

Summary
For understanding the rules and laws of adaptive immunity, high‐throughput profiling of T‐cell receptor (TCR) repertoires becomes a powerful tool. The structure of TCR repertoires is instructive even before the antigen specificity of each particular receptor becomes available. It embodies information about the thymic and peripheral selection of T cells; the readiness of an adaptive immunity to withstand new challenges; the character, magnitude and memory of immune responses; and the aetiological and functional proximity of T‐cell subsets. Here, we describe our current analytical approaches for the comparative analysis of murine TCR repertoires, and show several examples of how these approaches can be applied for particular experimental settings. We analyse the efficiency of different metrics used for estimation of repertoire diversity, repertoire overlap, V‐gene and J‐gene segments usage similarity, and amino acid composition of CDR3. We discuss basic differences of these metrics and their advantages and limitations in different experimental models, and we provide guidelines for choosing an efficient way to lead a comparative analysis of TCR repertoires. Applied to the various known and newly developed mouse models, such analysis should allow us to disentangle multiple sophisticated puzzles in adaptive immunity.
Keywords: aging, diversity, functional T‐cell subsets, T cell, T‐cell receptor repertoires
Introduction
T‐cell receptor (TCR) profiling with high‐throughput sequencing is becoming an indispensable tool used for studying the adaptive immune response in mouse models and human clinical research.1, 2, 3, 4 Even before sufficient data, methods and algorithms have been accumulated for massive identification of antigen‐specific TCR variants (although efforts are being made5, 6, 7, 8), rational comparative analysis of the structure of immune receptor repertoires can be highly informative. The informative features of TCR repertoire analysis include, but are not limited to:
estimation of repertoire diversity, for which different metrics may be used depending on the questions raised.
estimation of homology of compared repertoires, understood as a number, or relative abundance or correlation of frequencies of shared clonotypes, which may reflect functional (at amino acid level) or aetiological (at nucleic acid level) similarity of repertoires.
V‐gene and J‐gene segment usage statistics, which may reflect bulk functional features of repertoires.
statistics on the hypervariable region (CDR3), such as average length and distribution of lengths (in silico spectratyping), number of randomly added ‘N’ nucleotides (activity of TdT and closeness to germline, which largely determines the ‘publicity’ of TCR variants9), and, importantly, amino acid composition that reflects the biophysical characteristics of CDR3,10 providing the link between the general structure of TCR repertoires and the range of their potential antigenic specificities.
The quality of comparative repertoire analysis relies on the methods of TCR library preparation and sequencing (TCR‐seq) and the following software analysis algorithms (Fig. 1). Preferably, the method of library preparation for the high‐throughput sequencing and data analysis should be standardized, including the particular version of a software used for the repertoire extraction from raw sequencing data and further analysis. Unbiased methods of TCR libraries preparation and data analysis11 and minimization of cross‐sample contaminations12 are important. In many cases, comparative analysis requires accurate normalization, for which using unique molecular identifiers (UMI)13, 14, 15 is a method of choice.16, 17, 18
Figure 1.
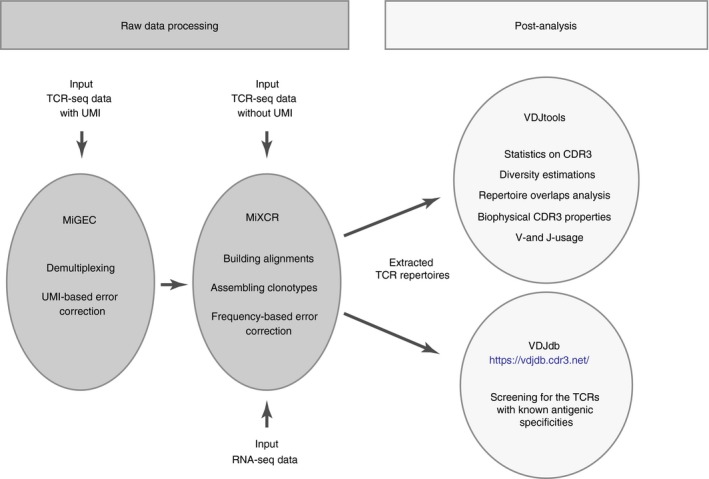
Extracting and comparing T‐cell receptor (TCR) repertoires. TCR repertoires can be extracted from targeted TCR sequencing (TCR‐seq) performed using genomic DNA or cDNA methods, with or without unique molecular identifiers (UMI), e.g. using MiGEC and MiXCR software tools. Alternatively, TCR repertoires can be extracted from bulk RNA‐seq data using MiXCR RNA‐seq mode. The latter approach works most efficiently for samples enriched with T cells or representing pure sorted T cells.
A very useful alternative to obtain TCR repertoires starts with the development of efficient software for extraction of CDR3 repertoires from bulk RNA‐seq of sorted T cells.19 The resulting TCR‐α and ‐β CDR3 repertoires are large in size and allow for the accurate comparison of diversity metrics, averaged CDR3 properties and even repertoire overlaps. Paired‐end and relatively long‐range sequencing (e.g. 100 + 100 nt, or better 150 + 150 nt) is preferable to obtain deep and unbiased TCR repertoires from RNA‐seq, although even single‐end 50‐nt sequencing may yield information on the repertoire.
Statistical metrics can be calculated either per clone (unweighted – per unique clonotype in a data set) or per T cell (weighted – per sequencing read, or per UMI‐labelled cDNA or DNA molecule), at the nucleotide or amino acid level, for the in‐frame (functional) or out‐of‐frame (containing statistical information on the repertoire before functional TCR selection20, 21, 22, 23) repertoires, resulting in a multitude of different conclusions that can be drawn from the same data set.
We demonstrate the capabilities of comparative TCR repertoire analysis using a number of examples from our current work. We focus on TCR‐α and TCR‐β repertoires of syngeneic mice, which have several specific features. The first difference from the human samples comes from genetic homogeneity, which makes syngeneic mice similar to genetically identical human twins. Although human twin TCR repertoires are highly different, they have higher similarity in V‐J segments usage frequencies and more pronounced overlap among the top‐frequency clonotypes compared with unrelated donors.23, 24 In mice, repertoire convergence is additionally strengthened due to a genetically lower entropy of TCR recombination – shorter CDR3 length and lower number of randomly added ‘N’ nucleotides (Fig. 2). As a result, even limited available T‐cell counts often create the possibility of making clear and statistically significant conclusions concerning the characteristics and similarity of syngeneic mouse TCR repertoires for the different T‐cell subsets, different tissues, before and after therapy, different age groups, and in various transgenic mouse models.
Figure 2.
