

Abstract
The Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative is developing and validating a mechanistic‐based assessment of the proarrhythmic risk of drugs. CiPA proposes to assess a drug's effect on multiple ion channels and integrate the effects in a computer model of the human cardiomyocyte to predict proarrhythmic risk. Unanticipated or missed effects will be assessed with human stem cell‐derived cardiomyocytes and electrocardiogram (ECG) analysis in early phase I clinical trials. This article provides an overview of CiPA and the rationale and design of the CiPA phase I ECG validation clinical trial, which involves assessing an additional ECG biomarker (J‐Tpeak) for QT prolonging drugs. If successful, CiPA will 1) create a pathway for drugs with hERG block / QT prolongation to advance without intensive ECG monitoring in phase III trials if they have low proarrhythmic risk; and 2) enable updating drug labels to be more informative about proarrhythmic risk, not just QT prolongation.
PROARRHYTHMIA RISK EVALUATION: PUBLIC HEALTH AND REGULATORY CONTEXT
In the 1990s to early 2000s, multiple noncardiac drugs were removed from the market because of their association with torsade de pointes (TdP), a potentially fatal ventricular arrhythmia.1 It was quickly recognized that these drugs usually block the potassium channel encoded by the human ether‐à‐go‐go related gene (hERG). The block of the hERG potassium channel (referred to as “hERG block”) reduces the delayed rectifier potassium current (IKr) and prolongs cardiac repolarization, which appears as prolongation of the heart rate corrected QT (QTc) interval on the electrocardiogram (ECG) and predisposes to arrhythmias. In response, two international regulatory guidelines for industry were established: the International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S7B2 and the ICH E14.3 The ICH S7B recommends an in vitro assay to assess whether a compound and its metabolites block hERG, while the ICH E14 guideline describes a specific clinical study, the thorough QT (TQT) study.
ICH S7B2 describes a nonclinical testing strategy for assessing the potential of an investigational new drug and its metabolites to prolong ventricular repolarization (Figure 1 a). The guideline recommends an integrated risk assessment that includes 1) an in vitro assay to evaluate the effects on late repolarization currents (e.g., hERG assay); 2) an in vivo (animal) QT assay; and 3) use of chemical and pharmacological class information as well as nonclinical and clinical information. Results of these studies can support the planning and interpretation of subsequent clinical studies.
Figure 1.
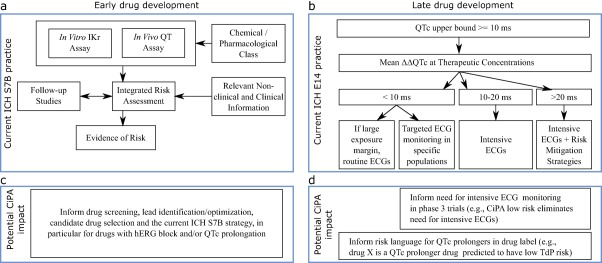
Potential impact of CiPA on early and late drug development. (a) Top, left panel shows current ICH S7B nonclinical testing strategy. (b) Under CiPA, early assessment of drug effects on multiple ion channels using high‐throughput systems coupled with the in silico model predictions and outcomes from human induced pluripotent stem cell (iPSC) derived cardiomyocytes assays could inform lead identification/optimization, candidate drug selection, and the current ICH S7B strategy, in particular for drugs with hERG block and/or QTc prolongation. (c) Top, right panel shows current ICH E14 ECG monitoring considerations for QTc prolonging drugs in clinical development. Intensity of ECG monitoring depends on pharmacokinetic characteristics, patient characteristics, and adverse events that increase proarrhythmic risk. (d) CiPA's mechanistic approach will provide additional information that could influence this ECG monitoring decision process and drug labeling (see FDA advisory committee9 responses to questions in the text). Top, left panel diagram A adapted from ICH S7B guideline.2 Top, right panel diagram C depicts examples described in ICH E14 Questions & Answers Document in Section 7 (Electrocardiograms Monitoring in Late Stage Clinical Trials).4 ΔΔQTc, QTc placebo adjusted change from baseline.
ICH E143 describes the clinical evaluation of QTc prolongation and proarrhythmic potential for nonantiarrhythmic drugs, the TQT study (Figure 1 c). The goal of the TQT study is not to classify drugs as being proarrhythmic per se. The primary goal is to determine whether a drug prolongs the QTc beyond a certain threshold that informs the necessary intensity of ECG monitoring in late‐stage clinical trials in patients and to cover the supratherapeutic concentration that would be obtained due to inhibition of metabolism or excretion. TQT studies are typically conducted in healthy volunteers and include dose levels that result in drug concentrations exceeding those expected in patients. The ICH E14 Questions & Answers (Q&A) document was revised in 2015 to allow exposure–response (concentration‐QTc) modeling to be used as a primary analysis for assessing the QTc interval prolongation risk of new drugs.4 The use of exposure–response analysis in early‐phase studies can serve as an alternative pathway to assess QTc prolongation of new drugs in lieu of a dedicated TQT study.5
The ICH S7B and E14 guidelines have been successful in that no new marketed drugs have been associated with unexpected QTc prolongation or unacceptable risk of TdP. However, this has had unintended effects on drug development. While these two biomarkers (hERG block and QTc prolongation) are sensitive for identifying drugs with TdP risk, they are not specific, as multiple drugs block hERG and/or prolong QTc but do not cause TdP. Drug developers routinely perform early screening for hERG potassium channel activity and may discontinue compounds from development if they block hERG.6 They could then select compounds to advance that may not have optimal on‐target or other pharmacological profile effects.7 Furthermore, the finding of QTc prolongation during clinical development can result in the termination of a drug despite the fact that QTc prolongation does not necessarily predict a significant proarrhythmic liability. In addition, numerous drugs contain labeling information about QTc prolongation (often quite moderate prolongation) but have an unclear risk of TdP. This may influence a physician's prescribing behavior, such as discouraging the use of certain drugs.
The Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative emerged from the need to develop a standardized, mechanistic‐based approach to determine actual TdP proarrhythmic risk (instead of the current approach of relying on nonspecific biomarkers) that could be applied early in drug development to aid in compound selection (Figure 1 b).8 Since 2013, when it was first introduced, the CiPA initiative has matured into a global effort among regulators (US Food and Drug Administration (FDA), European Medicines Agency, Health Canada, Japan Pharmaceuticals and Medical Devices Agency), industry, and academia coordinated by multiple public–private partnerships (Health and Environmental Science Institute, the Safety Pharmacology Society and the Cardiac Safety Research Consortium). The CiPA initiative proposes a new mechanistic, model‐informed approach to cardiac safety assessment of new drugs, an approach made possible by a more comprehensive understanding of the ionic currents that play a role in QTc prolongation and the development of TdP.
On March 15, 2017, the FDA held a Pharmaceutical Science and Clinical Pharmacology Advisory Committee public meeting to receive input on the CiPA initiative.9 The advisory committee voted on three questions related to the proposed approach and potential impact of CiPA as well as the FDA's role in facilitating implementation. The questions and voting results are summarized below:
-
For a QT‐prolonging drug, will this mechanistic, model‐based approach be fit for the following two applications:
Determining whether ECGs need to be collected in phase III: VOTE: 11 yes, 2 no;
Informing proarrhythmic risk language in drug labeling: VOTE: 11 yes, 2 no.
Does the committee agree with the proposed approach for validating the new paradigm that involves assessing 28 drugs classified into low, intermediate, and high risk by an expert panel? VOTE: 10 yes, 3 no.
As this new mechanistic, model‐based approach is implemented, should the FDA collect the world's experience (i.e., digital waveform data from in vitro experiments) to facilitate future enhancements, as was done by the FDA with the ECG warehouse for QT studies? VOTE: 13 yes, 0 no.
The advisory committee members also expressed interest in CiPA's ongoing validation efforts. This article provides a review of the CiPA initiative followed by the background and rationale of the ongoing validation clinical study for the ECG component that would be applied in early phase I clinical trials.
RATIONALE AND COMPONENTS OF CiPA
As depicted in Figure 2, a key tenet of the CiPA initiative is that ventricular repolarization and TdP risk are dependent on a “symphony” or “balance” of multiple inward and outward ion channel currents, not just reduced IKr by hERG block.10 In terms of drug‐induced TdP, the two most important currents in addition to IKr are the late sodium current and L‐type calcium current. IKr is an outward current that, when active, facilitates repolarization. Reduction of IKr caused by hERG block (Figure 2 a) delays repolarization, increasing the probability of L‐type calcium current triggering extra beats called early afterdepolarizations (EADs; Figure 2 b), which can initiate TdP (Figure 2 c). Because EADs are triggered by inward currents,11, 12 blocking inward currents (late sodium or calcium; Figure 2 d) can prevent EADs (Figure 2 e) and has antiarrhythmic effects (i.e., not leading to TdP; Figure 2 f).11, 12, 13, 14, 15, 16, 17, 18 Therefore, drugs that block hERG, but also have approximately equipotent late sodium or calcium channel blocking effects, are likely to have a low risk of TdP. These are referred to as balanced ion channel‐blocking drugs. Drugs illustrating this could include verapamil (hERG + calcium block), ranolazine (hERG + late sodium block), and some of the human immunodeficiency virus (HIV) protease inhibitors such as lopinavir and ritonavir, which block hERG, late sodium, and calcium.19 Amiodarone and its metabolite cause potent hERG block and QTc prolongation, often >60–90 ms; however, it has been associated with a low risk of TdP despite its profound effects on repolarization, probably because it also blocks both late sodium and calcium currents.19 Thus, late sodium and calcium block shorten the action potential duration of ventricular cells20 (and QTc13, 14, 15); and, in the presence of QTc prolongation, blockage of these inward currents can prevent the initiation of TdP.21
Figure 2.
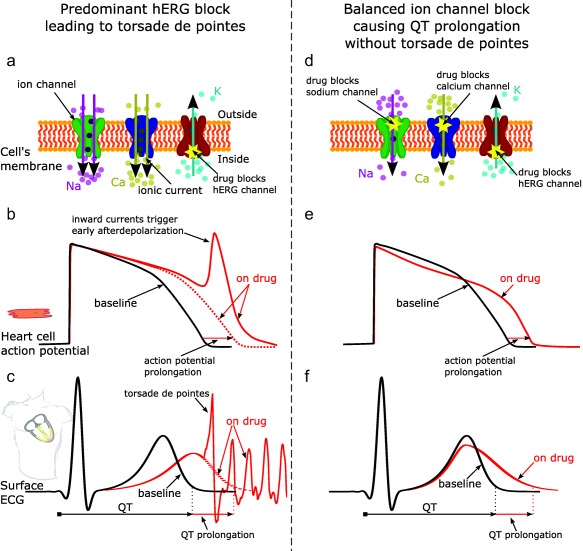
Effect of predominant hERG potassium channel block vs. balanced ion channel block on QT prolongation and generation of torsade de pointes. Illustration of predominant hERG block leading to torsade de pointes (left column) vs. balanced ion channel block causing QT prolongation without torsade de pointes (right column). Predominant hERG block reduces the hERG potassium channel current (panel a), which delays repolarization and prolongs the action potential duration of cardiomyocytes (panel b, red dotted line) and the QT interval on the ECG (panel c, red dotted line). Prolonged repolarization can result in early afterdepolarizations (panel b, red solid line), which are caused by inward currents through sodium and calcium channels11 (panel a, purple and yellow arrows) and can trigger torsade de pointes (panel c, solid red line). In addition to causing hERG block, balanced ion channel‐blocking drugs block the L‐type calcium and/or late sodium currents (panel d). While balanced ion channel block can prolong both the action potential duration of cadiomyocytes (panel e) and the QT interval on the ECG (panel f), the block of inward currents (calcium, late sodium) prevents the occurrence of early afterdepolarizations and has antiarrhythmic effects.11, 12, 13, 14, 15, 16, 17, 18 In addition, balanced ion channel block causes different morphology changes in the action potential (panel e, red solid line) and the ECG (panel f, red solid line) than predominant hERG block (panels b and c, respectively). The goal of the CiPA phase I ECG validation study described in this article is to show that exposure–response ECG analysis can differentiate predominant hERG‐blocking drugs from balanced ion channel‐blocking drugs. Na, sodium ions; Ca, calcium ions; K, potassium ions.
Other evidence of the importance of late sodium current block in preventing TdP comes from a canine model of TdP, where coadministration of mexiletine, a late sodium current blocker, reduced QTc prolongation and the number of TdP events associated with sotalol, a predominant hERG blocker.16 In addition, previous clinical studies have demonstrated that mexiletine can mitigate QTc prolongation caused by the strong hERG blocker quinidine.13, 14, 15 This has been confirmed in recent clinical studies demonstrating that late sodium current blockers (mexiletine, lidocaine) can reduce drug‐induced QTc prolongation caused by the selective hERG blocker dofetilide,22 and also that mexiletine can mitigate recurrent TdP caused by the acquired long QT syndrome.18
Because there is now a comprehensive understanding of the ionic currents that play a role in QTc prolongation and the development of TdP, the CiPA initiative proposes a new mechanistic, model‐informed approach to cardiac safety assessment of new drugs. Figure 3 illustrates the four components of CiPA's approach: 1) in vitro assessment of drug effects on multiple cardiac ion channels; 2) integration of the multi‐ion channel effects in an in silico computer model of the human ventricular cardiomyocyte to output a proarrhythmic risk score; 3) assessment of unanticipated effects in vitro using human induced pluripotent stem cell (iPSC) derived cardiomyocytes; and 4) phase I clinical studies with exposure–response analysis to assess whether the in vivo human electrophysiology differs from what would be expected based on the ion channel testing.
Figure 3.
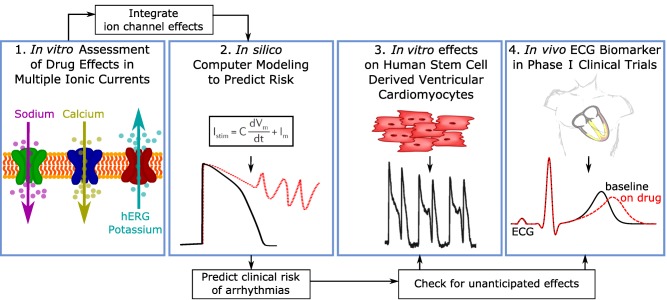
The four components of the Comprehensive in vitro Proarrhythmia Assay (CiPA). Illustration of the four components of the CiPA initiative. First, drug effects on multiple ion channel currents are assessed. Second, a proarrhythmic score is computed using an in silico model of the human ventricular cardiomyocyte, which integrates the individual ion channel effects assessed in the first component. The third component is a confirmatory in vitro study using human stem cell‐derived ventricular cardiomyocytes. The goal of component four is to use human phase I ECG data to determine if there are unexpected ion channel effects in humans compared to preclinical ion channel data.60
Four working groups have tackled each of the four components, developing intersecting validation plans. To support development and validation of the nonclinical CiPA assays, a set of drugs with well‐defined cardiac electrophysiology and known clinical characteristics was identified by a team of expert clinicians, safety pharmacologists, and cardiac electrophysiologists. This drug selection group discussed, selected, and categorized 28 drugs (12 for training and 16 for validation, Supplementary Table S1) into low, intermediate, and high risk of TdP based on published reports, analysis of the FDA adverse event reporting system database, other data sources, and expert opinion.6, 10, 23 These 28 drugs are being tested in vitro against the multiple ion channel currents and in human iPSC‐cardiomyocytes.
