

Abstract
Gemcitabine‐based therapy remains the mainstay of treatment for patients with biliary tract cancers (BTCs) with no second‐line treatment(s) established yet. Aberrant activation of the MAPK pathway in patients with BTC indicates its importance in BTC. Trametinib is a potent, highly selective, allosteric non‐competitive inhibitor of MEK1/MEK2. In this phase IIa open‐label, single‐arm study, we investigated the efficacy and safety of trametinib in Japanese patients with advanced BTC refractory to gemcitabine‐based therapy. All patients received oral trametinib 2 mg once daily until progressive disease (PD), death, or unacceptable toxicity. The primary objective was to determine the 12‐week non‐PD rate. Secondary assessments included safety, progression‐free survival (PFS), overall survival, and overall response rate. Targeted exome sequencing was used to identify biomarkers for sensitivity or resistance to trametinib monotherapy. Twenty patients (median age, 61.5 years) with carcinoma of gall bladder (40%), intrahepatic (25%) or extrahepatic (30%) bile duct, and ampulla of Vater (5%) were enrolled. The non‐PD rate at week 12 was 10% (95% confidence interval, 1.2‐31.7); it did not reach the threshold rate of 25%. Median PFS was 10.6 weeks (95% confidence interval, 4.6‐12.1) and 1‐year overall survival was 20.0%. Stable disease and PD were observed in 13 (65%) and seven (35%) patients, respectively. No new safety signals were reported. Although the primary end‐point was not met, prolonged PFS was observed in one patient having six somatic variants including synonymous NF1 exon 12 splice variant and a loss‐of‐function variant in ARID1A. Efforts to understand responsive mutations and sensitivity to targeted therapies are warranted. This trial was registered with ClinicalTrials.gov: NCT01943864.
Keywords: biliary tract cancer, gemcitabine, MEK inhibitor, refractory, trametinib
Abbreviations
- AE
adverse event
- BTC
biliary tract cancer
- CI
confidence interval
- OS
overall survival
- PD
progressive disease
- PFS
progression‐free survival
- PR
partial response
- RR
response rate
- S‐1
tegafur
- SAE
serious adverse event
- SD
stable disease
1. INTRODUCTION
Biliary tract cancer (BTC) is a highly fatal malignancy that arises from the epithelial lining of the gallbladder and bile ducts. It encompasses intrahepatic, perihilar, and extrahepatic cholangiocarcinoma as well as carcinomas arising from the gallbladder, and is characterized by regional lymph node metastasis, vascular encasement, and distant metastasis.1 Biliary tract cancer is the sixth leading cause of cancer‐related deaths in Japan.2 The prognosis of BTC is poor because most patients present at advanced stages and are thus diagnosed with unresectable tumors.3, 4 The standard first‐line treatment for locally advanced or metastatic BTC is systemic chemotherapy with cisplatin and gemcitabine.3, 5, 6 The alternative regimen with tegafur (S‐1), an oral fluoropyrimidine, has shown a comparable response rate (RR) as that with gemcitabine.7, 8 Moreover, S‐1 has been investigated as a second‐line treatment for patients with advanced BTC refractory to gemcitabine.9, 10 Molecular targeted agents, such as cetuximab, erlotinib, or sorafenib alone or in combination with chemotherapy have not shown promising results in small Phase Ib and Phase II trials.11 Owing to the limited benefits of these treatments, there are no standard second‐line treatments established for patients with BTC; this highlights the need for a new drug or chemotherapy regimens as a second‐line treatment option.12
Increasing evidence indicates that RAS and RAF proto‐oncogenes are mutated and are thus activated at a significant rate in BTC; this highlights the importance of the RAS/RAF/MEK/ERK (MAPK) pathway in the pathogenesis of this disease.13, 14, 15, 16 So far, two MEK inhibitors, selumetinib and MEK162 (ARRY‐438162), have demonstrated promising results as monotherapy in patients with unresectable, locally advanced, or metastatic BTC.17, 18 Trametinib, another MEK inhibitor, is a potent and highly selective allosteric non‐competitive inhibitor of MEK1/MEK2 activation and kinase activity.19 Preclinical data indicate inhibited cell growth in BTC models with KRAS‐mutated cell lines after treatment with trametinib.20 Therefore, based on the importance of the MAPK pathway in biliary carcinogenesis and potential activity shown by MEK inhibitor in the treatment of BTC, trametinib was investigated as a potential novel single agent treatment for unresectable BTC.
The present phase IIa study was undertaken to investigate the preliminary efficacy and safety of an oral MEK inhibitor, trametinib, in patients with advanced BTC refractory to a gemcitabine‐based regimen.
2. MATERIALS AND METHODS
2.1. Patients
Japanese patients aged ≥20 years with histologically or cytologically confirmed cholangiocarcinoma (intrahepatic or extrahepatic) or gallbladder cancer or ampulla of Vater cancer refractory to gemcitabine‐based anticancer therapy (first‐line chemotherapy), a performance status score of ≤1 on the ECOG scale, and adequate organ function were enrolled. Tumor samples were to be collected either from archived tissue or fresh biopsy for biomarker research before patients were assigned treatment. Measurable disease, that is, presenting with at least one measurable lesion per RECIST 1.1, was required.21
Key exclusion criteria were history of another malignancy, prior use of any MEK inhibitor, radiotherapy within 2 weeks before randomization, symptomatic or untreated leptomeningeal or brain metastases or spinal cord metastases, known HIV infection, and history or evidence of cardiovascular risk.