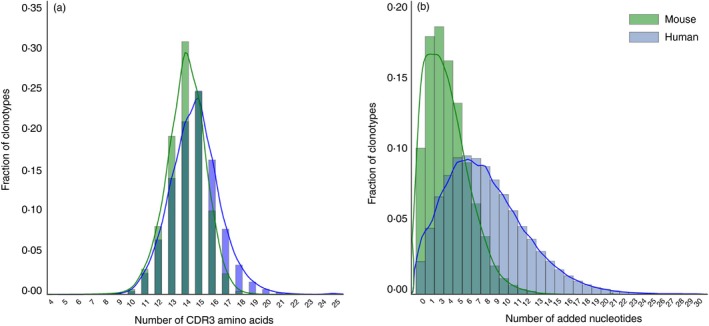
Comparison of the CDR3 length distributions for mice and humans. (a) T‐cell receptor‐β variable (TRBV) CDR3 length histogram for C57BL/6J mouse (3 months old) and human (male, 35 years old) peripheral blood mononuclear cells. CDR3 is defined starting from the last codon of TRBV (cysteine, position 92) and ending at the phenylalanine in the conserved TRBJ segment motif FGXG. (b) Added N‐nucleotides histogram. The data sets were processed using MiXCR, with correction for the probability of zero insertions.71
Comparative analysis of TCR repertoire diversity
Estimation of total diversity of TCR variants in a T‐cell subset or tissue of interest or within the whole animal is not a trivial task; it is hampered by the limited size of the analysed sample and limitations of the extrapolation methods.25 In practice, the task for the researcher is usually to compare the relative diversity and the evenness of clonal size distribution within multiple samples of interest. Such comparison shows the relative size of TCR repertoires and the extent of oligoclonal expansions, which is often informative.
However, all diversity metrics depend on the analysis depth,26 which always differs from sample to sample, even in the highly standardized experimental conditions. This introduces bias and high variance into comparisons, increasing one's uncertainty in the observed differences.
The best way to normalize the depth of high‐throughput sequencing analysis in targeted resequencing (of which TCR repertoire profiling is a special case) is to employ UMI.13, 14, 15 In this approach, each starting DNA or cDNA molecule is labelled with a unique barcode, which is used in downstream high‐throughput sequencing data analysis to assign each sequencing read to a particular template molecule. In the cDNA‐based RACE approach,27, 28, 29 sampling equal numbers of UMI‐labelled TCR‐β cDNA molecules allows us to efficiently normalize multiple samples for the comparison of diversity metrics.3, 4, 16, 17, 18, 30
To demonstrate the usefulness of the UMI approach for comparative analysis of mouse T‐cell repertoire diversity, we analysed peripheral blood TCR‐β CDR3 repertoires for eight young (3‐month‐old) and eight old (23‐month‐old) C57BL/6J mice. Total RNA isolated from 100 μl of peripheral blood was converted into cDNA and used for TCR‐β library preparation. For each sample, we obtained at least 200 000 sequencing reads, covering at least 15 000 distinct UMI‐labelled cDNA molecules (see Supplementary material, Table S1).
T‐cell receptor repertoire diversity was assessed using several widely used approaches: Shannon–Wiener,31 Simpson,32 Efron33 and D50 diversity indices, Chao1,34 extrapolated Chao estimate,35 and directly observed diversity (number of unique clonotypes in a sample) (Fig. 3). All metrics were obtained for the conventionally analysed sequencing data (MiXCR36) and for the UMI‐analysed data (MIGEC37 to group reads by UMI, then MiXCR to extract repertoires). In each case, all metrics were obtained for the total data sets and for the normalized data sets (downsampled to 15 000 randomly chosen sequencing reads or UMI). We also obtained flow cytometry data on the percentage of naive CD62L+ CD44− T cells for each sample. Since TCR diversity decreases with age both due to the drop of the percentage of the naive T cells and due to the decrease of diversity within the naive T‐cell pool,17, 38 we calculated Spearman correlation of various TCR diversity metrics with age and with naive T‐cell percentage and used this as a measure of accuracy of diversity metrics (Fig. 3a,b).
Figure 3.
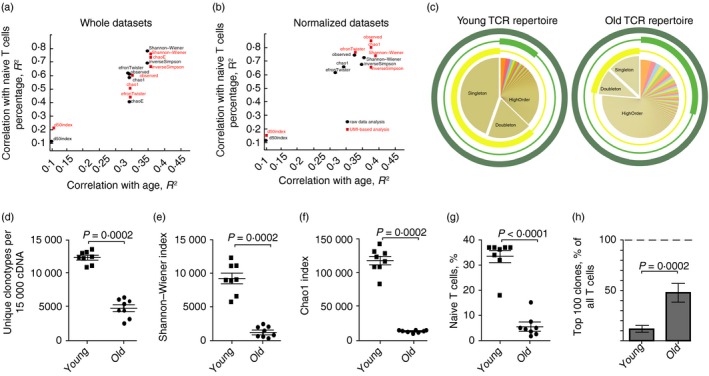
Analysis of T‐cell receptor‐β (TCR‐β) CDR3 repertoire diversity in young and old mouse peripheral blood mononuclear cells (PBMC). (a, b) Spearman correlations for the diversity metrics and age or naive T cells percentage in the blood of young (n = 8) and old (n = 8) mice. Black circles correspond to the MiXCR data sets. Red squares correspond to the unique molecular identifiers (UMI)‐based analysis (MIGEC+MiXCR). Correlations are shown for the non‐normalized data sets: all obtained UMI or sequencing reads (a), and normalized data sets (down‐sampled to 15 000 sequencing reads or 15 000 UMI) (b). (c) Schematic representation of the TCR repertoire diversity assessment using different diversity metrics. Examples of a young (left) and an old (right) mouse peripheral blood TCR‐β CDR3 repertoire are shown. Three circles represent observed diversity (dark green), Shannon–Wiener index (light green), and Chao1 (yellow) metrics. Segment of the TCR repertoire that contributes mainly to a metric is shown by the bold part of the circle. Observed diversity takes into account all clonotypes, Chao1 depends mostly on the representation of singletons and doubletons (clonotypes represented by one and two reads, correspondingly).70 Shannon–Wiener index characterizes structural complexity of the TCR repertoire based on assessment of the evenness of distribution of relatively abundant clonotypes. (d–f) Diversity metrics for TCR‐β CDR3 repertoires in young and old mice. Data sets were normalized by down‐sampling to 15 000 randomly chosen UMI. Two‐tailed unpaired Mann–Whitney test showed that all diversity estimates were significantly different between old and young mice. (g) Percentage of naive T cells of all CD3+ T cells in peripheral blood of young and old mice. Each symbol represents an individual mouse; small horizontal line indicates the mean. Two‐tailed unpaired Mann–Whitney test was applied. (h) Share of the whole repertoire occupied by the top 100 most frequent clonotypes.
As expected, diversity metrics correlated better with the percentage of naive T cells in a sample than with the animal's age. The diversity is directly affected by the percent of naive T cells, which in itself depends on age and physiological factors such as an ongoing immune response. The best performance was achieved with directly observed diversity and Chao1, but only for the UMI‐normalized data sets (Fig. 3b, red squares). Shannon–Wiener and Inverse Simpson indices weighted toward the evenness component showed relatively high correlation with age and percentage of naive T cells, being relatively independent, in this particular experiment, of the sample normalization or use of UMI. However, it should be noted that here we obtained all samples in parallel, in identical experimental conditions. Experiment‐to‐experiment and laboratory‐to‐laboratory differences in library preparation impair the consistency of repertoire diversity comparisons using a routine data analysis pipeline. Normalization based on UMI counts always reduces analysis to the fixed number of cDNA molecules, protecting from biases both within and between the experiments. On the condition of sampling of the excess numbers of cells compared with the total captured cDNA molecules (15 000 cDNA molecules from 200 000 T cells in our example), and taking into account relatively low dispersion in TCR mRNA expression levels between T cells,39, 40, 41 each cDNA molecule roughly represents a single T cell. Therefore, in this design, one is comparing TCR diversity metrics calculated for the equal T‐cell counts, which makes analysis highly resistant to experimental deviations.
Importantly, different diversity metrics may have intrinsically different meanings that describe various structural features of the repertoire (Fig. 3c). The latter can be illustrated by the next experiment, where we performed analysis of TCR‐β repertoires of eight 1‐year‐old BALB/c mice, separately for the lymph node, spleen, thymus and peripheral blood mononuclear cell tissues (see Supplementary material, Table S2). Relative values for the observed, Shannon–Wiener and Chao1 diversity estimates obtained for the UMI‐normalized data (30 000 randomly chosen UMI groups) differed notably (Fig. 4a–c). In particular, the Chao1 estimate, which depends on relative abundance and distribution of rare clonotypes, was highest for lymph node samples (Fig. 4c), reflecting the fact that lymph nodes are enriched with naive CD44− CD62L+ T cells (Fig. 4d). At the same time, directly observed diversity and Shannon–Wiener index, which depend also on the distribution of large and medium clonotypes, were the highest in thymus (Fig. 4a,b), reflecting the lower presence of large clonal expansions and more even distribution of clonotype sizes.
Figure 4.
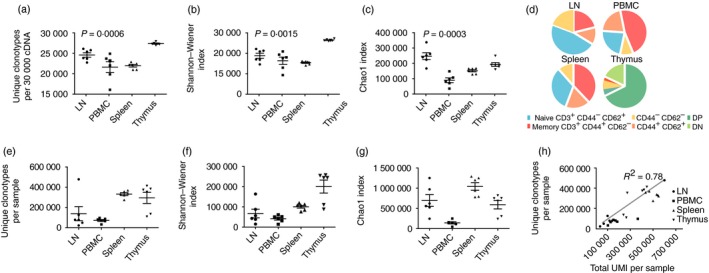
Analysis of T‐cell receptor‐β (TCR‐β) CDR3 repertoire diversity in mouse tissues. (a–c) Diversity metrics (n = 6 mice). Data sets obtained for the samples from lymph nodes (LN), spleen (SP), thymus (THY) or peripheral blood mononuclear cells (PBMC) were normalized by down‐sampling to 30 000 randomly chosen unique molecular identifier (UMI)‐labelled cDNA molecules. Observed diversity (number of clonotypes per 30 000 T cells (a), Shannon–Wiener index (b), and Chao1 (c) metrics are shown. Kruskal–Wallis test showed that diversity metric values were significantly different across various tissues on each plot. (d) The average percentages of T‐cell subsets according to flow cytometry analysis. CD3‐positive lymphocyte gates were defined using CD62 and CD44 staining among thymocytes, splenocytes, PBMC and lymphocytes isolated from lymph nodes. (e–g) The same diversity metrics calculated without data normalization. (h) Correlation of observed TCR‐β diversity with the number of analysed UMI‐labelled cDNA molecules.
Notably, diversity metrics obtained without previous data normalization (i.e. without extracting the same numbers of UMI) yielded drastically different results compared with the normalized data (Fig. 4e–g). The values obtained for the bulk data correlated with the relative depth of analysis (number of distinct UMI extracted from a sample, Fig. 4h). In particular, spleen data sets that contained high numbers of unique UMI (i.e. large numbers of analysed cDNA molecules) were characterized by high observed diversity and Chao1 values. This example vividly illustrates the critical importance of UMI‐based data normalization for the unbiased comparison of sample diversity.
In the analysis of TCR repertoires obtained from the paired‐end RNA‐seq data,19 it is desirable to remove PCR duplicates (cases when the same amplified RNA fragment was sequenced twice) and optical duplicates (cases when a single DNA cluster on Illumina was misidentified as a group of several identical DNA clusters18) characterized by the same starting nucleotide positions,42 and employ the same number of unique paired‐end sequencing reads in order to normalize samples for the accurate comparison of diversity metrics, by analogy with the UMI‐based analysis of targeted TCR cDNA libraries.
Visualizing repertoire overlaps: dendrograms and multi‐dimensional scaling
Relative similarity of TCR repertoires (presence of shared CDR3 sequences) in different functional T‐cell subsets, tissues or mice upon antigenic challenge, can be informative in T‐cell immunity studies. The relative overlap of any two TCR CDR3 repertoires can be calculated in different ways, for example: (i) as the normalized number of common clonotypes (normalization is critical as the overlap depends on the depth); (ii) as the correlation of frequencies for the common clonotypes in the two repertoires; (iii) as the average share that common clonotypes occupy in the two repertoires (considering the clonotype size). Such metrics, applied to multiple samples of comparison, can tell a lot about functional and aetiological proximity of T‐cell subsets. Overlap can be analysed for the TCR‐α, β, γ or δ chains, in‐frame or out‐of‐frame CDR3 repertoires, at the nucleotide or amino acid sequence level, with or without considering the identity of V‐ and J‐gene segments of the shared CDR3 variants.
According to our current experience, a rational combination of computational steps suitable to study the functional similarity of mouse TCR repertoires is:
To compare TCR‐α CDR3 repertoires. TCR‐α are generally less diverse than TCR‐β, making the TCR‐α repertoire more ‘public’. The presence of a relatively high number of identical amino acid TCR‐α CDR3 variants in different mice, tissues and T‐cell subsets provides larger overlaps that allow the clustering of samples with similar structure of TCR repertoire. At the same time, the extent of these overlaps differs, which reveals the differences in the organization of analysed repertoires of interest.
To compare overlaps of in‐frame clonotypes that carry identical amino acid CDR3 sequence, V‐ and J‐gene segments. This approach reports functional similarity of repertoires, whereas nucleotide overlaps are always lower and may contain insufficient information to group functionally similar samples.
In contrast, to study the aetiological proximity of T‐cell subsets within the same animal, the more rational approach is to analyse nucleotide TCR‐β CDR3 repertoires, which are more distinctive and so are more suitable as lineage tracking markers reporting, for example, the conversion of conventional CD4 T cells to regulatory T (Treg) cells.
The different methods of the TCR CDR3 overlap comparison are realized in VDjtools software43 as ‘F2’, ‘R’ and ‘D’ metrics.
F2 metrics is a clonotype‐wise sum of geometric mean frequencies, so mostly reflects the proximity of TCR repertoires in respect to relative share occupied by the common clonotypes within two repertoires of comparison. It therefore works reliably, e.g. in those experiments when common expanded clonotypes are expected within the group of similarly treated mice. On a multidimensional plot visualization, the closer two samples are, the higher the total frequency of the clonotypes they share (Fig. 5).
Figure 5.
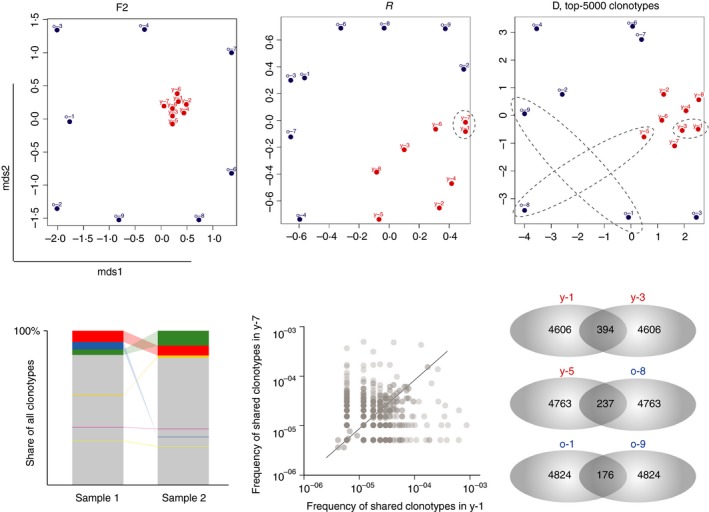
Visualizing repertoire overlaps. Multi‐dimensional scaling (MDS) analysis of the T‐cell receptor‐β (TCR‐β) CDR3 repertoires in young (red circles, n = 8) and old (blue circles, n = 8) mouse peripheral blood (upper panels). Metrics F2, R and D show that young (red dots) data sets show a high similarity to each other, whereas the old ones demonstrate distinctive features of the TCR‐β repertoires, due to the decreasing proportion of naive T cells and expansion of different antigen‐experienced clones with age. For the D metric, cluster analysis was restricted to the top 5000 clonotypes per sample. Bottom panels show schematic (for the F2 metric) and exemplary (for R and D metrics) pairwise analyses of samples overlap.
In contrast, metric R (Pearson correlation of clonotype frequencies, restricted to the shared clonotypes) reflects the overall similarity of repertoire organization. It takes into account both large and small shared clonotypes, and thus the R metric is less dependent on cross‐sample contaminations, which is almost inevitable in cell sorting. Notably, normalization (down‐sampling to the size of the smallest data set in terms of CDR3‐containing sequencing reads or UMI‐labelled cDNA molecules) may be deleterious for R metrics overlap analysis, as it drops information that otherwise could be rationally interpreted.
Metric D completely ignores clonotype frequencies and shows the number of shared clonotypes between the two samples, normalized on the product of the observed number of clonotypes in two samples. The normalization on the product of the diversities implied in the D metric supposedly neutralizes the difference in the depth of the sequencing of the two samples. However, such normalization is not ideal, because the structure of the TCR repertoire depends on the depth of analysis and on proportions of functional T‐cell subsets contained within the samples.16 Hence the most robust way to apply the D metric is to limit analysis on the top‐X (e.g. top‐5000, top‐20 000, etc.) most frequent clonotypes in each of the compared samples of interest. In the latter approach, the comparison is reduced to the simple but informative and normalized comparison of the number of shared clonotypes between samples.
In comparison of peripheral blood amino acid TCR‐β CDR3 repertoires for the young and old C57BL/6J mice discussed above, all overlap metrics showed the shortest distance between the samples of young mice (Fig. 5). This closeness reflects the fact that the peripheral blood of young mice carries a high percentage of naive T cells. Naive TCR repertoires share a large number of relatively public clonotypes, resulting from high frequency events and convergent recombination events,9, 17, 20, 44 especially characteristic for the T cells inherited from the TdT‐free prenatal period,16 potentially pre‐expanded in fetus.45 Hence, in spite of the generally high diversity of naive T cells, this diversity includes a core public component that makes TCR repertoires of young mice similar.
Fixed TCR‐β chain makes the narrowed TCR‐α repertoire highly ‘digital’
In the DO11.10 TCR‐β + Tcra +/− mouse model, the TCR‐β chain is fixed and so the whole TCR diversity and antigen specificity is determined by a TCR‐α chain. Furthermore, necessity for an α‐chain to pair with pre‐determined TCR‐β limits the TCR‐α diversity.46 Therefore, on a fixed TCR‐β background, different functional T‐cell subsets contain a large proportion of identical TCRs, providing rich information for the repertoire overlap metrics. At the same time, the relative frequencies of such shared TCR‐α variants may differ dramatically between the subsets, but are often similar for the same functional subsets in different animals.
In this model, the ‘R’ overlap metric that measures the correlation of frequencies for the shared clonotypes efficiently, almost ‘digitally’, differentiates functional T‐cell subsets based on TCR‐α repertoires (Fig. 6a). Note that in this example, the ‘R’ overlap metric not only shows that CD4 effector conventional T (Teff) cell and CD4 effector Treg cell repertoires are distinct (which was expected), but also clearly distinguishes Treg, Teff and naive CD4 T‐cell subsets from spleen, thymus and lymph nodes, reflecting the distinct homing preferences and, so the internal functional heterogeneity of these subsets. Note that TCR‐α repertoires of the same subsets obtained from different mice have much in common (Fig. 6a–c), contrasting dominant patient‐level clustering commonly observed in human TCR repertoires and, to a lesser extent, in wild‐type mouse TCR‐β repertoires. Importantly, the R metric demonstrates high efficiency in distinguishing and describing the functionally distinct T‐cell subsets, such as the CXCR3‐positive and CXCR3‐negative Treg cell subsets47 (Fig. 6d).
Figure 6.
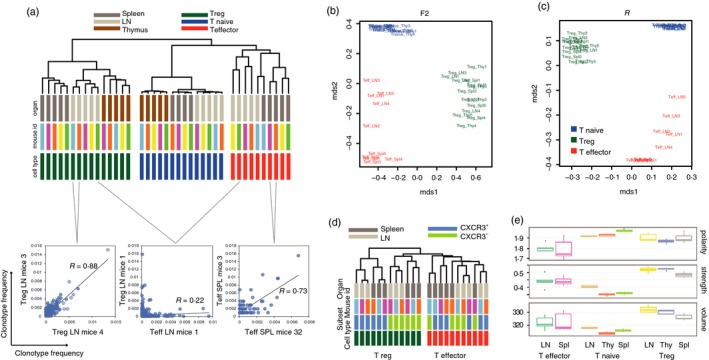
Analysis of the T‐cell receptor‐α (TCR‐α) repertoires on a fixed TCR‐β chain background. Amino acid TCR‐α CDR3 repertoires for the sorted T‐cell subsets of the Foxp3 gfp Tcra −/+ mice (n = 5) bearing the DO11.10 TCR‐β transgene were analysed. (a) Dendrogram showing ‘R’ overlap for the regulatory (green bars), naive (blue bars) and effector (red bars) CD4 T cells from spleen (dark grey bars), lymph nodes (LN, light grey) and thymus (brown). (b,c) Multi‐dimensional scaling (MDS) analysis of the TCR‐α repertoire overlaps using ‘F2’ (b) and ‘R’ (c) metrics. (d) Dendrogram showing ‘R’ overlap for the CXCR3‐positive and CXCR3‐negative T regulatory (green bars), and effector (red bars) CD4 T cells from spleen (dark grey bars) and lymph nodes (LN, light grey). (e) Amino acid composition characteristics of the CDR3 middle part. Analysis of the TCR repertoires derived from thymic (Thy), lymph node (LN) and splenic (Spl) CD4+ effector, CD4+ naive, and CD4+ regulatory T cells of Foxp3 gfp Tcra −/+ mice (n = 5) bearing the DO11.10 TCR‐β transgene. Weighted analysis is shown (i.e. the size of clonotypes was considered). (a–c) and (e) data are from ref. 46, (d) data are from ref. 47.
V‐ and J‐gene segments usage
A strategy that is simpler than the search for shared TCR sequences (but also quite useful and informative) is the comparison of V‐ and J‐segment usage frequencies, which may reflect the functional differences in TCR repertoires or biases in thymic recombination machinery. Comparison of segment usage frequencies can be performed in either a ‘weighted’ or ‘unweighted’ manner. Weighted gene‐segment usage analysis implies that each clonotype contributes proportional to its frequency and so largely depends on the nature of expanded clones in a sample. In unweighted analysis, each clonotype contributes equally and the clonotype frequencies are ignored. Such analysis mostly describes the average gene segments usage among numerous small clonotypes.
For example, the unweighted analysis of the TCR‐β variable (TRBV) segments usage distribution differentiated young from old C57BL/6J age peripheral blood TCR‐β repertoires (Fig. 7a), probably reflecting the age‐related difference in CD4 and CD8 and naive T‐cell ratios48 (Fig. 7b). At the same time, weighted analysis of segment usage poorly distinguished old from young mice, probably due to the presence of antigen‐experienced clonotypes that were expanded upon different challenges in a non‐deterministic manner, being intrinsically stochastic.
Figure 7.
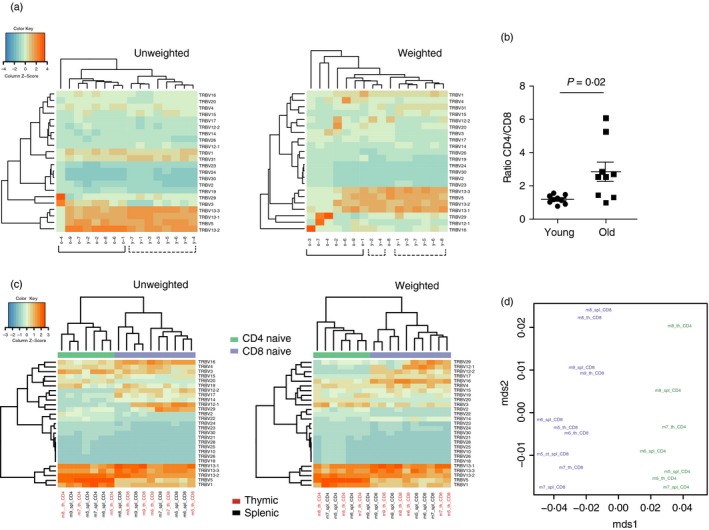
Analysis of the T‐cell receptor‐β variable (TRBV) gene segment usage. (a) Unweighted and weighted analysis of TRBV gene segment usage in old (n = 8) and young (n = 8) C57BL/6J mouse peripheral blood samples. (b) CD4/CD8 ratio in the young (n = 8) and old (n = 8) C57BL/6J mice. Two‐tailed Mann–Whitney test. (c) Unweighted and weighted analysis of TRBV gene segment usage in sorted thymic and splenic naive CD4 and CD8 T cells of 1‐year‐old C57BL/6J mice (n = 5). (d) Jensen–Shannon divergence of TRBV gene segment usage distributions in sorted thymic and splenic naive CD4 and CD8 T cells of 1‐year‐old C57BL/6J mice (n = 5).
Upon specific antigenic challenges, within functional T‐cell subsets in specific tissues, similar patterns of V‐ and J‐segment usage by expanding clones may be observed, making the weighted approach useful in revealing the functional differences between repertoires (our unpublished data). The above analysis of peripheral blood repertoires was performed on bulk, unsorted T cells. Hence, clonally diverse naive T cells mostly contributed to the case of unweighted analysis, while in the weighted analysis, naive T cells contribute proportionally to the percentage that they occupy in a T‐cell sample. Notably, for the sorted CD4 and CD8 naive T‐cell subsets of 1‐year‐old C57BL/6J mice, clearly distinct TRBV usage was observed using both weighted and unweighted approaches, without essential differences between the thymic and splenic naive T cells (Fig. 7c,d).
CDR3 characteristics
All CDR3 characteristics within the repertoire can also be analysed in a ‘weighted’ or ‘unweighted’ manner, i.e. considering or not considering the size of each clonotype. In the first case, the parameter is measured as average per CDR3‐containing sequencing read (or per UMI) in a sample. In the ‘unweighted’ mode, the parameter is measured as average per clonotype, where each clonotype contributes equally.
These two approaches reflect preferentially the characteristics of expanded clonotypes (or convergent clonotypes, i.e. independently recombined variants with the same nucleotide or amino acid sequence49, 50, 51) or of the expansion‐unaware repertoire composition, respectively.
Besides commonly used CDR3 characteristics related to a recombination process, such as distribution of CDR3 lengths within the TCR repertoire (in silico spectratyping) and a number of randomly added ‘N’ nucleotides (Fig. 2), it may be informative to explore the biophysical properties of amino acids within CDR3. For example, a relative number of strongly interacting amino acids,10 volume, polarity, or hydrophobicity of amino acids within CDR3 repertoires can be compared. To reflect functional differences in TCR repertoires with respect to potential recognition of antigenic peptides, such analysis may be limited to the middle portion of a CDR3 amino acid sequence, which mostly interacts with antigenic peptide presented by the MHC molecule.52 Such comparative analysis makes it possible to extract information on the general functional differences between the compared repertoires.
For instance, upon analysing different T‐cell subsets in Foxp3gfp Tcra −/+ mice we have found that Treg cell TCR‐α repertoires are characterized by distinct biophysical features, such as an increased volume and number of strongly interacting amino acids compared with effector/memory and naive T‐cell subpopulations (Fig. 6e). There are several factors that have been demonstrated to regulate the selection of Treg cells versus Tconv cells in the thymus. These include limiting niches of antigen‐presenting cells presenting self‐antigen, which dictate the overall numbers of Treg cells generated in the thymus;53, 54 expression of the AIRE transcription factor in medullary epithelial cells, which drives thymocytes with TCRs that recognize organ‐specific self‐peptides into the Treg cell lineage,55, 56, 57 and TCR affinity and signalling strength upon thymocyte engagement of self‐peptide–MHC complexes on thymic antigen‐presenting cells, with higher affinities and signalling strengths favouring Treg over Tconv cell selection.46, 58 TCR repertoire analysis could provide additional clues regarding the biophysical properties of Treg cell TCRs that may potentially affect thymic Treg cell selection. Bulky CDR3s containing strongly interacting amino acids were more prominent in the Treg cell TCR pool compared with Tconv T cells (Fig. 6e). Such characteristics of Treg cell CDR3 sequences may contribute to the previously observed higher TCR affinity of Treg cells for self‐peptide–MHC complexes, which may enable Treg cell thymocyte precursors to compete more efficiently for a limited niche of antigen found on thymic antigen‐presenting cells.
Discussion
Here we reviewed the typical examples of comparative post‐analysis of mouse TCR repertoires that can provide highly useful information for ongoing research on adaptive immunity. We hope that these examples and explanations will be helpful for investigators employing TCR repertoire analysis in fundamental studies and applied immunology. With the use of available software tools, many basic approaches to comparative analysis of deep sequenced immune repertoires become straightforward and feasible even for the laboratories with little bioinformatics experience. Applied to the variety of relevant mouse models ranging from infectious diseases, autoimmunity, cancer and vaccine development models to specific alterations in the immune system, it should provide a new level of understanding of diverse adaptive immune mechanisms, complementing decades of research in classical immunology.
Furthermore, rapid development of adaptive immunity profiling technologies indicates that possibilities of comparative repertoire analysis will also grow quickly in the near future. In particular, paired analysis of α and β chains using emulsion PCR,59, 60, 61, 62 stochastics63 and massive single‐cell sequencing5, 6 will join the ‘halves of the keys’, providing data on the fullly functional TCR repertoires. This will allow the performance of fully featured comparative analysis of functional TCR repertoire diversity and characteristics.
Development of single cell transcriptomics64, 65, 66 reveals new horizons for the scrupulous disentangling of immune cell social networks in multidimensional space. It should allow the creation of a merged picture of clonal and functional T‐cell diversity layers. By linking the TCR and functional properties of each cell,64, 67 we should be able to assess the functional heterogeneity of each T‐cell clone, build a landscape of such possible heterogeneities for a T‐cell repertoire, and compare these landscapes in health and upon various challenges.
Accumulation and systematization of the knowledge about TCRs with known antigenic specificities (https://vdjdb.cdr3.net/ 8) will allow us to screen analysed repertoires for the presence and abundance of particular antigen‐specific T cells. 1, 6, 7 For example, we have recently applied this approach to track the fate of cytomegalovirus‐ and Epstein–Barr virus‐specific T‐cell clones during human life, from birth to centenarians,16 revealing expansion of selected clones along with the decrease of antigen‐specific T‐cell diversity. Furthermore, a large knowledgebase of antigen‐specific TCR variants coupled with structural information can be used to develop de novo antigen specificity prediction methods that are not limited to a relatively small reference set of epitopes (currently ~100 epitopes with known TCRs in VDJdb) characterized so far.
Understanding the statistics behind recombinatorial events20, 68 and the selection pressure producing the functional TCR repertoires,21, 22, 23 makes it possible to assess the aetiology of studied TCR repertoires,45 to distinguish randomly shared clonotypes from the functionally derived convergence, and to distinguish unique clonotypes from independently produced identical public TCR variants.69
We are on the edge of attaining a much more comprehensive, global view on the whole mathematical beauty of adaptive immunity with the use of advanced bioinformatics analysis of high‐throughput TCR profiling data.
Disclosures
MiLaboratory LLC develops MiXCR, VDJtools and MiGEC software and has exclusive rights for its commercial distribution. DAB is employed by MiLaboratory LLC. DMC has a share in MiLaboratory LLC.
Funding
Supported by a grant from the Ministry of Education and Science of the Russian Federation No. 14.W03.31.0005. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supporting information
Table S1. Basic data and diversity metrics for the sequencing of young and old mice peripheral blood mononuclear cell TCR‐β repertoire.
Table S2. Basic data and diversity metrics for the sequencing of mice tissues TCR‐β repertoire.
Acknowledgements
The work was carried out in part using equipment provided by the IBCH core facility (CKP IBCH, equipment supported by the Russian Ministry of Education and Science, grant RFMEFI62117X0018).
References
- 1. Woodsworth DJ, Castellarin M, Holt RA. Sequence analysis of T‐cell repertoires in health and disease. Genome Med 2013; 5:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linnemann C, Mezzadra R, Schumacher TN. TCR repertoires of intratumoral T‐cell subsets. Immunol Rev 2014; 257:72–82. [DOI] [PubMed] [Google Scholar]
- 3. Greiff V, Miho E, Menzel U, Reddy ST. Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol 2015; 36:738–49. [DOI] [PubMed] [Google Scholar]
- 4. Heather JM, Ismail M, Oakes T, Chain B. High‐throughput sequencing of the T‐cell receptor repertoire: pitfalls and opportunities. Brief Bioinform 2017; doi: 10.1093/bib/bbw138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo XZ, Dash P, Calverley M, Tomchuck S, Dallas MH, Thomas PG. Rapid cloning, expression, and functional characterization of paired αβ and γδ T‐cell receptor chains from single‐cell analysis. Mol Ther Methods Clin Dev 2016; 3:15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dash P, Fiore‐Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A et al Quantifiable predictive features define epitope‐specific T cell receptor repertoires. Nature 2017; 547:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F et al Identifying specificity groups in the T cell receptor repertoire. Nature 2017; 547:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G et al VDJdb: a curated database of T‐cell receptor sequences with known antigen specificity. Nucleic Acids Res 2017; doi: 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venturi V, Quigley MF, Greenaway HY, Ng PC, Ende ZS, McIntosh T et al A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol 2011; 186:4285–94. [DOI] [PubMed] [Google Scholar]
- 10. Kosmrlj A, Jha AK, Huseby ES, Kardar M, Chakraborty AK. How the thymus designs antigen‐specific and self‐tolerant T cell receptor sequences. Proc Natl Acad Sci U S A 2008; 105:16671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bolotin DA, Mamedov IZ, Britanova OV, Zvyagin IV, Shagin D, Ustyugova SV et al Next generation sequencing for TCR repertoire profiling: platform‐specific features and correction algorithms. Eur J Immunol 2012; 42:3073–83. [DOI] [PubMed] [Google Scholar]
- 12. Seitz V, Schaper S, Droge A, Lenze D, Hummel M, Hennig S. A new method to prevent carry‐over contaminations in two‐step PCR NGS library preparations. Nucleic Acids Res 2015; 43:e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108:9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S et al Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 2012; 9:72–4. [DOI] [PubMed] [Google Scholar]
- 15. Casbon JA, Osborne RJ, Brenner S, Lichtenstein CP. A method for counting PCR template molecules with application to next‐generation sequencing. Nucleic Acids Res 2011; 39:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Britanova OV, Shugay M, Merzlyak EM, Staroverov DB, Putintseva EV, Turchaninova MA et al Dynamics of individual T cell repertoires: from cord blood to centenarians. J Immunol 2016; 196:5005–13. [DOI] [PubMed] [Google Scholar]
- 17. Britanova OV, Putintseva EV, Shugay M, Merzlyak EM, Turchaninova MA, Staroverov DB et al Age‐related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J Immunol 2014; 192:2689–98. [DOI] [PubMed] [Google Scholar]
- 18. Egorov ES, Merzlyak EM, Shelenkov AA, Britanova OV, Sharonov GV, Staroverov DB et al Quantitative profiling of immune repertoires for minor lymphocyte counts using unique molecular identifiers. J Immunol 2015; 194:6155–63. [DOI] [PubMed] [Google Scholar]
- 19. Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI et al Antigen receptor repertoire profiling from RNA‐seq data. Nat Biotechnol 2017; 35:908–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murugan A, Mora T, Walczak AM, Callan CG Jr. Statistical inference of the generation probability of T‐cell receptors from sequence repertoires. Proc Natl Acad Sci U S A 2012; 109:16161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Putintseva EV, Britanova OV, Staroverov DB, Merzlyak EM, Turchaninova MA, Shugay M et al Mother and child T cell receptor repertoires: deep profiling study. Front Immunol 2013; 4:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robins HS, Srivastava SK, Campregher PV, Turtle CJ, Andriesen J, Riddell SR et al Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med 2010; 2:47ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubelt F, Bolen CR, McGuire HM, Vander Heiden JA, Gadala‐Maria D, Levin M et al Individual heritable differences result in unique cell lymphocyte receptor repertoires of naive and antigen‐experienced cells. Nat Commun 2016; 7:11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zvyagin IV, Pogorelyy MV, Ivanova ME, Komech EA, Shugay M, Bolotin DA et al Distinctive properties of identical twins’ TCR repertoires revealed by high‐throughput sequencing. Proc Natl Acad Sci U S A 2014; 111:5980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. https://arxiv.org/abs/1604.00487.
- 26. Laydon DJ, Bangham CR, Asquith B. Estimating T‐cell repertoire diversity: limitations of classical estimators and a new approach. Philos Trans R Soc Lond B Biol Sci 2015; 370: 20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matz M, Shagin D, Bogdanova E, Britanova O, Lukyanov S, Diatchenko L et al Amplification of cDNA ends based on template‐switching effect and step‐out PCR. Nucleic Acids Res 1999; 27:1558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Douek DC, Betts MR, Brenchley JM, Hill BJ, Ambrozak DR, Ngai KL et al A novel approach to the analysis of specificity, clonality, and frequency of HIV‐specific T cell responses reveals a potential mechanism for control of viral escape. J Immunol 2002; 168:3099–104. [DOI] [PubMed] [Google Scholar]
- 29. Mamedov IZ, Britanova OV, Zvyagin IV, Turchaninova MA, Bolotin DA, Putintseva EV et al Preparing unbiased T‐cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol 2013; 4:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zvyagin IV, Mamedov IZ, Tatarinova OV, Komech EA, Kurnikova EE, Boyakova EV et al Tracking T‐cell immune reconstitution after TCRαβ/CD19‐depleted hematopoietic cells transplantation in children. Leukemia 2017; 31:1145–53. [DOI] [PubMed] [Google Scholar]
- 31. Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948; 27:379–423. [Google Scholar]
- 32. Simpson EH. Measurement of diversity. Nature 1949; 163:688. [Google Scholar]
- 33. Efron B, Thisted R. Estimating the number of unseen species: how many words did Shakespeare know?. Biometrika 1976; 63:435–47. [Google Scholar]
- 34. Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol 2001; 67:4399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colwell RK, Chao A, Gotelli NJ, Lin S, Mao CX, Chazdon RL et al Models and estimators linking individual‐based and sample‐based rarefaction, extrapolation and comparison of assemblages. J Plant Ecol 2012; 5:3–21. [Google Scholar]
- 36. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV et al MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015; 12:380–1. [DOI] [PubMed] [Google Scholar]
- 37. Shugay M, Britanova OV, Merzlyak EM, Turchaninova MA, Mamedov IZ, Tuganbaev TR et al Towards error‐free profiling of immune repertoires. Nat Methods 2014; 11:653–5. [DOI] [PubMed] [Google Scholar]
- 38. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY et al Diversity and clonal selection in the human T‐cell repertoire. Proc Natl Acad Sci U S A 2014; 111:13139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cho BK, Wang C, Sugawa S, Eisen HN, Chen J. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci U S A 1999; 96:2976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schrum AG, Wells AD, Turka LA. Enhanced surface TCR replenishment mediated by CD28 leads to greater TCR engagement during primary stimulation. Int Immunol 2000; 12:833–42. [DOI] [PubMed] [Google Scholar]
- 41. Schrum AG, Turka LA, Palmer E. Surface T‐cell antigen receptor expression and availability for long‐term antigenic signaling. Immunol Rev 2003; 196:7–24. [DOI] [PubMed] [Google Scholar]
- 42. Xu H, Luo X, Qian J, Pang X, Song J, Qian G et al FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 2012; 7:e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV et al VDJtools: unifying post‐analysis of T cell receptor repertoires. PLoS Comput Biol 2015; 11:e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Madi A, Poran A, Shifrut E, Reich‐Zeliger S, Greenstein E, Zaretsky I et al T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. Elife 2017; 6:e22057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pogorelyy MV, Elhanati Y, Marcou Q, Sycheva AL, Komech EA, Nazarov VI et al Persisting fetal clonotypes influence the structure and overlap of adult human T cell receptor repertoires. PLoS Comput Biol 2017; 13:e1005572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feng Y, van der Veeken J, Shugay M, Putintseva EV, Osmanbeyoglu HU, Dikiy S et al A mechanism for expansion of regulatory T‐cell repertoire and its role in self‐tolerance. Nature 2015; 528:132–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Levine AG, Medoza A, Hemmers S, Moltedo B, Niec RE, Schizas M et al Stability and function of regulatory T cells expressing the transcription factor T‐bet. Nature 2017; 546:421–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahmed M, Lanzer KG, Yager EJ, Adams PS, Johnson LL, Blackman MA. Clonal expansions and loss of receptor diversity in the naive CD8 T cell repertoire of aged mice. J Immunol 2009; 182:784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Venturi V, Kedzierska K, Price DA, Doherty PC, Douek DC, Turner SJ et al Sharing of T cell receptors in antigen‐specific responses is driven by convergent recombination. Proc Natl Acad Sci U S A 2006; 103:18691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H, Ye C, Ji G, Wu X, Xiang Z, Li Y et al Recombinatorial biases and convergent recombination determine interindividual TCRβ sharing in murine thymocytes. J Immunol 2012; 189:2404–13. [DOI] [PubMed] [Google Scholar]
- 51. Quigley MF, Greenaway HY, Venturi V, Lindsay R, Quinn KM, Seder RA et al Convergent recombination shapes the clonotypic landscape of the naive T‐cell repertoire. Proc Natl Acad Sci U S A 2010; 107:19414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang JC, Ober RJ, Ward ES. The central residues of a T cell receptor sequence motif are key determinants of autoantigen recognition in murine experimental autoimmune encephalomyelitis. Eur J Immunol 2005; 35:299–304. [DOI] [PubMed] [Google Scholar]
- 53. Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self‐reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol 2006; 7:401–10. [DOI] [PubMed] [Google Scholar]
- 54. Lee HM, Bautista JL, Scott‐Browne J, Mohan JF, Hsieh CS. A broad range of self‐reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity 2012; 37:475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ et al Projection of an immunological self shadow within the thymus by the AIRE protein. Science 2002; 298:1395–401. [DOI] [PubMed] [Google Scholar]
- 56. Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. AIRE enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 2016; 44:1102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Derbinski J, Gabler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M et al Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J Exp Med 2005; 202:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA et al Thymic selection of CD4+ CD25+ regulatory T cells induced by an agonist self‐peptide. Nat Immunol 2001; 2:301–6. [DOI] [PubMed] [Google Scholar]
- 59. Turchaninova MA, Britanova OV, Bolotin DA, Shugay M, Putintseva EV, Staroverov DB et al Pairing of T‐cell receptor chains via emulsion PCR. Eur J Immunol 2013; 43:2507–15. [DOI] [PubMed] [Google Scholar]
- 60. Munson DJ, Egelston CA, Chiotti KE, Parra ZE, Bruno TC, Moore BL et al Identification of shared TCR sequences from T cells in human breast cancer using emulsion RT‐PCR. Proc Natl Acad Sci U S A 2016; 113:8272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeKosky BJ, Ippolito GC, Deschner RP, Lavinder JJ, Wine Y, Rawlings BM et al High‐throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol 2013; 31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD et al In‐depth determination and analysis of the human paired heavy‐ and light‐chain antibody repertoire. Nat Med 2015; 21:86–91. [DOI] [PubMed] [Google Scholar]
- 63. Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW et al High‐throughput pairing of T cell receptor α and β sequences. Sci Transl Med 2015; 7:301ra131. [DOI] [PubMed] [Google Scholar]
- 64. Fan HC, Fu GK, Fodor SP. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 2015; 347:1258367. [DOI] [PubMed] [Google Scholar]
- 65. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M et al Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell 2015; 161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V et al Droplet barcoding for single‐cell transcriptomics applied to embryonic stem cells. Cell 2015; 161:1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Han A, Glanville J, Hansmann L, Davis MM. Linking T‐cell receptor sequence to functional phenotype at the single‐cell level. Nat Biotechnol 2014; 32:684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Elhanati Y, Murugan A, Callan CG Jr, Mora T, Walczak AM. Quantifying selection in immune receptor repertoires. Proc Natl Acad Sci U S A 2014; 111:9875–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nazarov VI, Minervina AA, Komkov AY, Pogorelyy MV, Maschan MA, Olshanskaya YV et al Reliability of immune receptor rearrangements as genetic markers for minimal residual disease monitoring. Bone Marrow Transplant 2016; 51:1408–10. [DOI] [PubMed] [Google Scholar]
- 70. Chiu CH, Chao A. Estimating and comparing microbial diversity in the presence of sequencing errors. PeerJ 2016; 4:e1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Elhanati Y, Marcou Q, Mora T, Walczak AM. repgenHMM: a dynamic programming tool to infer the rules of immune receptor generation from sequence data. Bioinformatics 2016; 32:1943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Basic data and diversity metrics for the sequencing of young and old mice peripheral blood mononuclear cell TCR‐β repertoire.
Table S2. Basic data and diversity metrics for the sequencing of mice tissues TCR‐β repertoire.