As was discussed during the FDA advisory committee deliberations, it is important to note that CiPA is not intended to inform risk for individual patients (i.e., personalized medicine) that may have rare genetic channelopathies that increase the risk for arrhythmias, but rather to rank the risk of a new drug relative to 28 drugs with well‐characterized effects based on years of clinical experience. Of note, the current paradigm (ICH S7B/E14) focuses on assessing hERG block and QT prolongation in healthy volunteers. As novel technologies advance (e.g., organs‐on‐chips and 3D human iPSC‐cardiomyocytes), their potential application to drug development will be considered, dependent on validation studies that can establish adequate reproducibility and predictive ability.10, 24, 25, 26, 27
Ion channel, in silico modeling, and human iPSC‐cardiomyocyte work streams
For the ion channel work stream, the 12 training drugs have been assessed against seven ion channel currents using manual patch clamp techniques to assess block potency19 (e.g., to determine percentage ion channel current block and half maximal inhibitory concentration (IC50)) and dynamic hERG block.28, 29 It was observed that hERG, late sodium, and L‐type calcium currents were the most frequently blocked ion channel currents at maximum free drug concentrations (Cmax) and that low‐risk drugs had equal or greater late sodium or calcium block compared to hERG block.19 The remaining 16 validation drugs are also being assessed via manual patch clamp techniques for their effects on these most critical ion channel currents: hERG, calcium, and sodium (peak and late currents). At the same time, all 28 CiPA drugs are part of an international, multicenter study using high‐throughput automated patch clamp techniques.
For the in silico modeling work stream, a dynamic model of the hERG potassium channel with temperature‐sensitive transitions between channel states was developed by modifying the Markov model published by Di Veroli et al. in 2013,30 which could reproduce temperature‐induced changes in gating processes.31 This hERG model was incorporated into the O'Hara–Rudy ventricular cardiomyocyte cell model32 and experimental human heart data was reproduced. Using this updated O'Hara–Rudy cell model, the initial 12 CiPA drugs were categorized into low, intermediate, and high risk for TdP (Figure 4).28, 31, 33 This categorization was made computing the area under the curve of the net current (i.e., the sum of outward potassium and inward calcium and late sodium currents (qNet)) during an action potential.33 The in silico group also developed a method to incorporate experimental uncertainty and investigated the need for simulating intersubject variability. The in silico model is now being tested with the 16 CiPA validation drugs.
Figure 4.
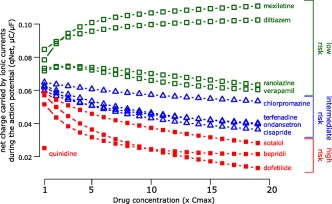
In silico proarrhythmia risk categorization of the 12 CiPA training set drugs. Mechanistic proarrhythmic prediction from the in silico cardiomyocyte model indicating how close a drug is to generating an early afterdepolarization, the trigger for torsade de pointes. The X axis indicates drug concentration in multiples of clinical Cmax for each drug separately (e.g., 1‐fold Cmax, 2‐fold Cmax, etc., up to 19‐fold Cmax); Y axis is the proarrhythmic metric, which is defined as the area under the curve of the net current during an action potential (qNet, see text and Dutta et al.33). Drugs associated with different TdP risk categories are labeled and color‐coded as high risk (red solid squares), intermediate risk (blue triangles), and low risk (green empty squares) based on the clinical risk categorization in Supplementary Table S1. For quinidine, qNet is reported only for Cmax because simulations for higher concentrations caused early afterdepolarizations. Adapted from Dutta et al.33
Similar to the ion channel and in silico work, the 28 CiPA drugs are being assessed in human iPSC‐cardiomyocyte assays in an international, multisite validation study. Two different laboratory device platforms are being studied: multielectrode arrays and voltage‐sensitive optical technologies. While that analysis is ongoing, multiple preliminary studies have demonstrated the potential of these technologies.34, 35, 36, 37 The exact role of the iPSC‐cardiomyocytes under CiPA will be guided by the results of these validation efforts.
Clinical phase I ECG biomarker work stream
The last component of CiPA involves an ECG assessment in early clinical phase I studies (single or multiple ascending dose studies) to determine if there are unexpected ion channel effects compared to the preclinical ion channel data, such as due to a human‐specific metabolite or differences in protein binding. The CiPA phase I ECG working group identified the following key criteria for any new CiPA ECG biomarker: the new ECG biomarker must 1) add additional information beyond PR, QRS, and QTc that allows for differentiating predominant hERG‐blocking drugs (with TdP risk) from drugs that block hERG along with calcium and/or late sodium (balanced ion channel‐blocking drugs with low TdP risk); 2) be corrected for heart rate if needed; 3) have sufficient power to detect changes in small sample sizes with exposure–response analysis; and 4) be available for widespread use.
The CiPA phase I ECG working group considered 12 potential ECG biomarkers.38 Through analysis of two prior FDA‐sponsored clinical trials including eight drugs (dofetilide, quinidine, ranolazine, verapamil, lidocaine, mexiletine, moxifloxacin, and diltiazem), along with multiple drug combinations, the working group identified the heart rate‐corrected J‐Tpeak interval (J‐Tpeakc; defined below) as the best biomarker to differentiate QTc‐prolonging drugs with predominant hERG block (with TdP risk) from QTc‐prolonging drugs with hERG and late sodium or calcium current block (balanced ion channel‐blocking drugs with low TdP risk).39
Figure 5 a defines the J‐Tpeak interval and shows its relation to the ventricular cell action potential. The QT interval can be divided into three components: 1) QRS interval that corresponds to ventricular cell depolarization; 2) end of QRS to peak of the T‐wave (J‐Tpeak interval) that corresponds to early repolarization; and 3) the peak of the T‐wave to end of the T‐wave (Tpeak‐Tend interval) that corresponds to late repolarization. A retrospective analysis of 34 TQT studies40 followed by two prospective clinical trials22, 41 including eight drugs demonstrated that drugs that predominantly block hERG prolong QTc by prolonging both J‐Tpeakc (early repolarization) and Tpeak‐Tend (late repolarization), while drugs that block hERG along with calcium and/or late sodium (balanced ion channel block) prolonged QTc by prolonging Tpeak‐Tend with no or limited effect on the J‐Tpeakc interval.22, 40, 41 Figure 5 b shows data from the first prospective clinical trial,41 where dofetilide (predominant hERG blocker) prolonged QTc by prolonging both J‐Tpeakc and Tpeak‐Tend. In contrast, ranolazine (balanced hERG and late sodium blocker) prolonged QTc by only prolonging Tpeak‐Tend, but not J‐Tpeakc.
Figure 5.
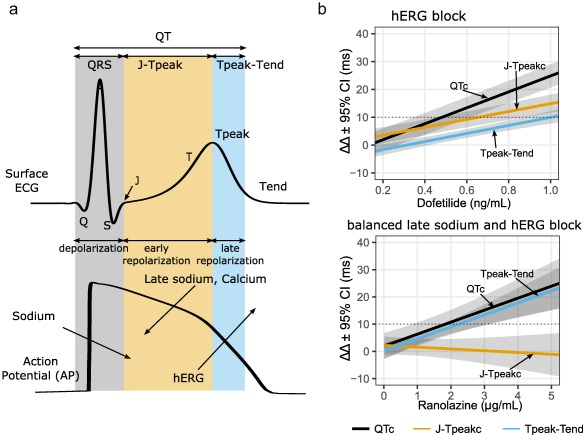
Relationship between components of the QT interval and underlying ion channel currents. (a) An illustration of the body‐surface ECG and a corresponding ventricular action potential. The QT interval can be divided into depolarization (QRS), early repolarization (J‐Tpeak), and late repolarization (Tpeak‐Tend). Arrows pointing into the action potential are inward currents (sodium and calcium) and arrows pointing out denote outward currents (hERG). The J‐Tpeak interval corresponds with the plateau of the action potential and the balance of inward vs. outward ionic currents. (b) Exposure–response models showing the effects of a predominant hERG blocker (dofetilide; top panel) vs. a balanced hERG and late sodium blocker (ranolazine; bottom panel). Both drugs prolong QTc and Tpeak‐Tend; however, only dofetilide prolongs J‐Tpeakc. The absence of J‐Tpeakc prolongation is a sign of balanced ion channel block between outward (hERG) and inward (late sodium) currents. Adapted from Johannesen et al.41
A second clinical trial22 studied the combined effects of dofetilide (predominant hERG blocker) with lidocaine and mexiletine, which are each predominant late sodium blockers. The goal of the study was to determine if the ECG “signature” of ranolazine (balanced hERG and late sodium block) could be recreated by combining two separate drugs (dofetilide + lidocaine and dofetilide + mexiletine). As shown in Figure 6, dofetilide alone prolonged QTc by prolonging both J‐Tpeakc and Tpeak‐Tend. When lidocaine and mexiletine were coadministered with dofetilide, QTc was shortened by exclusively shortening J‐Tpeakc, without an effect on Tpeak‐Tend. Thus, the ECG “signature” of ranolazine was recreated, where QTc is prolonged without prolonging J‐Tpeakc.
Figure 6.
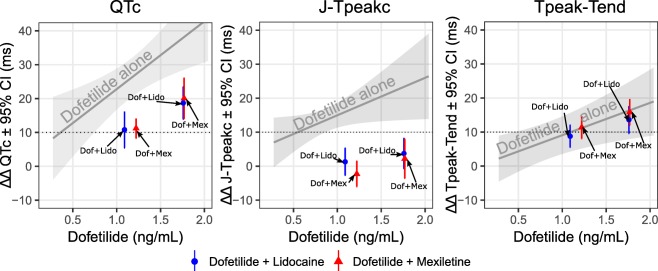
ECG effects of hERG block alone vs. combined hERG and late sodium block. Results from a clinical trial studying the effects of a hERG blocker alone (dofetilide) and a hERG blocker (dofetilide) in combination with a late sodium blocker (lidocaine or mexiletine). As in Figure 5 b, dofetilide alone prolongs QTc (left panel), J‐Tpeakc (middle panel), and Tpeak‐Tend (right panel). When lidocaine or mexiletine are coadministered with dofetilide, QTc shortens. Timepoints when lidocaine or mexiletine were coadministered are shown as blue and red, respectively, oriented on the x‐axis according to the dofetilide concentration. The QTc shortening effect of lidocaine and mexiletine (left panel) was entirely due to shortening the J‐Tpeakc interval (middle panel), with no effect on Tpeak‐Tend (right panel). Thus, the combination of late sodium current‐blocking drugs (lidocaine or mexiletine) with a hERG‐blocking drug (dofetilide) recreated the ECG “signature” of ranolazine as shown in Figure 5 b. Adapted from Johannesen et al.22
Most of the other studied ECG biomarkers had exposure–response relationships with hERG block, but paralleled QTc changes and did not provide independent information.38 In addition, the J‐Tpeak interval can be corrected for heart rate40 and has sufficient power to differentiate changes with exposure–response analysis in the small sample sizes (Section 2 in the statistical analysis plan in the Online Supplement) typical of phase I studies. The J‐Tpeakc analysis algorithm has been released as an open‐source code to facilitate widespread testing (https://github.com/FDA/ecglib),42 which has been performed by multiple independent groups as a part of an “ECG challenge” associated with the 2017 International Society of Computerized Electrocardiology annual conference.43, 44, 45, 46, 47
The CiPA phase I ECG working group held a public workshop sponsored by the Cardiac Safety Research Consortium in April 2016, where the current status, validation, and implementation strategy were discussed. Identified next steps included: 1) finalizing a statistical framework that can be applied in small sample sizes with exposure–response analysis similar to QTc; 2) extending analysis to a larger number of prior clinical studies with matching multi‐ion channel data; 3) performing a multisite reproducibility study of using the J‐Tpeakc interval; and 4) planning and completing a confirmatory prospective study in 2017 with a small sample size design similar to first‐in‐human single ascending dose or multiple ascending dose studies. The design of this validation study is described below.
Potential impact of CiPA
If CiPA is successful, it will enable compounds with hERG block and/or QTc prolongation, which might be terminated from development under the current paradigm, to have a clearer path to advance if they are shown to have low proarrhythmic risk. Clinical development would be streamlined as drugs with a low proarrhythmic potential, despite inhibiting hERG and prolonging the QTc interval, would not require intensive ECG monitoring during phase III clinical trials and could be expected to have labeling indicating they are low‐risk drugs (Figure 1 d). In addition, QTc prolonging drugs on the market that are not proarrhythmic could have labeling updated to reflect this.
As previously discussed, the primary goal of the TQT study described in ICH E14 is to determine the intensity of ECG monitoring in later‐stage clinical studies based on whether a drug prolongs QTc. If the drug prolongs QTc, ICH E14 and its Q&A provide an approach as to which intensity of monitoring should be applied in late‐stage studies (Figure 1 c). With CiPA, the integrated assessment of proarrhythmic risk described earlier could influence this monitoring decision process (Figure 1 d). In particular, the CiPA ECG component and the ECG biomarker clinical validation study discussed below could provide more clarity in cases where the QTc is prolonged by more than 10 ms.
CLINICAL VALIDATION STUDY BACKGROUND AND MOTIVATION
As outlined previously, at clinically relevant concentrations, drugs that predominantly block hERG without late sodium or L‐type calcium current block (predominant hERG‐blocking drugs) cause QTc prolongation19, 22, 40, 41 and have a high risk of TdP. However, at clinically relevant concentrations, drugs that block hERG with approximately equipotent late sodium and/or calcium current block (balanced ion channel‐blocking drugs) can cause QTc prolongation,19, 22, 40, 41 but have a low risk of TdP. Thus, not all hERG block and QTc prolongation is associated with TdP.
Under CiPA, the initial prediction of proarrhythmic risk will come from the in silico computational model (Figure 3). CiPA will utilize an ECG assessment in early clinical phase I studies to determine if there are unexpected ion channel effects, such as effects resulting from a human‐specific metabolite or differences in protein binding, compared to the preclinical ion channel data (Figure 3).23 As detailed previously, the CiPA phase I ECG working group identified the J‐Tpeakc as the best biomarker to differentiate balanced ion channel‐blocking drugs from predominant hERG‐blocking drugs.39 Drugs that exhibit “balanced ion channel” effects often have QTc prolongation, but do not exhibit J‐Tpeakc prolongation.22, 40, 41 These ECG “signatures” have been shown for multiple drugs and drug combinations blocking either hERG and late sodium or hERG and late sodium and calcium; however, the evidence is less clear for whether QTc prolongation caused by hERG and calcium block alone (with no late sodium block) exhibits J‐Tpeakc prolongation.22, 41
The current clinical study (clinicaltrials.gov: NCT03070470) was designed to: 1) validate the proposed role of phase I ECG assessment under CiPA, and 2) to address the important hERG and calcium block question. The study consists of two parts: a 50‐subject parallel part (Part 1) and a 10‐subject crossover part (Part 2). The primary objectives of the study are:
To confirm that exposure–response analysis of the electrocardiographic QTc and J‐Tpeakc intervals in phase I clinical pharmacology studies can be used to confirm that balanced ion channel‐blocking drugs do not cause J‐Tpeakc prolongation and predominant hERG‐blocking drugs cause QTc prolongation. This will be assessed in Part 1.
To test the hypothesis that calcium channel block can reduce the QTc prolongation from hERG block by shortening J‐Tpeakc. This will be assessed in Part 2.
The study design is summarized below. Complete and detailed versions of the study protocol and statistical analysis plan can be found in the online Supplement.
Part 1: Parallel study design
Part 1 of the study will assess primary objective 1. Part 1 will be a double‐blind, randomized, placebo‐controlled, one‐period parallel design to assess the effect of four marketed drugs and one placebo on the QTc and J‐Tpeakc intervals in 50 healthy subjects. Part 1 will include four drugs with well‐characterized ion channel effects, QTc effects, and TdP risk. Three drugs will be balanced ion channel blockers (approximately equipotent hERG and late sodium and/or calcium block) with low TdP risk (ranolazine, verapamil, and lopinavir + ritonavir combination) and one drug will be a predominant hERG blocker with TdP risk (chloroquine). The ion channel effects of these drugs are shown in Figure 7.19 A parallel design similar to an early phase I study will be used that will result in each study drug being administered to 10 subjects, and placebo to 10 subjects, on 3 consecutive days to achieve low and high exposure levels on Days 1 and 3, respectively.
Figure 7.

Ion channel profiles of drugs in Parts 1 and 2. Ion channel current effects for drugs in Part 1 (chloroquine, ranolazine, verapamil, lopinavir, and ritonavir) and Part 2 (dofetilide and diltiazem) of the clinical study. Each panel shows relative drug‐induced current block from patch clamp experiments; the error bars represent mean±SE of percentage current block measured in the experiments. Vertical lines represent clinical Cmax (solid line) from previous studies and 3‐fold Cmax (dashed line). Ion channel data from Crumb et al.19
Part 1: Treatments administered
On Days 1, 2, and 3 subjects will receive one of the treatments listed under Part 1 in Table 1 according to the study's randomization schedule.
Table 1.
Study treatments and dosing schedule
| Dosing schedule | Part 1 | Part 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | Ranolazine | Verapamil | Lopinavir + Ritonavir | Chloroquine | Dofetilide | Diltiazem + Dofetilide | ||
| Day 1 | Morning | Pbo | 1,500 mg | 120 mg IR | Lop: 800 mg; Rit: 200 mg | 1,000 mg | 0.125 mg | Dilt: 120 mg IR |
| Afternoon | Pbo | Pbo | 120 mg IR | Pbo | Pbo | Pbo | Pbo | |
| Evening | Pbo | 1,500 mg | 240 mg ER | Lop: 800 mg; Rit: 200 mg | Pbo | Pbo | Dilt: 240 mg ER | |
| Day 2 | Morning | Pbo | 1,500 mg | 120 mg IR | Lop: 800 mg; Rit: 200 mg | 500 mg | Pbo | Pbo |
| Afternoon | Pbo | Pbo | 120 mg IR | Pbo | Pbo | Pbo | Pbo | |
| Evening | Pbo | 1,500 mg | 240 mg ER | Lop: 800 mg; Rit: 200 mg | Pbo | Pbo | Dilt: 240 mg ER | |
| Day 3 | Morning | Pbo | 1,500 mg | 120 mg IR | Lop: 800 mg; Rit: 200 mg | 1,000 mg | 0.375 mg | Dilt: 120 mg IR; Dof: 0.25 mg |
| Ion channel block | none | hERG + calcium/late sodium | hERG | hERG | hERG + calcium | |||
Treatments administered for each study part and day as well as their corresponding ion channel effects. Pbo, placebo; Lop, lopinavir; Rit, ritonavir; Dof, dofetilide, Dilt, diltiazem; IR, immediate release; ER, extended release; hERG, predominant hERG block. Figure 7 shows specific ion channel effects for each drug. For more detailed information, see text and clinical study protocol in the online supplement.
Ranolazine
The dose selected for ranolazine is 1,500 mg twice a day for 2.5 days. In a prior study, a single dose of 1,500 mg resulted in 2.3 μg/mL Cmax and caused a mean QTc prolongation of ∼10 ms without prolonging J‐Tpeakc.41 Ranolazine causes approximately equipotent hERG and late sodium block (Figure 7). This balanced ion channel‐blocking drug prolongs QTc but has a low risk of TdP.19 Since the primary endpoint of this study is to test whether J‐Tpeakc is not prolonged at high exposure levels for balanced ion channel‐blocking drugs, 1,500 mg twice a day for 2.5 days will be administered. This supratherapeutic dose has been administered previously to 14 healthy subjects for up to 5 days48 and 191 patients for 1 week.49 The selected dose is expected to cause QTc prolongation with no prolongation of the J‐Tpeakc interval at high exposure levels on Day 3.
Verapamil
Verapamil predominantly blocks the L‐type calcium current, and to a somewhat lesser degree it also blocks hERG at clinical concentrations (Figure 7). A single oral dose of verapamil 120 mg prolonged the PR interval on the ECG, but did not prolong QTc or J‐Tpeakc in a previous study.41 To achieve high verapamil concentrations on Day 3 while minimizing the risk for potential adverse events in healthy subjects (e.g., heart block), the dose selected for verapamil is 120 mg immediate release for the morning and afternoon doses on Days 1 and 2, a 240 mg extended release evening dose on Days 1 and 2, and a 120 mg immediate release morning dose on Day 3. These doses are within the maximum dose of the drug label (480 mg daily), have been previously administered to healthy subjects,50 and will result in substantially higher concentrations than those in Johannesen et al.41 No prolongation of the J‐Tpeakc interval with potential QTc prolongation is expected at high exposure levels on Day 3.
Lopinavir + Ritonavir
Lopinavir is available only in a fixed dose combination with ritonavir, which is used to increase and maintain adequate lopinavir concentrations. The dose selected for lopinavir + ritonavir is 800/200 mg twice a day for 2.5 days. This dose was selected to be equivalent to the supratherapeutic dose administered in a TQT study in 39 healthy subjects.51, 52 Both lopinavir (hERG and calcium block) and ritonavir (hERG, calcium and late sodium block) block inward currents in addition to hERG (Figure 7). This dose is expected to cause ∼10 ms QTc prolongation at low exposure levels on Day 1 and 20 ms QTc prolongation and no J‐Tpeakc prolongation at high exposure levels on Day 3.
Chloroquine
Chloroquine is a predominant hERG blocker (Figure 7) that prolongs QTc. The dose selected for chloroquine is 1,000 mg on Day 1, 500 mg on Day 2, and 1,000 mg on Day 3. This dose is consistent with the labeled dose of 2.5 g chloroquine phosphate over 3 days and has been previously administered to 40 healthy subjects.53 It is expected that this dose will cause QTc prolongation of ∼10 ms at low exposure levels on Day 1 and 20 ms at high exposure levels on Day 3.53
Part 1: Statistical methods
Sample size
The sample size of 10 subjects per arm in a parallel design was selected to mirror the sample size in standard single or multiple ascending dose studies. For example, a waiver for a TQT study was recently granted based on exposure–response analysis of an early phase I clinical study.5 That study had 20 subjects on placebo, 10 subjects in the single ascending dose portion, and 10 subjects in the multiple ascending dose portion. In addition, resampling of the data from the ranolazine and moxifloxacin arms from Johannesen et al.22, 41 using previously published methodologies54, 55 suggests that this sample size and study design would be sufficient to detect QTc prolongation from predominant hERG block by moxifloxacin and to exclude J‐Tpeakc prolongation from balanced ion channel block with ranolazine. Of note, this was with a single dose of ranolazine 1,500 mg, while the current study will include ranolazine 1,500 mg twice a day for 2.5 days. Similar analysis of previously conducted TQT studies suggests that this sample size and study design will also be sufficient to exclude J‐Tpeakc prolongation for the balanced ion channel‐blocking drugs in this study. This is also consistent with the method used in the Darpo et al. study.56 That study was able to detect QTc prolongation for the predominant hERG‐blocking drugs and to exclude QTc prolongation for the drug with no ion channel effects.57
Primary analysis
The primary variable for the exposure–response analysis will be the change‐from‐baseline in QTc (ΔQTc) for the predominant hERG‐blocking drug (chloroquine) and change from baseline in J‐Tpeakc (ΔJ‐Tpeakc) for the balanced ion channel‐blocking drugs (ranolazine, verapamil, lopinavir+ritonavir combination), where the mean of the three predose ECG readings on Day 1 will be used as the baseline. The concentration of the drug (set to “0” for placebo) will be used as a covariate. Data will be analyzed using linear mixed‐effects exposure–response models as follows:
where ΔECG is change from baseline in the ECG biomarker (e.g., ΔQTc, ΔJ‐Tpeakc), concentration is the drug concentration (set to “0” for placebo), time is the nominal timepoint, treatment is coded as “0” for placebo and “1” for the active drug, subjid is the subject identifier and “(1 + concentration|subjid)” indicates between‐subject random effects on the intercept and the slope for concentration.
Supplementary Table S2 summarizes the statistical tests and corresponding hypotheses for each primary endpoint. Briefly, the criterion for the three balanced ion channel‐blocking drugs (ranolazine, verapamil, lopinavir + ritonavir combination) will be based on the predicted J‐Tpeakc effect on the third day of dosing. To demonstrate a lack of J‐Tpeakc prolongation for each of the three drugs:
The upper bound of the two‐sided 90% confidence interval (CI) of the predicted mean placebo adjusted change from baseline ΔΔJ‐Tpeakc must be <10 ms at the observed geometric mean of the maximum concentration (Cmax) on Day 3.
The criterion for the predominant hERG‐blocking drug (chloroquine) will be based on the predicted QTc effect on the first day of dosing. To demonstrate the presence of QTc prolongation for chloroquine:
The upper bound of the two‐sided 90% CI of the predicted mean placebo adjusted change from baseline QTc (ΔΔQTc) must be ≥10 ms at the observed geometric mean of Cmax on Day 1.
Part 2: Crossover study design
Part 2 will test whether calcium block (diltiazem) can reduce the QTc prolongation from hERG block (dofetilide) by shortening J‐Tpeakc. Part 2 will be a double‐blind, randomized, two‐period crossover design to assess the effect of hERG block vs. calcium block on the QTc and J‐Tpeakc intervals in 10 healthy subjects. Part 2 will include two oral drugs (dofetilide and diltiazem) with well‐characterized individual ion channel effects and TdP risk. Dofetilide is a predominant hERG blocker, while diltiazem is a predominant L‐type calcium channel blocker.19 The ion channel effects of these drugs are shown in Figure 7. A crossover design similar to a drug–drug interaction phase I study will be used that will result in each study drug or a combination of both being administered to 10 subjects in two periods of 3 consecutive dosing days each, with a 5‐day washout period in between. This washout period is enough to prevent carryover effects because it is greater than 5 times the half‐lives for dofetilide58 and diltiazem.59
Part 2: Treatments administered
In a crossover design, subjects will receive each of the treatments listed under Part 2 in Table 1 according to a randomization schedule with a 5‐day washout between treatment assignments.
Dofetilide
Dofetilide is a predominant hERG blocker (Figure 7). The dose selected for dofetilide is 0.125 mg on Day 1, placebo on Day 2, and 0.375 mg on Day 3. This dose is less than the labeled dose (0.5 mg twice a day) and less than the dose of 0.5 mg previously administered to healthy subjects.41 It is expected that this dosing regimen will cause QTc prolongation of ∼15 ms and ∼45 ms on Days 1 and 3, respectively.22, 41 While it is not part of the primary endpoint, it is expected that dofetilide will prolong QTc by prolonging both the J‐Tpeakc and Tpeak‐Tend intervals.22, 41
Diltiazem and dofetilide
Diltiazem is a predominant L‐type calcium blocker (Figure 7). The dose selected for diltiazem is 120 mg immediate release in the morning on Day 1, 240 mg extended release in the evening on Day 1 and 2, and 120 mg immediate release on Day 3. This dose is within the maximum labeled dose of 480 mg daily for immediate release and 540 mg daily for extended release. On Day 3, 0.25 mg of dofetilide will be coadministered with diltiazem. This dose is different from the dose administered when dofetilide is given alone because there may be a pharmacokinetic interaction causing an increase in dofetilide concentration. When diltiazem is given in combination with dofetilide, QTc prolongation greater than 10 ms with no J‐Tpeakc prolongation is expected on Day 3.
Part 2: Statistical methods
Sample size
For Part 2, the sample size of 10 subjects, with five subjects per treatment sequence in a crossover design was selected by resampling and further simulation of the data from Johannesen et al.22 This analysis demonstrated that, with eight subjects, the use of exposure–response modeling could detect the QTc shortening by J‐Tpeakc shortening caused by inward current block on the QTc and J‐Tpeakc prolongation associated with hERG block using a crossover design. For Part 2, we anticipate that the selected dose for the calcium blocker (diltiazem) will result in similar or larger QTc and J‐Tpeakc effects than those observed with mexiletine and lidocaine in Johannesen et al.22 because the calcium current is larger than the late sodium current. Therefore, and to account for potential dropouts, Part 2 of the study will use a crossover design with 10 subjects.
Primary analysis
For Part 2, the primary variable for the exposure–response analysis will be the change‐from‐baseline in QTc (ΔQTc) for the pooled treatment sequences of dofetilide alone, diltiazem alone, and dofetilide + diltiazem, where the mean of the three predose ECG readings on Day 1 will be used as the baseline. Data will be analyzed using linear mixed‐effects exposure–response models where the concentration of dofetilide and diltiazem will be used as covariates. The following model is an example for QTc:
where ΔQTc is change from baseline in QTc, dof is dofetilide concentration, dilt is diltiazem concentration and subjid is the subject identifier and “(1 + dof|subjid)” and “(1 + dilt|subjid)” indicate between‐subject random effects on the intercept and the slope for dofetilide and diltiazem concentrations, respectively.
Supplementary Table S2 summarizes the statistical tests and corresponding hypotheses for each primary endpoint. Briefly, to demonstrate that calcium channel block (diltiazem) reduces the QTc prolongation from hERG block (dofetilide) by shortening J‐Tpeakc:
It will be assessed whether the projected QTc effect of dofetilide alone is significantly greater (i.e., P < 0.05) than the projected QTc effect of the combination of dofetilide + diltiazem. This will be assessed at the dofetilide peak plasma level on Day 3 (computed from the combination of dofetilide + diltiazem) on the pooled dofetilide alone, diltiazem alone, and dofetilide + diltiazem data using a linear mixed‐effects model.
If the previous test is statistically significant for QTc, the same test will be performed for J‐Tpeakc.
Overall, this two‐part clinical study will significantly contribute to knowledge regarding multi‐ion channel effects on the QTc and the J‐Tpeakc intervals. This study will also provide a test of the hypothesis that calcium channel block reduces the QTc prolongation from hERG block by shortening J‐Tpeakc. If successful, this clinical study will validate a strategy to differentiate drugs that are predominant hERG blockers from balanced ion channel‐blocking drugs using ECG data from small sample size clinical trials that could be used under CiPA.
CONCLUSIONS AND FUTURE DIRECTIONS
CiPA is intended to be a fit‐for‐purpose paradigm that will utilize an in silico computational model of the human ventricular cardiomyocyte to serve as the primary prediction of proarrhythmic risk of new drugs. There will be additional checks to ensure that drug effects on repolarization are not missed by assessing human iPSC‐cardiomyocytes as well as phase I clinical ECG data using exposure–response modeling. As part of the validation of CiPA, a clinical validation study with small sample size was developed to confirm that exposure–response analysis of the QTc and J‐Tpeakc intervals in clinical phase I studies can be used to differentiate between predominant hERG‐blocking drugs and balanced ion channel‐blocking drugs as well as to test the hypothesis that calcium channel block can reduce QTc prolongation from hERG block by shortening J‐Tpeakc.
If successful, CiPA will 1) enable application of mechanistic studies to inform drug screening early in drug development, in particular for drugs with hERG block and/or QTc prolongation, to better inform which compounds should be selected to advance in development; 2) create a pathway for QTc prolonging drugs with low proarrhythmic risk to advance without the need for intensive ECG monitoring in clinical phase III trials if they are shown to have low proarrhythmic risk; and 3) enable updating drug labels to be more informative about proarrhythmic risk, not just QT prolongation. As discussed in this article, in March 2017, an FDA advisory committee voted that CiPA would be fit for impacting clinical drug development (advisory committee questions 2 and 3), pending completion of the described nonclinical and clinical validation studies.
The CiPA Steering Committee has provided the ICH S7B/E14 Discussion Group with regular updates on the ongoing validation efforts. The results of the nonclinical and clinical validation studies described in this article will inform how CiPA may result in modification to the ICH guidelines to shift from focusing on hERG block and QT prolongation to informing proarrhythmic risk, as not all hERG‐blocking and QT prolonging drugs cause TdP. Moreover, CiPA's mechanistic‐based, multicomponent and multistakeholder approach can serve as a model for how to assess safety of new drugs.
DISCLAIMER
This article reflects the views of the authors and should not be construed to represent FDA's views or policies.
AUTHOR CONTRIBUTIONS
D.G.S., J.V., R.Z., L.J., J.W.M., P.S., V.P., M.K.M., Z.L., J.L., C.G., N.S., and I.Z. wrote the article. D.G.S., J.V., R.Z., L.J., J.W.M., P.S., V.P., M.K.M., J.L., C.G., and N.S. designed the clinical study.
CONFLICT OF INTEREST
P.T. Sager has consulting agreements with Biomedical Systems, Charles River, and iCardiac. The other authors report no conflicts of interest.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by the FDA Safety Research Interest Group and Critical Path Initiative in the Center for Drug Evaluation and Research and the FDA's Office of Women's Health.
References
- 1. Roden, D. Drug‐induced prolongation of the QT interval. N. Engl. J. Med. 350, 1013–1022 (2004). [DOI] [PubMed] [Google Scholar]
- 2. International Council on Harmonisation . ICH Topic S7B the Non‐clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals. <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf> (2005). [PubMed]
- 3. International Council on Harmonisation . Guideline for Industry E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐antiarrhythmic Drugs. <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf> (2005). [PubMed]
- 4. International Council on Harmonisation . ICH E14 Guideline: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs Questions & Answers (R3). <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Q_As_R3__Step4.pdf> (2015).
- 5. Nelson, C.H. et al A quantitative framework to evaluate proarrhythmic risk in a first‐in‐human study to support waiver of a thorough QT study. Clin. Phamacol. Ther. 98, 630–638 (2015). [DOI] [PubMed] [Google Scholar]
- 6. Fermini, B. et al A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J. Biomol. Screen. 21, 1–11 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Stockbridge, N. , Morganroth, J. , Shah, R. & Garnett, C. Dealing with global safety issues: Was the response to QT‐liability of non‐cardiac drugs well coordinated? Drug Saf. 36, 167–182 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Sager, P. , Gintant, G. , Turner, R. , Pettit, S. & Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300 (2014). [DOI] [PubMed] [Google Scholar]
- 9. FDA . Pharmaceutical Science and Clinical Pharmacology Advisory Committee Meeting Announcement. <https://www.fda.gov/AdvisoryCommittees/Calendar/ucm535513.htm> (2017). [DOI] [PubMed]
- 10. Gintant, G. , Sager, P. & Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 15, 457–471 (2016). [DOI] [PubMed] [Google Scholar]
- 11. January, C.T. & Riddle, J.M. Early afterdepolarizations: mechanism of induction and block. A role for L‐type Ca2+ current. Circ. Res. 64, 977–990 (1989). [DOI] [PubMed] [Google Scholar]
- 12. January, C. & Moscucci, A. Cellular mechanisms of early afterdepolarizations. Ann. N. Y. Acad. Sci. 644, 23–32 (1992). [DOI] [PubMed] [Google Scholar]
- 13. Duff, H.J. , Roden, D. , Primm, R.K. , Oates, J.A. & Woosley, R.L. Mexiletine in the treatment of resistant ventricular arrhythmias: enhancement of efficacy and reduction of dose‐related side effects by combination with quinidine. Circulation 67, 1124–1128 (1983). [DOI] [PubMed] [Google Scholar]
- 14. Duff, H. , Mitchell, B. , Manyari, D. & Wyse, G. Mexiletine‐quinidine combination: Electrophysiologic correlates of a favorable antiarrhythmic interaction in humans. J. Am. Coll. Cardiol. 10, 1149–1156 (1987). [DOI] [PubMed] [Google Scholar]
- 15. Giardina, E.‐G.V. & Wechsler, M.E. Low dose quinidine‐mexiletine combination therapy versus quinidine monotherapy for treatment of ventricular arrhythmias. J. Am. Coll. Cardiol. 15, 1138–1145 (1990). [DOI] [PubMed] [Google Scholar]
- 16. Chézalviel‐Guilbert, F. , Davy, J.‐M. , Poirier, J.‐M. & Weissenburger, J. Mexiletine antagonizes effects of sotalol on QT interval duration and its proarrhythmic effects in a canine model of torsade de pointes. J. Am. Coll. Cardiol. 26, 787–792 (1995). [DOI] [PubMed] [Google Scholar]
- 17. Guo, D. , Zhao, X. , Wu, Y. , Liu, T. , Kowey, P. & Yan, G.‐X. L‐type calcium current reactivation contributes to arrhythmogenesis associated with action potential triangulation. J. Cardiovasc. Electrophysiol. 18, 196–203 (2007). [DOI] [PubMed] [Google Scholar]
- 18. Badri, M. et al Mexiletine prevents recurrent torsades de pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin. Electrophysiol. 1, 315–322 (2015). [DOI] [PubMed] [Google Scholar]
- 19. Crumb, W.J., Jr. , Vicente, J. , Johannesen, L. & Strauss, D.G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 81, 251–262 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Wu, L. et al Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Heart Rhythm 5, 1726–1734 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu, L. et al Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res. 77, 481–488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johannesen, L. et al Late sodium current block for drug‐induced long QT syndrome: Results from a prospective clinical trial. Clin. Phamacol. Ther. 99, 214–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colatsky, T. et al The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative — Update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20 (2016). [DOI] [PubMed] [Google Scholar]
- 24. Vunjak Novakovic, G. , Eschenhagen, T. & Mummery, C. Myocardial Tissue Engineering: In Vitro Models. Cold Spring Harb. Perspect. Med. 4, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hirt, M.N. et al Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J. Mol. Cell. Cardiol. 74, 151–161 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Gintant, G. , Fermini, B. , Stockbridge, N. & Strauss, D. The evolving roles of human iPSC‐derived cardiomyocytes in drug safety and discovery. Cell Stem Cell 21, 14–17 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Strauss, D.G. & Blinova, K. Clinical trials in a dish. Trends Pharmacol. Sci. 38, 4–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li, Z. et al Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether‐à‐go‐go‐related gene) channel‐drug binding kinetics and multichannel pharmacology. Circ. Arrhythmia Electrophysiol. 10, e004628 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Windley, M.J. , Abi‐Gerges, N. , Fermini, B. , Hancox, J.C. , Vandenberg, J.I. & Hill, A.P. Measuring kinetics and potency of hERG block for CiPA. J. Pharmacol. Toxicol. Methods 87, 99–107 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Di Veroli, G. , Davies, M. , Zhang, H. , Abi‐Gerges, N. & Boyett, M. High‐throughput screening of drug‐binding dynamics to HERG improves early drug safety assessment. Am. J. Physiol. Heart Circ. Physiol. 304, H104–H117 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Li, Z. , Dutta, S. , Sheng, J. , Tran, P.N. , Wu, W. & Colatsky, T. A temperature‐dependent in silico model of the human ether‐a‐go‐go‐related (hERG) gene channel. J. Pharmacol. Toxicol. Methods 81, 233–239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Hara, T. , Virag, L. , Varro, A. & Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7, e1002061 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dutta, S. et al Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Front. Physiol. 8, 616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blinova, K. et al Comprehensive translational assessment of human‐induced pluripotent stem cell derived cardiomyocytes for evaluating drug‐induced arrhythmias. Toxicol. Sci. 155, 234–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hortigon‐Vinagre, M.P. , Zamora, V. , Burton, F.L. , Green, J. , Gintant, G.A. & Smith, G.L. The use of ratiometric fluorescence measurements of the voltage sensitive dye Di‐4‐ANEPPS to examine action potential characteristics and drug effects on human induced pluripotent stem cell‐derived cardiomyocytes. Toxicol. Sci. 154, 320–331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ando, H. et al A new paradigm for drug‐induced torsadogenic risk assessment using human iPS cell‐derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 84, 111–127 (2017). [DOI] [PubMed] [Google Scholar]
- 37. Nozaki, Y. et al CSAHi study: Validation of multi‐electrode array systems (MEA60/2100) for prediction of drug‐induced proarrhythmia using human iPS cell‐derived cardiomyocytes—assessment of inter‐facility and cells lot‐to‐lot‐variability. Regul. Toxicol. Pharmacol. 77, 75–86 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Vicente, J. et al Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J. Am. Heart Assoc. 4, e001615 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vicente, J. et al Electrocardiographic biomarkers for detection of drug‐induced late sodium current block. PLoS One 11, e0163619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johannesen, L. et al Improving the assessment of heart toxicity for all new drugs through translational regulatory science. Clin. Phamacol. Ther. 95, 501–508 (2014). [DOI] [PubMed] [Google Scholar]
- 41. Johannesen, L. et al Differentiating drug‐induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Phamacol. Ther. 96, 549–558 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Johannesen, L. , Vicente, J. , Hosseini, M. & Strauss, D. Automated algorithm for J‐Tpeak and Tpeak‐Tend assessment of drug‐induced proarrhythmia risk. PLoS One 11, e0166925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Couderc, J.‐P. et al An evaluation of multiple algorithms for the measurement of the heart rate corrected JTpeak interval. J. Electrocardiol. <https://doi.org/10.1016/j.jelectrocard.2017.08.025> (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vicente, J. , Hosseini, M. , Johannesen, L. & Strauss, D.G. Electrocardiographic Biomarkers to Confirm Drug's Electrophysiological Effects Used for Proarrhythmic Risk Prediction under CiPA. J. Electrocardiol. <https://doi.org/10.1016/j.jelectrocard.2017.08.003> (2017). [DOI] [PubMed] [Google Scholar]
- 45. Badilini, F. , Libretti, G. & Vaglio, M. Automated JTpeak analysis by BRAVO. J. Electrocardiol. <https://doi.org/10.1016/j.jelectrocard.2017.07.010> (2017). [DOI] [PubMed] [Google Scholar]
- 46. Chien, S.C. & Gregg, R.E. The algorithmic performance of J‐Tpeak for drug safety clinical trial. J. Electrocardiol. <https://doi.org/10.1016/j.jelectrocard.2017.08.018> (2017). [DOI] [PubMed] [Google Scholar]
- 47. Chiu, W.B. , de Bie, J. & Mortara, D.W. The J to T‐peak interval as a biomarker in drug safety studies: A method of accuracy assessment applied to two algorithms. J. Electrocardiol. <https://doi.org/10.1016/j.jelectrocard.2017.07.011> (2017). [DOI] [PubMed] [Google Scholar]
- 48. FDA . Ranolazine's Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021526_s000_Ranexa_BioPharmr.pdf> (2005).
- 49. Chaitman, B.R. et al Anti‐ischemic effects and long‐term survival during ranolazine monotherapy in patients with chronic severe angina. J. Am. Coll. Cardiol. 43, 1375–1382 (2004). [DOI] [PubMed] [Google Scholar]
- 50. Hartter, S. , Sennewald, R. , Nehmiz, G. & Reilly, P. Oral bioavailability of dabigatran etexilate (Pradaxa((R)) after co‐medication with verapamil in healthy subjects. Br J Clin Pharmacol 75, 1053–1062 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. FDA . Lopinavir/ritonavir Clinical Pharmacology and Biopharmaceutics Review. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/021906s000_ClinPharmR.pdf> (2005).
- 52. AbbVie Inc . Lopinavir/ritonavir label. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021251s052_021906s046lbl.pdf> (2016).
- 53. Miller, A.K. et al Pharmacokinetic interactions and safety evaluations of coadministered tafenoquine and chloroquine in healthy subjects. Br. J. Clin. Pharmacol. 76, 858–867 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferber, G. , Zhou, M. & Darpo, B. Detection of QTc effects in small studies—implications for replacing the thorough QT study. Ann. Noninvas. Electrocardiol. 20, 368–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garnett, C. , Needleman, K. , Liu, J. , Brundage, R. & Wang, Y. Operational characteristics of linear concentration‐QT models for assessing QTc interval in the thorough QT and phase I clinical studies. Clin. Phamacol. Ther. 100, 170–178 (2016). [DOI] [PubMed] [Google Scholar]
- 56. Darpo, B. et al The IQ‐CSRC prospective clinical phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?”. Ann. Noninvas. Electrocardiol. 19, 70–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Phamacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 58. Pfizer Labs . TIKOSYN® (dofetilide) capsules. Product information. Revised January 2014. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020931s012s013lbl.pdf> (2016).
- 59. Valeant Pharmaceuticals North America LLC . CARDIZEM (diltiazem) label. Revised November 2016. <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018602s067lbl.pdf> (2016).
- 60. Vicente, J. , Stockbridge, N. & Strauss, D. Evolving regulatory paradigm for proarrhythmic risk assessment for new drugs. J. Electrocardiol. 49, 837–842 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information