2.2. Standard protocol approvals, registrations, and patient consent
The protocol and amendments were reviewed and approved by the local independent ethics committees of the participating institutions. The study was carried out in accordance with Good Clinical Practice and ethical principles of the Declaration of Helsinki. All patients provided written informed consent to participate in the study. This study is registered with ClinicalTrials.gov (ClinicalTrials.gov identifier: NCT01943864).
2.3. Study design
This was a phase IIa, open‐label, single‐arm, multicenter study carried out at five centers in Japan. The study comprised a 21‐day screening phase, followed by a treatment phase and a follow‐up phase. All patients were randomized to receive oral trametinib 2 mg once daily and were required to visit the site every 4 weeks (treatment phase). Patients received trametinib until disease progression, death, or an unacceptable AE. Tumor response was evaluated every 8 weeks. All patients who permanently discontinued the study treatment without disease progression were followed up for progression until initiation of new anticancer therapy, disease progression, or death. All patients who permanently discontinued the study treatment were followed up for survival and new anticancer therapy every 8 weeks until death or until the patient had been followed up for 1 year from study initiation (day 1), whichever was earlier.
2.4. Concomitant medication
Supportive care therapies, including blood transfusion, treatment with antibiotics, antiemetics, antidiarrheal medications, and analgesics, were permitted during the study as clinically indicated. Other anticancer therapies (e.g., radiation therapy, surgery, and/or tumor embolization) and use of other investigational drugs were prohibited within 28 days (or five half‐lives, whichever was shorter) preceding the first dose of trametinib and during the study.
2.5. Study objectives
The primary objective of the study was to determine the 12‐week non‐PD rate, defined as the proportion of patients without progression at week 12, as assessed by RECIST 1.1.
Secondary objectives of the study were to assess the safety profile of trametinib and to determine PFS, OS, and overall RR in the overall population. Potential of blood‐ or tumor tissue‐derived biomarkers to predict biological activity and sensitivity or resistance to trametinib treatment were evaluated as exploratory end‐points of the study.
2.6. Targeted exome sequencing
Formalin‐fixed, paraffin‐embedded tumor samples were obtained from patients and macrodissected to enrich for tumor content. DNA was isolated using the QIAamp DNA FFPE kit (Qiagen, Valencia, CA, USA). Target gene regions were captured using a custom TargetSeq 409 cancer‐related gene panel (AltheaDx, San Diego, CA, USA) and sequenced on the Ion Proton System (Thermo Fisher Scientific, Waltham, MA, USA). The Torrent Suite (version 4.0.2; Thermo Fisher Scientific) was used for alignment against the hg19 reference genome as well as for variant calling. Variants were filtered for strand ratio and coverage. Subsequently, the sequences were annotated using annovar (annovar.openbioinformatics.org), Sorting Intolerant From Tolerant (http://sift.bii.a-star.edu.sg), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), Catalogue of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic; Wellcome Trust Sanger Institute, Hinxton, UK), and 1000 Genomes (http://www.internationalgenome.org; European Bioinformatics Institute, Hinxton, UK). Nine samples passed the filters for tumor content, sufficient DNA isolation, and sequencing quality control. A set of 31 genes was selected for validation on the GeneRead sequencing platform (SABiosciences, Qiagen). To distinguish germline from the somatic variants, matched peripheral blood samples were sequenced in parallel to their corresponding tumors.
2.7. Statistical analysis
The study was designed as a single‐arm trial to evaluate the clinical utility of trametinib monotherapy compared with historical single‐agent data for S‐1, a second‐line therapy, and selumetinib, used both as first‐ and second‐line therapy.9, 18 Considering the median PFS with S‐1 and selumetinib was 10.8 and 16.1 weeks, respectively, in the reference study,10, 18 the null hypothesis for threshold and the alternative hypothesis for expected non‐PD rate at week 12 were specified as 25% and 60%, respectively. Based on the null hypothesis and to achieve power >90%, 20 subjects were to be enrolled in the study. Exact binomial 90% and 95% CIs were computed for non‐PD rate, and one‐sided P‐value of the exact binomial test to reject the null hypothesis (≤25%) was provided at a one‐sided 5% level of significance. Progression‐free survival was summarized using Kaplan–Meier curves. The 1‐year OS was estimated using the Kaplan–Meier curves of OS. For overall RR, the exact 95% CI for RR was calculated. The duration of response was summarized for patients with confirmed complete response or partial response (PR). Primary assessments were based on imaging data. Investigator assessments were considered as primary assessments, whereas independent reviewer assessments were considered supplementary. All treated patients were included in the safety evaluations.
3. RESULTS
3.1. Patient demographics and baseline characteristics
In total, 20 Japanese patients were enrolled in this study, all of whom (100%) completed it. At the end of the study, all patients discontinued the study treatment. Primary reasons for treatment discontinuation were disease progression (n = 16, 80%) and AEs (n = 4, 20%; five events including drug‐induced liver injury, small intestinal obstruction, hepatic function abnormal and pyelonephritis, and retinal artery occlusion). Most patients were men (70%), and the median patient age was 61.5 years (range, 50‐83 years). Overall, 14 (70%) patients had an ECOG score of 0. The primary tumor types in most patients were gallbladder (n = 8, 40%) and extrahepatic bile duct (n = 6, 30%). All 20 patients had received prior anticancer therapy (chemotherapy [n = 20, 100%] or surgery [n = 10, 50%]). The most common drugs used as the first‐ (n = 13) and second‐line (n = 5) chemotherapy regimens for malignant tumor were cisplatin and gemcitabine combination therapy (Table 1).
Table 1.
Demographics and baseline characteristics of Japanese patients with advanced biliary tract cancers refractory to gemcitabine
| All treated patients n = 20 | |
|---|---|
| Age, years, n (%) | |
| Median (range) | 61.5 (50–83) |
| Male, n (%) | 14 (70) |
| ECOG at baseline, n (%) | |
| 0 | 14 (70) |
| 1 | 6 (30) |
| Primary tumor type, n (%) | |
| Gallbladder | 8 (40) |
| Extrahepatic bile duct | 6 (30) |
| Intrahepatic bile duct | 5 (25) |
| Ampulla of Vater | 1 (5) |
| Time since diagnosis, days | |
| n | 16 |
| Median (range) | 320 (120–2260) |
| Stage at diagnosis, n (%) | |
| Missing | 1 (5) |
| IV | 2 (10) |
| IVa | 1 (5) |
| IVb | 16 (80) |
| No. of prior chemotherapy regimens, n (%) | |
| 1 | 12 (60) |
| 2 | 8 (40) |
| Prior anticancer regimen, n | |
| First‐line regimen | 20 |
| Cisplatin + gemcitabine | 13 |
| S‐1 | 5 |
| Gemcitabine + S‐1 | 1 |
| Cisplatin + gemcitabine + S‐1 | 1 |
| Second‐line regimen | 7 |
| Cisplatin + gemcitabine | 5 |
| S‐1 | 2 |
| Adjuvant | 1 |
| Cisplatin + gemcitabine | 1 |
S‐1, tegafur.
3.2. Efficacy results
The non‐PD rate at week 12 was 10% (90% CI, 1.8‐28.3; 95% CI, 1.2‐31.7) per investigator assessment (primary) and 15% (90% CI, 4.2‐34.4; 95% CI, 3.2‐37.9) per independent radiologist assessment (Figure S1; Table 2). The primary end‐point did not reach the threshold rate of 25%. The one‐sided P‐value of exact binomial test to reject the null hypothesis was above the prespecified significance level of 0.05 (P = .976).
Table 2.
Non‐progressive disease rate at week 12 in phase IIa study of trametinib in Japanese patients with advanced biliary tract cancers refractory to gemcitabine (all treated patients)
| Investigator assessment n = 20 | Independent assessment n = 20 | |
|---|---|---|
| Response at week 12, n (%) | ||
| CR | 0 (0) | 0 (0) |
| PR | 0 (0) | 0 (0) |
| SD | 2 (10) | 3 (15) |
| Non‐CR/non‐PD | – | 0 (0) |
| PD | 3 (15) | 2 (10) |
| PD before week 12, n (%) | 12 (60) | 11 (55) |
| Censored before week 12,a n (%) | 3 (15) | 4 (20) |
| Non‐PD rate | ||
| CR + PR + SD, n (%) | 2 (10) | 3 (15) |
| 90% CI | (1.8‐28.3) | (4.2‐34.4) |
| 95% CI | (1.2‐31.7) | (3.2‐37.9) |
| P‐value | 0.976 | 0.909 |
–, Not applicable; CI, confidence interval; CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Patients censored before week 12 because of missing response assessment at week 12. Independent radiologist assessment was carried out using same computed tomography scans as those used for investigator assessment.
Progression or death was reported in 18 (90%) and 16 (80%) patients per investigator and independent radiologist assessment, respectively, and the corresponding median PFS was estimated as 10.6 weeks (95% CI, 4.6‐12.1) and 10.6 weeks (95% CI, 4.6‐12.7), respectively (Figure 1). Of the 20 patients enrolled, 16 (80%) patients died within 1 year from initiation of the study treatment. The 1‐year OS was estimated to be 20.0% (95% CI, 6.2‐39.3). The investigator‐assessed best response was SD in 13 patients (65%) and PD in seven patients (35%), whereas the independent radiologist‐assessed best response was PR in one patient (5%) and SD in 10 patients (50%; Table 3). Maximum reduction in tumor size compared to baseline was observed in seven patients per investigator assessment and in six patients per independent radiologist assessment (Figure 2).
Figure 1.
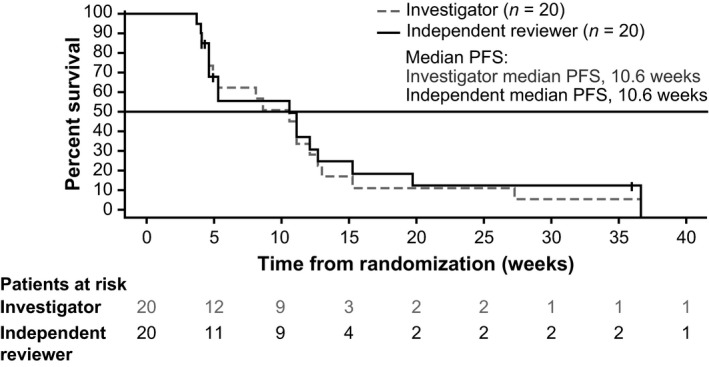
Kaplan–Meier curve of progression‐free survival (PFS) among Japanese patients with advanced biliary tract cancers refractory to gemcitabine, treated with trametinib (all treated patients)
Table 3.
Best response with confirmation in Japanese patients with advanced biliary tract cancers refractory to gemcitabine, treated with trametinib (all treated patients)
| Investigator assessment n = 20 | Independent assessment n = 20 | |
|---|---|---|
| Best response, n (%) | ||
| CR | 0 (0) | 0 (0) |
| PR | 0 (0) | 1 (5) |
| SD | 13 (65) | 10 (50) |
| Non‐CR/non‐PD | – | 1 (5) |
| PD | 7 (35) | 8 (40) |
| RR | ||
| CR + PR, n (%) | 0 (0) | 1 (5) |
| 95% CI | (0.0‐16.8) | (0.1‐24.9) |
–, Not applicable; CI, confidence interval; CR, complete response; PD, progressive disease; PR, partial response; RR, response rate; SD, stable disease.
Figure 2.
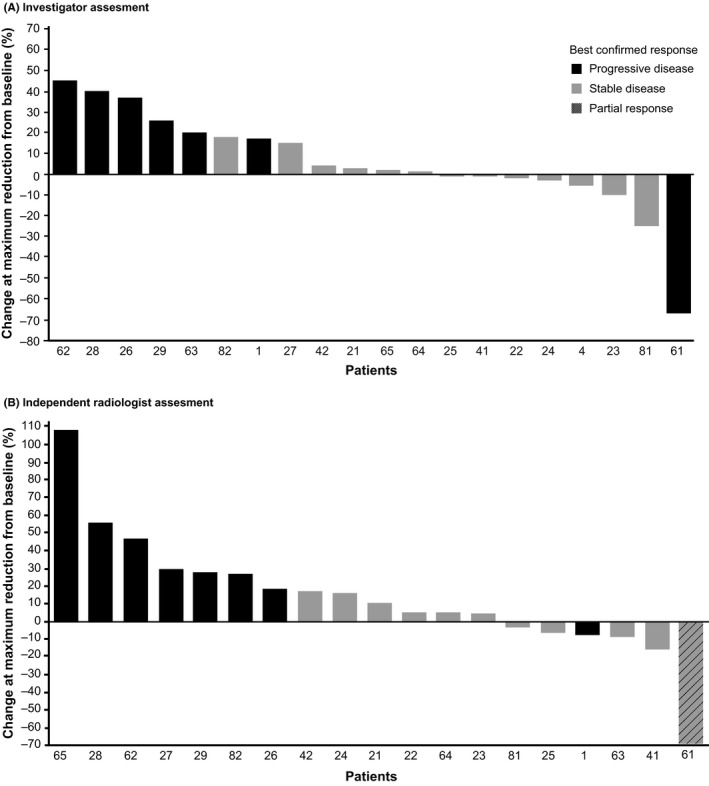
Percentage change at maximum reduction from baseline in tumor measurement among Japanese patients with advanced biliary tract cancers refractory to gemcitabine, treated with trametinib (all treated patients)
Most patients discontinued the study treatment within 12 weeks owing to disease progression (Figure 3). Three patients showed longer PFS over 12 weeks, particularly, one patient showed PR at week 13 and had confirmed PR at week 21. This patient continued on the study treatment for approximately 120 weeks, and the time to response was 20.1 weeks.
Figure 3.
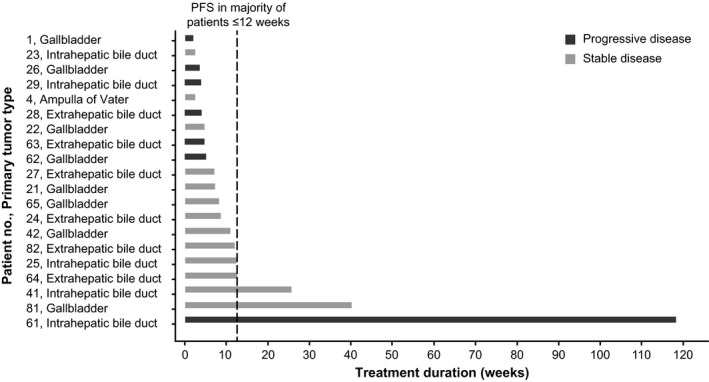
Duration of treatment in phase IIa study of trametinib in Japanese patients with advanced biliary tract cancers refractory to gemcitabine. PFS, progression‐free survival
3.3. Safety results
All patients experienced at least one AE (Table 4). The most common drug‐related AEs were dermatitis acneiform (n = 15, 75%), stomatitis (n = 6, 30%), fatigue (n = 5, 25%), and increase in blood creatine phosphokinase (n = 5, 25%). Most of the AEs reported were of grade 2 or 3 severity. Twelve (60%) and three (15%) patients reported grade 3 and 4 AEs, respectively. No grade 5 AEs were reported. Grade 3 drug‐related AEs included increase in blood creatine phosphokinase (n = 2, 10%), dermatitis acneiform, rash, retinal artery occlusion, decreased appetite, drug‐induced liver injury, and hepatic function abnormality (n = 1, 5% each). Grade 4 AEs included decreased platelet count and increased levels of blood bilirubin and amylase; however, these events were not related to the study treatment. Overall, 16 patients (80%) died during the study. The primary reason for death was disease progression. No fatal SAEs were reported. Non‐fatal SAEs were reported in 13 patients (65%). Non‐fatal SAEs related to the study treatment were reported in four patients (20%): chorioretinopathy, retinal detachment, hepatic function abnormal, and retinal artery occlusion. Four patients (20%) discontinued study treatment because of AEs, among which, drug‐induced liver injury, abnormal hepatic function, and retinal artery occlusion were considered related to the study treatment. Adverse events leading to dose interruption and dose reduction were reported in 15 (75%) and 10 (50%) patients, respectively.
Table 4.
Summary of adverse events (AEs) (>10%) in Japanese patients with advanced biliary tract cancers refractory to gemcitabine, treated with trametinib
| System organ class, preferred term | All grades n (%) | Grade 3 or highera n (%) |
|---|---|---|
| Any event, n (%) | 20 (100) | 15 (75) |
| Skin and subcutaneous tissue disorders | ||
| Dermatitis acneiform | 15 (75) | 1 (5) |
| Rash | 4 (20) | 1 (5) |
| Dry skin | 3 (15) | 0 (0) |
| Palmar–plantar erythrodysesthesia syndrome | 2 (10) | 0 (0) |
| Rash maculopapular | 2 (10) | 0 (0) |
| Gastrointestinal disorders | ||
| Diarrhea | 8 (40) | 0 (0) |
| Vomiting | 7 (35) | 0 (0) |
| Nausea | 6 (30) | 1 (5) |
| Stomatitis | 6 (30) | 0 (0) |
| Constipation | 3 (15) | 0 (0) |
| Anal hemorrhage | 2 (10) | 0 (0) |
| General disorders and administration site conditions | ||
| Fatigue | 7 (35) | 0 (0) |
| Pyrexia | 7 (35) | 0 (0) |
| Edema peripheral | 6 (30) | 0 (0) |
| Malaise | 5 (25) | 0 (0) |
| Edema | 2 (10) | 0 (0) |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 11 (55) | 2 (10) |
| Hypoalbuminemia | 5 (25) | 0 (0) |
| Infections and infestations | ||
| Pharyngitis | 4 (20) | 0 (0) |
| Paronychia | 4 (20) | 0 (0) |
| Biliary tract infection | 2 (10) | 2 (10) |
| Upper respiratory tract infection | 2 (10) | 0 (0) |
| Investigations | ||
| Blood creatine phosphokinase increased | 5 (25) | 2 (10) |
| Aspartate aminotransferase increased | 4 (20) | 1 (5) |
| Blood alkaline phosphatase increased | 3 (15) | 2 (10) |
| Blood creatinine increased | 3 (15) | 1 (5) |
| Alanine aminotransferase increased | 2 (10) | 1 (5) |
| Platelet count decreased | 2 (10) | 1 (5) |
| Hepatobiliary disorders | ||
| Cholangitis | 5 (25) | 3 (15) |
| Eye disorders | ||
| Vision blurred | 2 (10) | 0 (0) |
| Nervous system disorders | ||
| Dizziness | 3 (15) | 0 (0) |
| Dysgeusia | 2 (10) | 0 (0) |
| Respiratory, thoracic, and mediastinal disorders | ||
| Cough | 2 (10) | 0 (0) |
| Epistaxis | 2 (10) | 0 (0) |
| Neoplasms benign, malignant, and unspecified (including cysts and polyps) | ||
| Cancer pain | 2 (10) | 0 (0) |
| Tumor‐associated fever | 2 (10) | 0 (0) |
| Psychiatric disorders | ||
| Insomnia | 3 (15) | 0 (0) |
| Blood and lymphatic system disorders | ||
| Anemia | 3 (15) | 2 (10) |
Two grade 4 AEs (platelet count decreased and blood bilirubin increased) and no grade 5 AEs.
3.4. Targeted exome sequencing
Owing to tumor biopsy quality, targeted exome sequencing was possible for only nine subjects. A total of eight variants from three subjects were identified and validated as somatic (Table 5); of these, six occurred uniquely in the exceptional responder, including a synonymous NF1 exon 12 splice variant and a loss‐of‐function variant in ARID1A.
Table 5.
Ion Proton/GeneRead‐validated somatic variants in patients with biliary tract cancer
| Pt. no. | Chr | Position | Ref | Alt | Gene | Codon change | Amino acid location | Impact | dbSNP Database ID | 1000 Genomes allele frequency |
|---|---|---|---|---|---|---|---|---|---|---|
| 41 | 3 | 12647708 | C | T | RAF1 | atG/atA | M2241 | Missense | – | 0.0000 |
| 42 | 2 | 48026066 | C | G | MSH6 | tCt/tGt | S315C | Missense | rs63750491 | 0.0000 |
| 61 | 1 | 27094478 | TG | T | ARID1A | – | G1063 | Frameshift | – | 0.0000 |
| 61 | 1 | 120465007 | T | A | NOTCH2 | Att/Ttt | I1689F | Missense | rs38648601, rs60854092 | 0.0100 |
| 61 | 3 | 138461407 | T | C | PIK3CB | aAc/aGc | N205S | Missense | – | 0.0000 |
| 61 | 1 | 120468425 | G | A | NOTCH2 | tcC/tcT | S1338 | Synonymous | rs17024525 | 0.0500 |
| 61 | 7 | 6043386 | G | A | PMS2 | gcC/gcT | A96 | Synonymous | rs12532895 | 0.1100 |
| 61 | 17 | 29533389 | G | A | NF1 | ccG/ccA | P464 | Synonymous | rs201604273 | 0.0005 |
–, Not applicable; Alt, alternative; Chr, chromosome; dbSNP, Single Nucleotide Polymorphism Database; Pt, patient; Ref, reference.
4. DISCUSSION
To date, the combination of gemcitabine and cisplatin has been the only first‐line palliative treatment for advanced BTC with no clear second‐line therapy options.3, 5 The present study is the first proof‐of‐concept study evaluating the clinical activity of trametinib in patients with advanced BTC refractory to gemcitabine. As there is no established second‐line treatment for BTC, the present study was designed as a single‐arm study to evaluate the clinical utility of trametinib compared with historical single‐agent data for S‐1 and selumetinib. The median PFS with S‐1 and selumetinib based on the reference studies was 10.8 and 16.1 weeks, respectively, whereas the 12‐week non‐PD rate was estimated to be 25% and 60%, respectively. The primary analysis was carried out to determine whether the non‐PD rate at week 12 was greater than or equal to the threshold rate (25%). In this study, investigator‐assessed non‐PD rate at week 12 was 10% (P = .976), which means the primary end‐point was not met. The investigator‐assessed median PFS was estimated to be 10.6 weeks. Three patients in this study had longer PFS (>12 weeks), one of whom showed PR at week 13, which was confirmed at week 21; this patient continued study treatment for approximately 120 weeks. No new safety signals were observed in this study.
Results from previous trials have shown limited efficacy of second‐line chemotherapy for advanced BTC. A phase II study was carried out in 32 patients with BTC refractory to 5‐fluorouracil‐based palliative chemotherapy. Of 29 patients whose tumor response was evaluated, two patients achieved a PR (RR, 6.9%), six patients (20.7%) achieved SD, and 21 patients (72.4%) reported disease progression. The median time to progression and median OS after treatment with gemcitabine as second‐line treatment was reported to be 1.6 months and 4.1 months, respectively. Most commonly reported grade 3/4 AEs included thrombocytopenia and neutropenia.22 Treatment with a combination of capecitabine and celecoxib resulted in an overall RR of 9% and a median survival duration of 19 weeks; this combination was reported to be safe with moderate efficacy in patients with pancreatic cancer/BTC.23 Another study (n = 20) evaluated the feasibility of gemcitabine and cisplatin combination therapy as second‐line treatment for patients with advanced BTC refractory to gemcitabine and S‐1. Although no tumor response was observed, moderate prolongation of OS (5.9 months) and time to progression (3.6 months) was observed.24 Two phase II studies evaluating S‐1 monotherapy as second‐line chemotherapy for advanced BTC showed modest efficacy.9, 10 A systematic review reported by Lamarca et al25 evaluated 25 studies (n = 761) to determine the level of evidence for use of second‐line chemotherapy in patients with advanced BTC. With a reported mean OS of 7.2 months, a mean PFS of 3.2 months, and an RR of 7.7%, the evidence was considered insufficient (level C). Another retrospective pooled analysis of 603 patients reported a limited value of second‐line chemotherapy after progression with first‐line platinum and gemcitabine combination in patients with advanced BTC. Even though second‐line treatment resulted in disease control in 50% of the cases, it was not maintained for long because of short median PFS.26 In an open‐label, single‐arm study, combination therapy with oral vascular endothelial growth factor receptor tyrosine kinase inhibitor pazopanib and trametinib, reported modest clinical activity with signs of possible cumulative toxicity in patients with advanced cholangiocarcinoma. The study did not achieve statistically significant improvement in 4‐month PFS over the prespecified null hypothesized 4‐month PFS (P = .063).27 Although the aforementioned phase II trials present some scientific evidence, adequately powered trials are needed to identify the most suitable regimen and patient population who may benefit from such treatment.
Genomic profiling studies of BTC have reported a highly diverse collection of underlying mutations with a relatively low overall mutational burden in individual patients, the exception being a small cohort whose tumors are hypermutated due to errors in DNA mismatch repair.28, 29, 30, 31, 32 The results of the targeted exome sequencing in our clinical cohort are consistent with those findings, with six of nine patients failing to show any somatic mutations in our panel of genes and two of the remaining patients, in which somatic variants were validated, harboring alterations in DNA mismatch repair genes (Table 5). Missense variants were observed in RAF1, MSH6, NOTCH2, and PIK3CB, with one patient showing a frameshift variant in ARID1A. Variants in all of these genes have been observed previously in BTC in larger cohorts, the most prevalent being in ARID1A with a mutation frequency of 10%‐20%.28, 29, 30 Although no KRAS or TP53 variants were identified in our clinical samples, the cohort size was small and such negative findings likely reflect the lack of dominant oncogenic drivers in this disease.29
The long PFS observed in one patient was notable in two respects. First, six of the eight validated somatic mutations across the nine‐patient cohort were in this patient, which suggests that the patient's carcinoma may be hypermutated. Although the patient hosted a synonymous variant in the mismatch repair endonuclease, PMS2, this variant is also present in the healthy population (rs12532895) and hypermutation cannot be verified within our dataset. The result, however, is intriguing because MEK inhibition has recently been shown to protect against T‐cell apoptosis driven by chronic antigen stimulation33 and such a mechanism is expected to be especially relevant in hypermutated tumors with a higher incidence of antigenic epitopes. Given that immunotherapy has reported benefit in patients with BTC42 and that the above mechanism appears to work in parallel to the programmed cell death 1 axis (at least in preclinical syngeneic models), it may be worthwhile to explore the combination of MEK inhibition and immunotherapy in maximizing clinical response in patients with BTC. Second, this patient also hosted a notable somatic variant: a synonymous NF1 exon 12 splice variant. Published data implicate this specific NF1 splice variant in loss of gene function,34 which in turn has been shown to drive MEK dependence and trametinib sensitivity in BRAF/RAS wild‐type melanoma cell lines.35 If a similar functional dependence could be disclosed in BTC, this could potentially explain the exceptional response to trametinib observed in this patient (Figure S1). Other somatic variants, a frameshift loss‐of‐function mutation in ARID1A, which may be expected to increase activation of the PI3K/AKT pathway,36 and missense mutations in NOTCH2 and PIK3CB, have no known link to MEK inhibition sensitivity.
A limitation of this study was that patients were enrolled regardless of their mutation status. Furthermore, the small number of enrolled patients, inclusion of subtypes of BTC, and inclusion of mixed patients receiving second‐ and third‐line treatment, may have contributed to the lack of efficacy observed in this trial. Biliary tract cancer is reported to have varied biological behavior and sensitivity to chemotherapies among primary tumor types. Moreover, differences in terms of prior treatment regimens and/or number of prior regimens administered may affect treatment sensitivity of chemotherapy as well as PFS/OS. Further investigation is required to study the relationship between efficacy and the aforementioned factors.37
In conclusion, the primary end‐point of this study, non‐PD rate at week 12, did not reach statistical significance. Although the primary end‐point was not met, prolonged PFS was observed in one patient. The overall safety profile was comparable to that of trametinib monotherapy to date. Given the limited number of biopsies evaluable by next‐generation sequencing, the significance of the PIK3CB, NOTCH2, ARID1A, and NF1 mutations identified in the subject with a PR is not known; however, efforts to understand the role of these mutations and sensitivity to trametinib or other targeted therapies in BTC are warranted.
CONFLICT OF INTEREST
Masafumi Ikeda received research funding from GlaxoSmithKline, AstraZeneca, Yakult, Ono Pharmaceutical, and Eisai. Tatsuya Ioka received honoraria from Taiho Pharmaceutical and research funding from Merck Serono, AstraZeneca, Taiho Pharmaceutical, GlaxoSmithKline, and Zeri Shinyaku. Chigusa Morizane has received research funding from Pfizer, Yakuruto, Eisai, Taiho Pharmaceutical, and Ono Pharmaceutical. Hideaki Takahashi received research funding from Bayer Pharmaceuticals and Bristol‐Myers Squibb and travel support from Pfizer. Junji Furuse has received honoraria from Taiho Pharmaceutical, Fujifilm, Eisai, Astellas, Yakult, Sumitomo Dainippon, Eli Lilly Japan, and Kyowa Hakko Kirin, and research funding from J‐Pharma, Taiho Pharmaceutical, Ono Pharmaceutical, MSD, Janssen, and Daiichi Sankyo. Caretha L. Creasy and Shelby Gorman are employees of GlaxoSmithKline and hold stock options. Daniel J. Felitsky is a former employee of GlaxoSmithKline. Fanghong Zhang and Mikiro Kobayashi are employees of Novartis Pharma. The other authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The study was funded by GlaxoSmithKline (NCT01943864). The authors thank Preetinder Kaur (Novartis Healthcare) for providing medical writing support. Editorial support was funded by Novartis Pharma. The authors thank all patients, their families, investigators, and everyone else who participated in this clinical trial.
Ikeda M, Ioka T, Fukutomi A, et al. Efficacy and safety of trametinib in Japanese patients with advanced biliary tract cancers refractory to gemcitabine. Cancer Sci. 2018;109:215–224. https://doi.org/10.1111/cas.13438
Funding information
GlaxoSmithKline
REFERENCES
- 1. Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist. 2008;13:415‐423. [DOI] [PubMed] [Google Scholar]
- 2. National Cancer Center . Cancer statistics in Japan 2009. [Accessed January 2016.] Available from: http://www.fpcr.or.jp/publication/statistics.html.
- 3. Benavides M, Antón A, Gallego J. Biliary tract cancers: SEOM clinical guidelines. Clin Transl Oncol. 2015;17:982‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marcano‐Bonilla L, Mohamed EA, Mounajjed T, Roberts LR. Biliary tract cancers: epidemiology, molecular pathogenesis and genetic risk associations. Chin Clin Oncol. 2016;5:61. [DOI] [PubMed] [Google Scholar]
- 5. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273‐1281. [DOI] [PubMed] [Google Scholar]
- 6. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer. 2010;103:469‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furuse J, Okusaka T, Boku N, et al. S‐1 monotherapy as first‐line treatment in patients with advanced biliary tract cancer: a multicenter phase II study. Cancer Chemother Pharmacol. 2008;62:849‐855. [DOI] [PubMed] [Google Scholar]
- 8. Morizane C, Okusaka T, Mizusawa J, et al. Randomized phase II trial of gemcitabine plus S‐1 combination therapy versus S‐1 in advanced biliary tract cancer: A Japan Clinical Oncology Group trial (JCOG 0805). Cancer Sci. 2013;104:1211‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sasaki T, Isayama H, Nakai Y, et al. Multicenter phase II study of S‐1 monotherapy as second‐line chemotherapy for advanced biliary tract cancer refractory to gemcitabine. Invest New Drugs. 2012;30:708‐713. [DOI] [PubMed] [Google Scholar]
- 10. Suzuki E, Ikeda M, Okusaka T, et al. A multicenter phase II of S‐1 in gemcitabine refractory biliary tract cancer. Cancer Chemother Pharmacol. 2013;71:1141‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Furuse J, Okusaka T. Targeted therapy for biliary tract cancer. Cancers. 2011;3:2243‐2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walter T, Horgan AM, McNamara M, et al. Feasibility and benefits of second‐line chemotherapy in advanced biliary tract cancer: a large retrospective study. Eur J Cancer. 2013;49:329‐335. [DOI] [PubMed] [Google Scholar]
- 13. Tannapfel A, Sommerer F, Benicke M, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52:706‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reeves ME, DeMatteo RP. Genes and viruses in hepatobiliary neoplasia. Semin Surg Oncol. 2000;19:84‐93. [DOI] [PubMed] [Google Scholar]
- 15. Saetta AA, Papanastasiou P, Michalopoulos NV, et al. Mutational analysis of BRAF in gallbladder carcinomas in association with K‐ras and p53 mutations and microsatellite instability. Virchows Arch. 2004;445:179‐182. [DOI] [PubMed] [Google Scholar]
- 16. Xu RF, Sun JP, Zhang SR, et al. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed Pharmacother. 2011;65:22‐26. [DOI] [PubMed] [Google Scholar]
- 17. Finn RS, Javle MM, Tan RR Jr, et al. A Phase 1 study of MEK inhibitor MEK162 (ARRY‐438162) in patients with biliary tract cancer. J Clin Oncol. 2012;30(suppl 4). abstr 220. [Google Scholar]
- 18. Bekaii‐Saab T, Phelps MA, Li X, et al. Multi‐institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29:2357‐2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP‐74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989‐1000. [DOI] [PubMed] [Google Scholar]
- 20. Cavalloni G, Peraldo‐Neia C, Varamo C, et al. Preclinical activity of EGFR and MEK1/2 inhibitors in the treatment of biliary tract carcinoma. Oncotarget. 2016;7:52354‐52363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumors: revised RECIST guidelines (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 22. Oh SY, Jeong CY, Hong SC, et al. Phase II study of second line gemcitabine single chemotherapy for biliary tract cancer patients with 5‐fluorouracil refractoriness. Invest New Drugs. 2011;29:1066‐1072. [DOI] [PubMed] [Google Scholar]
- 23. Pino MS, Milella M, Gelibter A, et al. Capecitabine and celecoxib as second‐line treatment of advanced pancreatic and biliary tract cancers. Oncology. 2009;76:254‐261. [DOI] [PubMed] [Google Scholar]
- 24. Sasaki T, Isayama H, Nakai Y, et al. Feasibility study of gemcitabine and cisplatin combination chemotherapy for patients with refractory biliary tract cancer. Invest New Drugs. 2011;29:1488‐1493. [DOI] [PubMed] [Google Scholar]
- 25. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second‐line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol. 2014;25:2328‐2338. [DOI] [PubMed] [Google Scholar]
- 26. Brieau B, Dahan L, De Rycke Y, et al. Second‐line chemotherapy for advanced billiary tract cancerafter failure of the gemcitabine‐platinum combination: a large multicenter study by the Association des Gastro‐Entérologues Oncologues. Cancer. 2015;121:3290‐3297. [DOI] [PubMed] [Google Scholar]
- 27. Shroff RT, Yarchoan M, O'Connor A, et al. The oral VEGF receptor tyrosine kinase inhibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma. Br J Cancer. 2017;116:1402‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of billiary tract cancer. Nat Genet. 2015;47:1003‐1010. [DOI] [PubMed] [Google Scholar]
- 29. Chaisaingmongkol J, Budhu A, Dang H, et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell. 2017;32:57‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou S, Li J, Zhou H, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun. 2014;5:5696. [DOI] [PubMed] [Google Scholar]
- 31. Farshidfar F, Zheng S, Gingras M‐C, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH‐mutant molecular profiles. Cell Rep. 2017;18:2780‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinoma. Nat Genet. 2013;45:1470‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ebert PJR, Cheung J, Yang Y, et al. MAPK kinase inhibition promotes T cell and anti‐tumor activity in combination with PD‐L1 checkpoint blockade. Immunity. 2016;44:609‐621. [DOI] [PubMed] [Google Scholar]
- 34. Beauchamp EM, Woods BA, Dulak AM, et al. Acquired resistance to dasatinib in lung cancer cell lines conferred by DDR2 gatekeeper mutation and NF1 loss. Mol Cancer Ther. 2014;13:475‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Can Res. 2014;74:2340‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Samatzis EP, Gutsche K, Dedes KJ, et al. Loss of ARID1A expression sensitizes cancer cells to PI3K‐ and AKT‐inhibition. Oncotarget. 2014;5:5295‐5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cereda S, Belli C, Rognone A, Mazza E, Reni M. Second‐line therapy in advanced biliary tract cancer: what should be the standard? Crit Rev Oncol Hematol. 2013;88:368‐374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials