NanoLuc is useful for analyzing the early stage of HBV life cycle. However, it is expected that HBV/NL can also be used to evaluate the entire HBV cycle by co‐production of HBV core and pol genes in HBV/NL infected cells. In fact, our preliminary work showed the production of recombinant HBV/NL in PXB cells when co‐infected with HBV (Figure S2). It is thought that the association of hepadnavirus pgRNA with polymerase followed by reverse transcription of HBV pgRNA is efficient if polymerase is supplied in a cis‐acting manner.16 In this process, co‐translated polymerase has a more efficient role compared with being supplied in trans from a helper virus. If this is the case, the efficiency of progeny virus production from HBV/NL may be low because of the lack of a pol gene. We constructed a plasmid to produce recombinant HBV carrying the entire pol gene and NL gene, HBV/NL(S+pol) (Figure 1). HBV pol gene with a deletion in its spacer region was obtained from a patient treated with adefovir.13 This deleted pol is functional in regard to catalyzing the synthesis of HBV DNA.13 Thus, we also constructed pHBV/NL(S+polS) that has a deletion in the spacer region (Figure 1).
The expression of virus RNA from plasmids transfected into HepG2 cells is shown in Figure 6A. Cells transfected with the plasmid expressing wild‐type HBV produced virus RNA with the expected size corresponding to 3.3 kb (pgRNA) and 2.1‐2.4 kb (HB RNA). X gene RNA was visible after long exposure (data not shown). The size of the pgRNA of HBV/NL was similar to that of wild‐type HBV. However, pgRNA transcribed from pHBV/NL(S+pol) was longer than that of wild‐type HBV, in agreement with the expected sizes of reporter genomes encoded in the recombinant plasmids.
Figure 6.
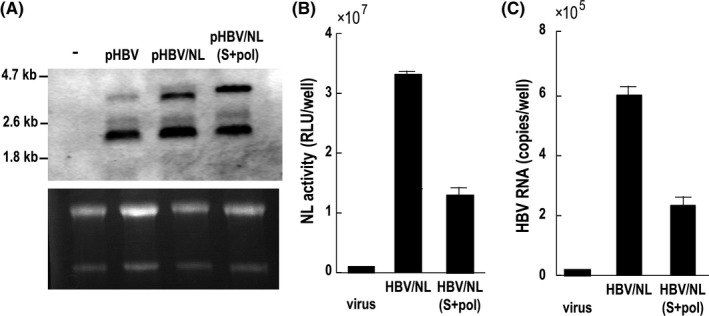
Comparative analysis of virus RNA in transfected cells and the amount of viruses produced from those cells. (A) Six days after transfection, 30 μg RNA from wild pHBV, pHBV/NL and pHBV/NL(S+pol)‐transfected cells was analyzed by northern blot using digoxigenin (DIG) HBV RNA probes covering the full‐length HBV genome. The gel was stained with ethidium bromide to visualize ribosomal RNA. The efficiency of transfection was similar (data not shown). (B,C) Six days after transfection by wild pHBV, pHBV/NL and pHBV/NL(S+pol), virus fractions from culture medium were harvested. One‐tenth of the volume of culture medium was used to infect HepG2/NTCP#22. NL activity and the amount of virus RNA in cell lysates in (B) and (C), respectively, were measured after 6 days of infection. HBV, hepatitis B virus; NL, NanoLuc
Amounts of pgRNA produced from plasmids expressing HBV/NL and HBV/NL(S+pol) were similar (Figure 6A). We analyzed the amount of reporter HBV secreted into culture medium from HepG2 transfected with HBV/NL and HBV/NL(S+pol). One‐tenth of the volume of culture medium from cells was used to infect HepG2/NTCP#22 cells. NL activity as well as HBV RNA in cell lysates were measured 6 days after infection. The amount of NL activity of HBV/NL(S+pol) was approximately 30% of that of HBV/NL and this ratio coincided with the relative amount of HBV RNA (Figure 6C,D), indicating that production of HBV/NL(S+pol) was one‐third of that of HBV/NL. It is likely that the size of the HBV/NL(S+pol) genome affects the productivity of the recombinant virus.
3.6. Verification of hepatitis B virus polymerase activity in hepatitis B virus/NanoLuc (S+pol) and hepatitis B virus/NanoLuc (S+polS)
We examined whether polymerase in pHBV/NL(S+pol) and pHBV/NL(S+polS) was functional with regard to producing infectious recombinant viruses. First, pHBV/NL was transfected with a helper plasmid, pHBV/D, pHBV/D/MDH or pHBV/D/MHD. pHBV/D/MDH and pHBV/D/MHD have mutations in the catalytic domain of the polymerase gene and, therefore, do not produce functional polymerase. Production of infectious HBV/NL in supernatants was analyzed by infecting HepG2/NTCP#22 with supernatant followed by measuring NL activity in cell lysates. Infectious virus was detected in culture medium obtained from cells transfected with pHBV/NL and pHBV/D but not from cells transfected with pHBV/NL and pHBV/D/MDH or pHBV/D/MHD (Figure 7), indicating that pHBV/D/MDH or pHBV/D/MHD have defective polymerase. However, cells transfected with pHBV/NL(S+pol) together with pHBV/D, pHBV/D/MDH or pHBV/D/MHD produced infectious viruses, although the level of NL activity of the recovered viruses from pHBV/D/MDH or pHBV/D/MHD was 75‐80% of that produced from cells transfected with pHBV/D. Moreover, HepG2 cells transfected with pHBV/NL(S+polS) and pHBV/D or pHBV/D/MDH produced infectious reporter HBV. These results indicate that polymerase produced from pHBV/NL(S+pol) or pHBV/NL(S+polS) is functional. This also suggests that HBV/NL(S+pol) and HBV/NL(S+polS) can repeat the virus replication cycle if HBV proteins not expressed in these viruses are supplemented.
Figure 7.
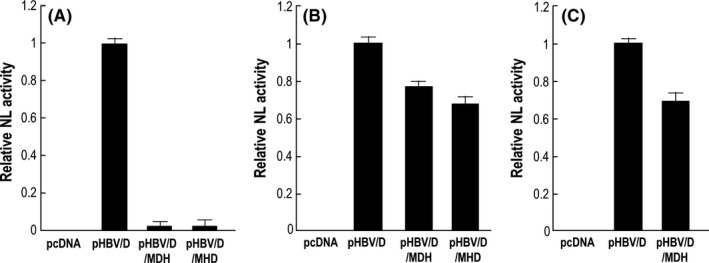
Production of reporter hepatitis B virus (HBV) by co‐transfecting pHBV/D or a helper HBV lacking polymerase activity. HepG2 cells were transfected with pHBV/NL (A), pHBV/NLS+pol (B) and pHBV/NLS+polS (C) with respective helper plasmids that expressed the entire virus proteins (pHBV/D), or expressed all proteins except pol (pHBV/MDH and pHBV/MHD). Six days after transfection, the virus fraction was harvested and the same volume of virus was used to infect HepG2/NTCP#22 cells. Six days after infection, NL activity was analyzed and indicated by relative NanoLuc activity. NL, NanoLuc
3.7. Production of infectious reporter hepatitis B virus under co‐existence of helper hepatitis B virus
Deletion of the core/pol coding sequence in HBV/NL, deletion of a core coding sequence in HBV/NL(S+pol), and deletion of core and HB coding sequences in HBV/NL(S+polS) demonstrated the lack of a complete replication cycle by these reporter viruses alone. However, these reporter HBV may mimic the entire life cycle of wild‐type HBV under the condition of a supply of HBV proteins that are not present in the reporter viruses. Based on this assumption, human primary hepatocytes (PXB cells) were infected with HBV/NL, HBV/NL(S+pol) and HBV/NL(S+polS) in the presence or absence of wild‐type HBV. After thoroughly washing wells with PBS to remove the remaining residual inoculum, the culture was continued. Virus fractions from the medium were used to infect HuH7/NTCP. Cell lysates at days 4, 7, 12, 16 and 22 after infection were prepared to measure NL activity.
The relative NL activities in the lysate from each reporter HBV infected cells are shown in Figure 8. NL activity in HuH7/NTCP infected with virus fractions obtained from PXB co‐infected with wild‐type HBV reached a plateau at 10 days and remained for more than 3 weeks. However, no NL activity was detected in HuH7/NTCP infected with the virus fraction from culture medium obtained from PXB cells without co‐infection with wild HBV (Figure 8). When cells were treated with 80 nM entecavir at day 16 of infection, NL activity was reduced thereafter. This suggests that NL activity in infected cells was the consequence of a balance between the levels of transcription and the reduced amount of virus cccDNA by entecavir. Taken together, these data indicate that infectious reporter viruses from reporter HBV propagated in PXB cells in the presence of wild‐type HBV were secreted into culture medium and infected naive cells.
Figure 8.
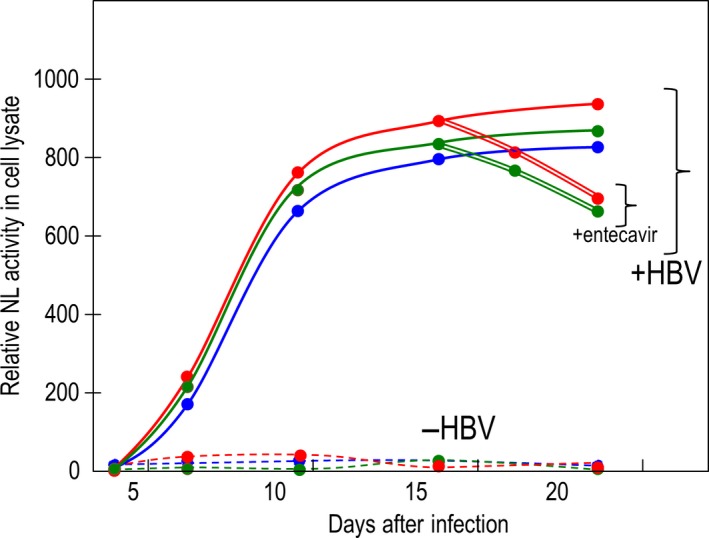
Trans‐rescue experiment of reporter hepatitis B virus (HBV) production. Reporter viruses were harvested from HepG2 transfected with the indicated plasmids together with pHBV/D. The viruses were used to infect 2 × 105 PXB cells in the presence or absence of 100 copies per cell of HBV obtained from primary human hepatocytes (PHH) maintained in urokinase‐type plasminogen activator transgenic/SCID mice. Virus was prepared from the medium and used to infect 1 × 105 HuH7/NTCP cells. NanoLuc (NL) activity was measured at the indicated times. On day 16, an aliquot of the cells was treated with 80 nmol/L entecavir. Solid lines indicate relative NL activity in HuH7/NTCP infected with culture medium recovered from PXB cells infected with HBV/NL (red), HBV/NLS+pol (green) and HBV/NLS+polS (blue) together with wild‐type HBV. Dotted lines indicate relative NL activity in cells infected with culture medium recovered from PXB cells infected with HBV/NL (red), HBV/NLS+pol (green) and HBV/NLS+polS (blue) without wild‐type HBV. Double lines with red (HBV/NL) and with green (HBV/NLS+pol) show NL activity after entecavir treatment. Experiments were conducted twice and the mean value is plotted in this figure
4. DISCUSSION
Previously, we generated a reporter HBV, HBV/NL, which expressed the NL gene upon infecting target cells. Therefore, HBV/NL can be used to analyze quantitatively the HBV life cycle with high sensitivity. To construct HBV/NL and its derivatives, the upstream pol coding sequence was deleted. Because this region is part of the core gene and AUG codons for methionine in this region are utilized as IRES functions for translation of the pol gene,14, 17, 18 it was uncertain whether polymerase in HBV/NL(S+pol) and HBV/NL(S+polS) would be expressed at a sufficient amount to produce progeny virus even if they lacked the authentic AUG codons to generate IRES function for translation of pol. To address this, we transduced HepG2 with plasmids that produced reporter viruses together with helper plasmids that produced all virus proteins except functional polymerase. HBV/NL(S+pol) and HBV/NL(S+polS) were produced by cells co‐transfected with the relevant plasmids together with helper plasmids that lacked production of functional polymerase. This result clearly showed that deletion of the upstream pol coding sequence in these reporter HBV did not interfere with polymerase production. Translation of the pol gene is likely to utilize the IRES‐like function using AUG codons in the C‐terminal region of the core protein.14, 17, 18 However, because the C‐terminal core sequence was deleted in HBV/NL and its derivatives, these reporter HBV cannot utilize authentic IRES‐like functions. NL gene‐derived AUG codons are followed by short ORF at 371 and 200 nucleotides upstream from the initiation codon of the pol gene in the genomes of HBV/NL(S+pol) and HBV/NL(S+polS). It is not known whether these AUG codons in the NL gene can generate a new IRES‐like function to translate the polymerase gene. It may be necessary to explore this possibility or to examine the presence of another mechanism of pol translation in HBV/NL and its derivatives, which may contribute to clarifying the translational mechanism of polymerase in wild‐type HBV.
It is thought that the association of hepadnavirus pgRNA with polymerase followed by reverse transcription of HBV pgRNA is efficient if polymerase is supplied in a cis‐acting manner.16 In this process, co‐translated polymerase has a more efficient role compared with being supplied in trans from a helper virus. If this is the case, the efficiency of progeny virus production from HBV/NL may be lower than the production of HBV/NL(S+pol) and HBV/NL(S+polS) because HBV/NL lacks pol expression whereas HBV/NL(S+pol) and HBV/NL(S+polS) produce functional polymerase themselves. We examined the production of HBV/NL, HBV/NL(S+pol) and HBV/NL(S+polS) under the presence of wild‐type HBV (Figure 8) and found all reporter viruses were produced. In the absence of wild‐type HBV, no reporter virus production was observed. Treatment of entecavir suppressed NL activity indicating NL activity is a consequence of transcription from the HBV cccDNA that was reduced by entecavir.
Interestingly, there was no significant difference in the kinetics of NL activity among HBV/NL, HBV/NL(S+pol) and HBV/NL(S+polS) (Figure 8). This suggests that supplying polymerase in a cis‐acting or trans‐acting manner does not affect the production of progeny viruses from these reporter HBV, indicating that the efficiency of HBV replication does not depend on the method of supplying polymerase. HBV does not require another sequence in addition to the sequence termed “epsilon” for the encapsidation of pgRNA.19 However, duck hepatitis B virus (DHBV) requires the epsilon sequence as well as its downstream sequence for encapsidation.20 This suggests that the mechanism of the encapsidation of pgRNA may be influenced by the downstream sequence of epsilon in the HBV genome and may affect the efficiency of encapsidation. Our reporter HBV lacked some sequences between epsilon and the N‐terminal region of the pol gene. Therefore, the encapsidation reaction in our reporter HBV might be modified and may affect the efficiency of pgRNA encapsidation.
Alternatively, if the encapsidation reaction of pgRNA is rate limited by the amount of polymerase, supplying an excess amount of polymerase from a helper virus might enhance the efficiency of the trans‐acting encapsidation reaction.
Hepatitis B virus X has a pivotal function in the transcription of virus RNA from HBV cccDNA.21 Without the expression of HBV X, transcription is barely detectable. However, HBV/NL lacking X expression showed NL activity that was one‐third or one‐quarter that of HBV/NL in HepG2/NTCP#22. It is not known whether cellular factors that suppress transcription of HBV RNA are not recruited to the HBV/NL cccDNA nucleosome structure22 or whether HBV X incorporated into recombinant virus functions to support transcription. Further work is needed to clarify this point.
In summary, HBV/NL and its derivatives described here are useful for screening anti‐HBV agents as well as exploring a variety of HBV replication strategies.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
Supporting information
ACKNOWLEDGMENTS
We thank Ms Hiromi Yamamoto and Ritsuko Shiina for technical assistance. This work was partly supported by Grants‐in‐Aid for Scientific Research from the Ministry of Health, Labor, and Welfare of Japan, by Grants‐in‐Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the Research Program on Hepatitis from Japan Agency for Medical Research and Development, AMED.
Nishitsuji H, Harada K, Ujino S, et al. Investigating the hepatitis B virus life cycle using engineered reporter hepatitis B viruses. Cancer Sci. 2018;109:241–249. https://doi.org/10.1111/cas.13440
REFERENCES
- 1. WHO . Hepatitis B. WHO Hepatitis B Fact sheet no 204, 2008.
- 2. Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res. 2008;134:235‐249. [DOI] [PubMed] [Google Scholar]
- 3. Gripon P, Rumin S, Urban S, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Nat Acad Sci U S A. 2002;99:15655‐15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishitsuji H, Ujino S, Shimizu Y, et al. Novel reporter system to monitor early stages of the hepatitis B virus life cycle. Cancer Sci. 2015;106:1616‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Protzer U, Nassal M, Chiang PW, Kirschfink M, Schaller H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild‐type virus infection. Proc Nat Acad Sci U S A. 1999;96:10818‐10823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Z, Wu L, Cheng X, et al. Replication‐competent infectious hepatitis B virus vectors carrying substantially sized transgenes by redesigned viral polymerase translation. PLoS One. 2013;8:e60306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paran N, Geiger B, Shaul Y. HBV infection of cell culture: evidence for multivalent and cooperative attachment. EMBO J. 2001;20:4443‐4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J, Zong L, Sureau C, Barker L, Wands JR, Tong S. Unusual features of sodium taurocholate cotransporting polypeptide as a hepatitis B virus receptor. J Virol. 2016;90:8302‐8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi S, Sainz B Jr, Corcoran P, Uprichard S, Jeong H. Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)‐treated Huh7 hepatoma cells. Xenobiotica. 2009;39:205‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizuguchi T, Mitaka T, Hirata K, Oda H, Mochizuki Y. Alteration of expression of liver‐enriched transcription factors in the transition between growth and differentiation of primary cultured rat hepatocytes. J Cell Physiol. 1998;174:273‐284. [DOI] [PubMed] [Google Scholar]
- 12. Seeger C, Zoulim F, Mason W. Hepadonaviruses In: Knipe D, Howley P, eds. Fields Virology, 6th edn New York, NY: Wolters Kluwer; 2013:2185‐2221. [Google Scholar]
- 13. Hong R, Bai W, Zhai J, et al. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J Virol. 2013;87:6615‐6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang LJ, Ganem D, Varmus HE. Mechanism of translation of the hepadnaviral polymerase (P) gene. Proc Nat Acad Sci U S A. 1990;87:5158‐5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Belloni L, Pollicino T, De Nicola F, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Nat Acad Sci U S A. 2009;106:19975‐19979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang MJ, Summers J. Infection initiated by the RNA pregenome of a DNA virus. J Virol. 1991;65:5435‐5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ou JH, Bao H, Shih C, Tahara SM. Preferred translation of human hepatitis B virus polymerase from core protein‐ but not from precore protein‐specific transcript. J Virol. 1990;64:4578‐4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fouillot N, Tlouzeau S, Rossignol JM, Jean‐Jean O. Translation of the hepatitis B virus P gene by ribosomal scanning as an alternative to internal initiation. J Virol. 1993;67:4886‐4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Junker‐Niepmann M, Bartenschlager R, Schaller H. A short cis‐acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990;9:3389‐3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ostrow KM, Loeb DD. Chimeras of duck and heron hepatitis B viruses provide evidence for functional interactions between viral components of pregenomic RNA encapsidation. J Virol. 2004;78:8780‐8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lucifora J, Arzberger S, Durantel D, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. 2011;55:996‐1003. [DOI] [PubMed] [Google Scholar]
- 22. Murphy CM, Xu Y, Li F, et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016;16:2846‐2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noguchi M, Hirohashi S. Cell lines from non‐neoplastic liver and hepatocellular carcinoma tissue from a single patient. In Vitro Cell Dev Biol Anim. 1996;32:135‐137. [DOI] [PubMed] [Google Scholar]
- 24. Aly HH, Watashi K, Hijikata M, et al. Serum‐derived hepatitis C virus infectivity in interferon regulatory factor‐7‐suppressed human primary hepatocytes. J Hepatol. 2007;46:26‐36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials