

Summary
Natural killer (NK) cells are large granular lymphocytes largely recognized for their importance in tumour surveillance and the host response to viral infections. However, as the major innate lymphocyte population, NK cells also coordinate early responses to bacterial infections by amplifying the antimicrobial functions of myeloid cells, especially macrophages, by production of interferon‐γ (IFN‐γ). Alternatively, excessive NK cell activation and IFN‐γ production can amplify the systemic inflammatory response during sepsis resulting in increased physiological dysfunction and organ injury. Our understanding of NK cell biology during bacterial infections and sepsis is mostly derived from studies performed in mice. Human studies have demonstrated a correlation between altered NK cell functions and outcomes during sepsis. However, mechanistic understanding of NK cell function during human sepsis is limited. In this review, we will review the current understanding of NK cell biology during sepsis and discuss the challenges associated with modulating NK cell function during sepsis for therapeutic benefit.
Keywords: inflammation, innate lymphoid cells, natural killer cell, sepsis
Introduction
Sepsis remains one of the leading causes of death in critically ill patients worldwide. In the USA, approximately 750 000 people develop sepsis each year and 20–30% of those die due to sepsis‐associated complications.1, 2 However, no targeted treatment for sepsis exists despite decades of efforts.3 As a dysregulated host response to infection, sepsis induces complex and multifactorial cellular, immunological, metabolic and circulatory abnormalities, which are not well understood. Extensive studies on innate myeloid cells such as macrophages, neutrophils and dendritic cells have resulted in a better understanding of their roles in propagating the inflammatory responses of sepsis and participating in sepsis‐induced immunosuppression. However, natural killer (NK) cells, another important component of the innate immune system, remain an ongoing enigma regarding their contribution to the pathophysiology of sepsis.4 Recent advances have revealed a non‐redundant role of NK cells during sepsis and reignited the interest in NK cells as a prognostic marker of sepsis and septic shock. Here, an overview of recent developments regarding the immunobiology of NK cells in both experimental and human sepsis will be provided including a discussion of the limitations and challenges of translating findings from animal studies to human patients. New insights and opportunities to apply NK cell‐based immunotherapy for sepsis are also discussed.
Definition of sepsis
The term ‘sepsis’ has been used for centuries to describe a critical illness caused by infections. However, the germ theory could not fully explain the pathobiology of sepsis, as antibiotic treatment in modern medicine failed to save many patients with sepsis even when microbes in the host were completely eliminated.5 This prompted the notion that sepsis was propagated through a dysfunctional host response to infection. In 2016, the Society for Critical Care Medicine and the European Society of Intensive Care Medicine re‐defined sepsis as ‘life‐threatening organ dysfunction caused by a dysregulated host response to infection’ (Fig. 1). Septic shock was characterized as ‘a subset of sepsis in which particularly profound circulatory, cellular, and metabolic abnormalities are associated with a greater risk of mortality than with sepsis alone’.6 The new clinical definition no longer uses the systemic inflammatory response syndrome as a criterion for diagnosis of sepsis as it lacks sufficient specificity in defining sepsis and strategies that target systemic inflammatory response syndrome have failed in the treatment of patients with septis.6 However, acute inflammation remains an essential component of the multifaceted host response during sepsis and septic shock and extensive studies have been performed to depict the nature of sepsis.
Figure 1.
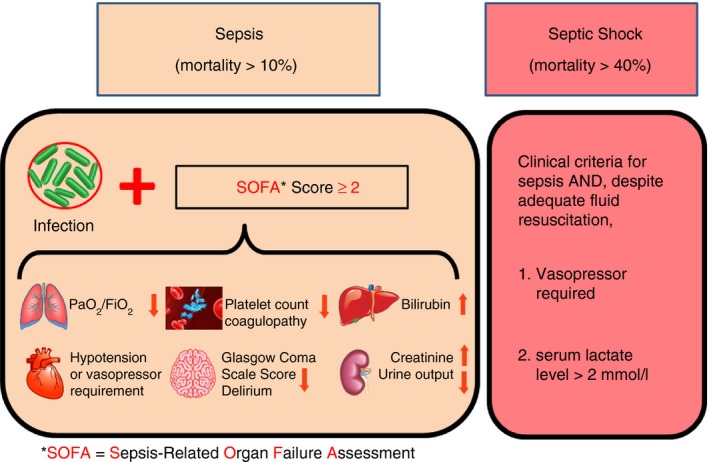
Definitions of sepsis and septic shock. Sepsis is currently defined as life‐threatening organ dysfunction caused by a dysregulated host response to infection. Organ dysfunction can be clinically represented by an increase in the Sepsis‐related Organ Failure Assessment (SOFA) score of 2 points or more, which is related to an in‐hospital mortality in an excess of 10%. Septic shock is defined as a subset of sepsis in which profound circulatory, cellular, and metabolic abnormalities are associated with a greater risk of mortality (> 40%) than with sepsis alone. Despite adequate fluid resuscitation, patients with septic shock require vasopressors to maintain mean arterial pressure (MAP) of 65 mmHg or greater and exhibit evidence of anaerobic metabolism as indicated by serum lactate concentration of > 2 mmol/l.
Pathophysiology of sepsis
Sepsis is a complicated and multifactorial disease process that affects almost all the systems in the host (Fig. 1). The characteristic pathophysiological alternations during sepsis include inflammation, microvascular damage and coagulopathy, haemodynamic instability, multiple organ dysfunction and immunosuppression. Septic shock is defined as persistent haemodynamic instability despite adequate fluid resuscitation and the presence of inadequate oxygen delivery or utilization as reflected by anaerobic metabolism and elevated plasma lactate concentration (Fig. 1).
Inflammation
Dysregulated inflammatory responses at sites of infections can directly cause tissue damage and dysfunction.7 An example is the hypoxaemia and pulmonary dysfunction associated with pneumonia. If the infection is severe, a systemic inflammatory cascade can be initiated and lead to haemodynamic instability, cardiovascular collapse, coagulopathy and multiple end organ failure, which are characteristics of sepsis.8 The initial ‘cytokine storm’ during the acute phase is a hallmark of sepsis.9 Excessive production of pro‐inflammatory cytokines including interleukin‐6 (IL‐6), tumour necrosis factor‐α (TNF‐α), IL‐1β and interferon‐γ (IFN‐γ) is thought to actively propagate systemic inflammatory cascades during acute sepsis.10, 11, 12, 13, 14 However, the functional contribution of the cytokine storm to the pathogenesis of sepsis is not well understood. Large clinical trials that have targeted cytokines during sepsis have not shown significant benefit. Acute inflammation is characterized by vasodilatation, fluid exudation and leucocyte migration.15 Those processes are mediated through the actions of numerous pro‐inflammatory factors including histamine, platelet‐activating factor, bradykinin, nitric oxide, prostaglandins and complement components. Most of these mediators have been targeted in sepsis trials with no benefit.3 Hence, although systemic inflammation is clearly present during sepsis, its contribution to the pathophysiology of sepsis remains controversial.
Microvascular dysfunction and coagulopathy
Compromised microvascular blood flow and oxygen delivery are common and serious alterations in patients with septis.16 Impaired capillary blood flow is observed during sepsis due to systemic hypotension and microvascular obstruction caused by marginating white blood cells adhering to the endothelium and microthrombosis caused by localized activation of the clotting cascade.17 Activated leucocytes, particularly neutrophils, can mediate damage to the endothelial lining, causing capillary leak and activation of coagulation pathways.18 Capillary leak can result in interstitial oedema accumulation in the lungs and other tissues which can lead to acute lung injury and generalized tissue oedema.19 If severe, acute lung injury will result in impaired gas exchange and the development of hypoxaemia. Tissue oedema contributes to organ dysfunction by altering key parenchymal cell functions. Disseminated intravascular coagulation, caused by systemic activation of the clotting cascade, is also a relatively common manifestation of sepsis.20 As a consumptive coagulopathy, disseminated intravascular coagulation remarkably increases the risk of severe bleeding and microthrombosis and is a marker of poor prognosis in patients with sepsis.21
Multiple organ dysfunction
The new consensus conference definitions highlight organ dysfunction as a hallmark of sepsis.6 Organ dysfunction may be among the first clinical signs of sepsis but the underlying causes are not well understood. Provoking factors include the direct effects of the inciting organism(s) (usually bacteria or bacterial components) on cellular metabolism and homeostasis and the impact of the inflammatory response as decreased in the last section.22 Microcirculatory failure, leucocytosis, poor tissue perfusion and metabolic dysfunction disrupt the normal functions of individual organs.23, 24 No organ system is invulnerable to the malignant systemic inflammatory cascades of sepsis.22 The development of haemodynamic collapse,25 circulatory insufficiency,23 cardiac dysfunction,26 acute lung injury,27 liver damage,28 delirium and acute kidney injury29 are common among patients with sepsis. Multiple organ failure is the major cause of morbidity and mortality in patients with sepsis.30
Sepsis‐associated immunosuppression
A paradigm shift in understanding the pathophysiology of sepsis has been triggered by mounting evidence for immunosuppression in patients with sepsis.31, 32 With improved intensive care management and supportive care, early mortality has significantly decreased.31, 32 However, prolonged sepsis has been shown to promote innate and adaptive immune cell dysfunctions including anergy, enduring inflammation, altered cytokine production, increased apoptosis, and reduced proliferation and effector functions of lymphoid and myeloid cells.33 Patients who have ‘recovered’ from acute sepsis are more likely to develop recurrent opportunistic infections, viral reactivation and late death.33 In particular, low HLA‐DR expression on monocytes and dendritic cells has been shown to correlate with poor outcomes during sepsis.34, 35 Blood monocytes isolated from patients with sepsis exhibit reduced ability to secrete pro‐inflammatory cytokines such as TNF‐α, IL‐1, IL‐6 and IL‐12.36 CD4+ T cells exhibit increased apoptotic cell death and have impaired ability to secrete both type 1 (Th1) and type 2 (Th2) T helper cytokines.37 Increased regulatory T‐cell ratios have been observed in patients with sepsis and correlate with the septic lethality.38
The combination of sustained inflammation, organ dysfunction and immunosuppression in critically ill patients, including those with sepsis, has been termed chronic critical illness.2 Chronic critical illness results in a prolonged need for intensive or palliative care and costs the US healthcare system an estimated $20 billion per year. Characteristics of chronic critical illness include prolonged ventilator dependence, delirium and brain dysfunction, neuromuscular weakness, malnutrition, skin breakdown and general inability to return to baseline function.39 These alterations lead to late mortality defined as a 1‐year mortality rate of > 50%. Hence, although more patients are surviving the classic 28‐day period, a large number of the survivors die within the first year after discharge from the hospital and many of the survivors are unable to regain a functional standard of life.40
Need for new therapy for human sepsis
Treatment for sepsis is largely supportive and emphasizes early antibiotic administration, adequate fluid resuscitation and treatments to support haemodynamic and organ functions.41 There have been more than 100 Phase II and III clinical trials aimed at developing effective and targeted therapy for human sepsis. Most trials used agents that demonstrated promising efficacy in animal models. Unfortunately, those trials have been largely disappointing. Most trials targeted systemic inflammation by blocking specific and non‐specific mediators such as TNF‐α, IL‐1, platelet‐activating factor and nitric oxide.42 Agents that non‐specifically block inflammation such as corticosteroids and non‐steroidal anti‐inflammatory agents have also been trialled without success.43 Studies targeting the clotting cascade using activated protein C showed some promise in decreasing sepsis mortality.44 However, extensive post‐market testing ultimately did not show efficacy and the agent was removed from the market.45 More recently, trials have been launched with the goal of reversing sepsis‐associated immunosuppression by administering immunomodulating agents such as granulocyte–macrophage colony‐stimulating factor or IFN‐γ.46 Many of the trials targeting immunosuppression are still in progress. More recently, immunoprophylaxis with Toll‐like receptor (TLR) ligands and immunomodulation with Fms‐like tyrosine kinase‐3 ligand (FLT‐3 ligand) have shown efficacy against infection and sepsis in animal studies, bringing hope to clinical prevention and treatment of sepsis.47, 48, 49, 50, 51
The failure of targeted interventions in patients with sepsis could be due to the complex and interdependent network of pro‐inflammatory responses during sepsis.52 Also, patients with sepsis that were recruited to trials exhibited considerable heterogeneity in genetic background, nature of the infecting microorganisms, site of infections, magnitude of pro‐inflammatory responses and co‐morbidities.53 To address these problems, a better understanding of immunological alterations during sepsis is necessary to identify plausible new targets for intervention.
Natural killer cells: important frontline of defence against infection and cancer
Natural killer cells are bone‐marrow‐derived large granular lymphocytes possessing a ‘natural’ ability to kill tumours without prior priming.54, 55 Most recently, NK cells have been classified among a cohort of lymphocytes termed innate lymphoid cells.56 Although NK cells and other innate lymphoid cells share lineage origin with T and B lymphocytes of the adaptive immune system, NK cells lack the expression of germ‐line rearranged immunoglobulin and T‐cell receptor genes.57 Due to this unique phenotype, NK cells possess the capability to mount a rapid, non‐specific innate immune response against cancer cells and cells infected with intracellular pathogens such as viruses.58 Moreover, NK cells play an important role in initiating host defence and coordinating innate and adaptive immune responses (Fig. 2). Comprising 5–15% of peripheral blood mononuclear cells and residing in tissues such as liver, spleen, lung and bone marrow, NK cells are derived from haematopoietic stem cells in bone marrow and undergo coordinated maturation and differentiation steps that confer functional specificity and competency.59
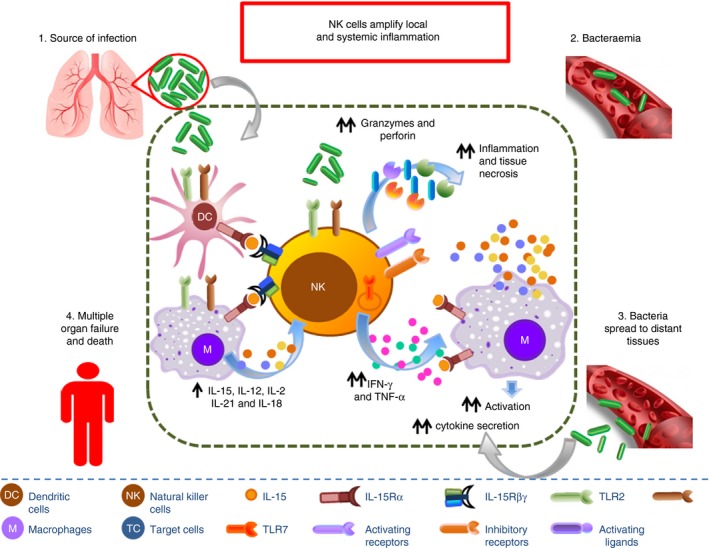
Figure 2.

Natural killer (NK) cells coordinate the early host response to infection. NK cells are activated by pathogen‐associated molecular patterns (PAMPs) through Toll‐like receptors (TLRs), loss of inhibition due to down‐regulation of MHC‐I on target cells, engagement of activating receptors with activating ligands on target cells, and by NK cell‐activating cytokines [interleukin‐ 2 (IL‐2), IL‐12, IL‐15, IL‐18, IL‐21] produced by antigen‐presenting cells such as dendritic cells and macrophages. Activated NK cells mediate cytotoxic granule‐dependent cytotoxicity of infected cells and promote activation of macrophage‐mediated microbial killing by secretion of interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α).
Activation of NK cells is governed by a balance between signals delivered through inhibitory and activating receptors.60 Inhibitory receptors include Ly49A/C/I/P in mice and KLR2DL1/2/3/5 in humans. The inhibitory receptors deliver signals to NK cells upon engagement with the mouse H‐2 and human leucocyte antigen (HLA) class I major histocompatibility complex (MHC‐I).61 During infections or tumour surveillance, NK cells recognize and destroy cells that down‐regulate expression of MHC‐I, such as virus‐infected or tumour cells (‘missing self’ hypothesis).62 In some cases, NK cells can mediate killing of cells expressing MHC‐I upon engagement of activating or stress‐induced ligands, like NKG2B/C/D/E and NKp30/44/46, expressed by transformed or virus‐infected cells.63 Direct activation of NK cells can also be achieved by stimulation with cytokines such as type I IFNs, IL‐2, IL‐12, IL‐15, IL‐18, IL‐21 and IL‐27 for transient or long‐lasting non‐antigen‐specific effector functions.64, 65, 66, 67 Activated NK cells use two primary strategies to eliminate target cells: death ligand engagement and the cytotoxic secretory granule pathway. Engagement of cell death receptor Fas on target cells by Fas ligand on NK cells causes caspase‐dependent apoptotic cell death.68 Cytotoxic granule‐mediated cell death is mediated by perforin and granzymes, a family of pro‐apoptotic serine proteases.69 NK cells can also induce cytotoxicity through antibody‐dependent cellular cytotoxicity, in which FcγRIII receptors expressed on NK cells recognize infected cells that are opsonized by antibodies.70
In addition to their cytotoxic functions, NK cells are endowed with the capability of secreting pro‐inflammatory cytokines, such as TNF‐α and IFN‐γ, which amplify the pro‐inflammatory and anti‐microbial functions of other leucocyte populations. IFN‐γ plays a central role in NK cell‐mediated activation of myeloid cells during infection and inflammation. In response to myeloid‐derived cytokines such as IL‐12, IL‐15 and IL‐18 as well as through direct activation by engagement of tumour cells, virus‐infected cells and microbial products, NK cells secrete large amounts of IFN‐γ.12 IFN‐γ subsequently amplifies the antimicrobial functions of myeloid cells, such as macrophages and dendritic cells, and facilitates further secretion of IFN‐γ‐inducing cytokines and sets up a positive feedback loop that serves to ramp up the activation of NK cells and myeloid cells during periods of infection and inflammation.13, 71
NK cells: friend or foe during sepsis?
Detrimental role of NK cells during experimental sepsis
Independent of tumour immunosurveillance and viral clearance, NK cells play an important role in facilitating inflammation during infection (Fig. 3). Recent advances have shown that, similar to other innate immune cells, NK cells employ TLR 2/3/4/9 to engage pathogen‐associated molecular patterns including bacteria‐associated peptidoglycan, lipopolysaccharide, virus‐derived dsRNA, and DNA with CpG motifs after encounter with invading pathogens.72 The subsequent activating signals are capable of directly activating NK cells. However, NK cell activation during infection is most potently induced by cytokines secreted by myeloid cells that co‐exist with NK cells at sites of infection.12 In response to lipopolysaccharide (LPS) or heat‐killed bacteria, macrophage‐derived IL‐12 is the major factor inducing NK cell IFN‐γ secretion with IL‐18 serving as the most potent co‐stimulus followed by IL‐15 and B7 proteins. NK cell‐derived IFN‐γ then facilitates the activation of myeloid cells to augment phagocytosis, respiratory burst, microbial killing and further secretion of IFN‐γ‐inducing cytokines resulting in a positive feedback loop that amplifies infection‐induced activation of NK cells and myeloid cells. Although these interactions facilitate beneficial antimicrobial functions among macrophages and neutrophils, the excessive cytokine production is likely to amplify inflammation during systemic infection.
Figure 3.
During the acute phase of sepsis, dysregulated activation of natural killer (NK) cells contributes to the unbridled pro‐inflammatory cytokine storm by secreting large amounts of interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α). IFN‐γ and TNF‐α subsequently facilitate further secretion of IFN‐γ/TNF‐α‐inducing cytokines by myeloid cells such as dendritic cells and macrophages and set up a positive feedback loop that serves to ramp up the activation of NK cells and myeloid cells, amplifying inflammation during systemic infection and causing multiple organ failure and mortality. In addition, secretory cytotoxic granules including granzymes and perforins by NK cells directly mediate tissue cell necrosis and aggravate systemic inflammation.
Several studies have demonstrated a role for NK cells as facilitators of systemic inflammation during infection or endotoxin challenge. Mice depleted of NK cells by treatment with anti‐asialoGM1 or anti‐NK1.1 antibodies are resistant to mortality during sepsis caused by caecal ligation and puncture.73, 74, 75 NK cell‐depleted mice showed decreased cytokine production, less organ injury and attenuated physiological dysfunction compared with controls. Bacterial burden was not different between groups and treatment with antibiotics greatly augmented the survival benefit conferred by NK cell depletion.76 These findings imply that the deleterious impact of NK cells during sepsis is mediated through their ability to amplify the pro‐inflammatory response or direct contributions to organ injury, possibly through cytotoxic mechanisms.
The importance of NK cells for facilitating shock caused by injection of microbial products, such as LPS, was recognized more than 20 years ago. Heremans et al. showed that mice depleted of NK cells are resistant to LPS‐induced shock and proposed that NK cell‐derived IFN‐γ has a pivotal role for facilitating mortality in their model.77 Further studies showed that selective antibody‐mediated NK cell depletion attenuates systemic inflammation and hypothermia, enhances microbial clearance, restores acid–base balance, and improves survival in experimental models of sepsis caused by polymicrobial peritonitis, pneumococcal pneumonia78 and Ehrlichia chaffeensis, systemic Escherichia coli and Streptococcus pyogenes infections.79, 80 Heinzel et al. confirmed the importance of IFN‐γ during LPS‐induced shock in mice and showed that IL‐12, probably derived from macrophages, is important for eliciting IFN‐γ secretion by NK cells.81 These findings were confirmed by Jansen et al. using a non‐human primate model. They demonstrated that secretion of IL‐12 is essential, but not sufficient, as a mediator of lethal IFN‐γ secretion during LPS‐induced shock.82 A co‐stimulatory role for NK cells during LPS‐induced shock was demonstrated by Fehniger et al.83 Subsequently, several investigators confirmed the essential role of NK cell‐derived IFN‐γ during LPS‐induced shock.84, 85
As noted above, early work identified IL‐15 as a co‐regulator of LPS‐induced shock. Our group investigated the role of IL‐15 in more detail and discovered that IL‐15 enables the pathogenesis of septic shock by maintaining and activating NK cells.86 IL‐15 is a small cytokine molecule essential for the development and differentiation of NK cells.87 In a previous study from our laboratory, overdose of IL‐15 caused sepsis‐like inflammatory pathology and mortality via hyperproliferation of activated NK cells and hyperproduction of IFN‐γ.88 In another study from our group, IL‐15‐deficient mice, which lack NK cells, exhibited improved survival, attenuated hypothermia, and less pro‐inflammatory cytokine production during septic shock caused by caecal ligation and puncture or endotoxin‐induced shock.86 IL‐15 superagonist (IL‐15SA, IL‐15/IL‐15Ra complex) regenerated NK cells and re‐established mortality of IL‐15 knockout mice during septic shock, while preventing NK cell regeneration attenuated the restoration of mortality caused by IL‐15SA. The crucial role of NK cell depletion in providing protection from sepsis in IL‐15‐deficient mice was supported by the use of IL‐15‐neutralizing IgG M96 in wild‐type mice. If given immediately before septic challenge, M96 failed to deplete NK cells and did not protect against septic shock. However, M96 caused NK cell depletion if given 4 days before septic challenge and then conferred protection. In separate experiments, IL‐15SA treatment amplified septic shock of wild‐type mice, which was prevented by NK cell or IFN‐γ depletion. Hence, our studies conclude that IL‐15 enables the development of septic shock by facilitating early systemic inflammation. In contrast, Inoue et al.89 reported that exogenous IL‐15SA treatment after caecal ligation and puncture attenuated sepsis‐induced apoptosis of multiple cell types, reversed immune dysfunction and improved survival during sepsis. The reasons for the different observations between the two studies are not completely clear, but could be due to the different dosages and delivery routes of IL‐15SA as well as differences in mouse strains and severity of the sepsis models used.
Natural killer cells rapidly migrate to sites of bacterial infection. CXCR3‐expressing NK cells quickly migrate from secondary lymphoid organs to the peritoneal cavity within 4–6 hr after the initiation of intra‐abdominal sepsis, in a manner regulated by the chemokines CXCL9 and CXCL10.90, 91 CXCR3+ NK cells are the murine equivalent of human CD56bright NK cells, both of which produce significantly more IFN‐γ than CXCR3− CD56dim NK cells.92 Our laboratory subsequently performed phenotypic characterization of these CXCR3+ mouse NK cells in the context of sepsis. In intra‐abdominal sepsis induced by caecal ligation and puncture, CXCR3+ NK cells that were recruited to the peritoneal cavity exhibited an activated phenotype with increased CD69 expression and highly augmented production of the pro‐inflammatory cytokines TNF‐α and IFN‐γ.93, 94 In further studies, blockade of CXCR3 or CXCL10 decreased local and systemic inflammation and provided survival benefit during caecal ligation and puncture‐induced septic shock.94 Taken together, these studies imply that CXCR3 activation contributes to the pathogenesis of septic shock either by facilitating NK cell migration to sites of infection or through direct NK cell activation.
The studies outlined above have defined the ability of NK cells to amplify the pro‐inflammatory response during sepsis, primarily through the secretion of IFN‐γ.13, 95 The other major mechanism by which NK cells could cause organ injury and physiological dysfunction during systemic inflammatory syndromes, such as sepsis, is through cytotoxicity. Interestingly, there is little evidence to indicate that cytolytic mechanisms contribute to NK cell‐mediated augmentation of septic shock. As described earlier in this review, the mechanisms by which NK cells mediate cytolysis is through secretion of perforin and granzymes as well as surface expression of FasL. Anthony et al. reported that perforin‐, granzyme M‐ and granzyme A‐deficient mice are resistant to LPS‐induced shock.96 Granzyme B‐deficient mice did not show resistance. Their data indicate that NK cell‐derived granzymes amplify the early inflammatory response during sepsis rather than facilitating cytotoxicity. To our knowledge, no other experimental studies have demonstrated a role for granzymes in the pathogenesis of sepsis although several observational studies in humans have shown a correlation between plasma granzyme concentrations and sepsis severity.
Although NK cells are predominantly found in the peripheral blood and secondary lymphoid organs such as spleen, emerging evidence shows that NK cells also exist in local tissues such as liver, lung, kidney and placenta and are distinct from conventional NK cells in their lineage development, surface marker expression and unique effector functions.97 As opposed to conventional NK cells in the blood and spleen, tissue‐resident NK subsets serve as the host first‐line of defence against invading microorganisms, and also play a critical role in modulating the magnitude of inflammation in a tissue‐specific manner. Rasid et al. found that, in the setting of sepsis, it is the local microenvironment that mediated the functional adaptation of tissue‐resident NK cells. In this study, tissue‐resident NK cells displayed organ‐specific thresholds of maximum activation and effector functions in lung, spleen, peritoneum, bone marrow and blood.98 In another study, Victorino et al.99 showed that NK cells resident in kidney directly mediated ischaemic kidney injury, which is common during septic shock and an indicator of poor prognosis. It is therefore noteworthy to perform more studies to examine how tissue‐resident NK cells could modulate compartment‐specific inflammation and the multiple organ dysfunctions in lethal sepsis.
Protective role of NK cells during infection
Although a significant number of animal studies have focused on the deleterious role of NK cells in propagation of systemic inflammation during sepsis, several independent studies have reported that NK cells confer protection against infection by diverse organisms. A study by Nilsson et al.100 showed that NK cells are crucial for orchestrating the host response to septic arthritis caused by Staphylococcus aureus. Other studies show that impaired NK cell function contributes to sepsis‐induced immunosuppression (Fig. 4). Hirsh et al. reported that the peak of acute lung injury induced by caecal ligation and puncture was highly associated with dysfunctions of NK cells in the lung.101 In that study, the lung‐resident NK cells displayed impaired cytotoxic responses and IFN‐γ production, which failed to initiate early inflammatory responses necessary for containment of microbes, resulting in unrestrained infection and concomitant organ injury mediated by dysregulated inflammation. Other studies report significantly reduced production of IFN‐γ by NK cells isolated from septic mice in response to ex vivo stimulation with IL‐18, or TLR2, TLR4 or TLR9 agonists indicating that NK cell dysfunction may contribute to sepsis‐associated immunosuppression.102 A study by Hiraki et al.103 showed that neutralization of IL‐10 significantly restored the capability of NK cells to secrete IFN‐γ, thereby improving survival of septic mice. Depletion of NK cells worsened infection severity during systemic infection with Citrobacter rodentium and in a Pseudomonas aeruginosa pneumonia model, implying a protective role of NK cells against infection caused by some Gram‐negative pathogens. In the study examining infection of Citrobacter rodentium, NK cells were essential to mount an effective antimicrobial response by direct killing, recruitment of innate immune cells and secretion of essential cytokines.104 During Pseudomonas aeruginosa pneumonia, host NK cells orchestrate neutrophil recruitment to the lung, thereby augmenting microbial clearance and improving survival.102
Figure 4.
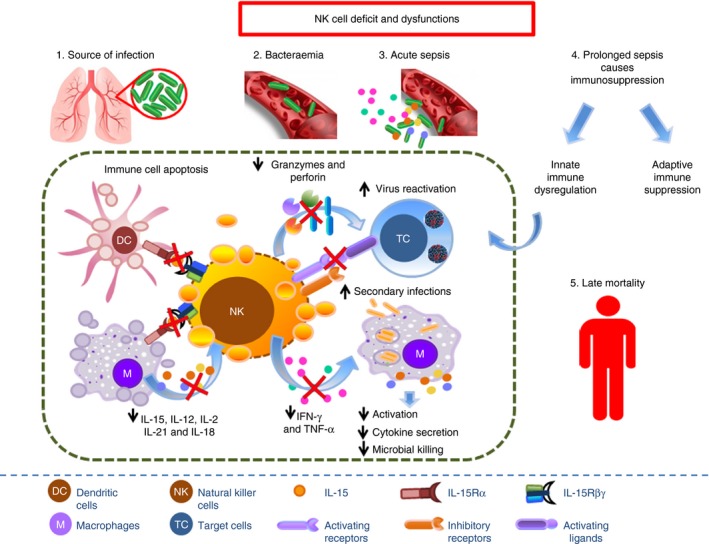
Apoptosis and reduced immune functions of natural killer (NK) cells during prolonged sepsis contribute to the increased susceptibility of patients with sepsis to secondary/nosocomial infections and viral reactivation, leading to worsened life quality and long‐term mortality.
Taken together, the role of NK cells in the pathogenesis of experimental sepsis is multi‐faceted and dependent on the severity and location of infection. Evidence indicates that NK cells are important for orchestrating local responses to infection and depletion of NK cells impairs effective microbial clearance resulting in propagation of infection with the possible development of severe infection and sepsis. Alternatively, activation of NK cells during periods of severe infection can amplify systemic inflammation and accelerate the development of sepsis and organ injury. Given the disconnection between animal and human models of sepsis, we next focus on recent clinical studies that phenotypically characterized NK cells in patients with sepsis, in an attempt to provide insights into the association of NK cell phenotype with outcomes during sepsis.
NK cells in human sepsis
Most of our knowledge about human NK cells during sepsis is limited to those in peripheral blood due to their accessibility. Human NK cells comprise 5–15% of lymphocytes in peripheral blood and can be divided into two distinct subsets based on surface expression of CD56 and CD16.105 CD56, also called neural cell adhesion molecule, is a homophilic binding glycoprotein expressed on NK cells, neurons, glia and skeletal muscle cells. CD16, a low‐affinity receptor for the Fc fragments of IgG (FcRIIIa), mediates the antibody‐dependent cell‐mediated cytotoxicity reaction leading to NK cell specific killing of target cells coated with IgG antibodies. The CD56dim CD16bright NK cell subset composes approximately 90% of all NK cells in peripheral blood, while the CD56bright CD16−/dim NK cell subset constitutes approximately 10%.106 The two human NK subsets display distinct effector functions. The CD56dim CD16bright NK cell subset is more naturally cytotoxic and expresses higher levels of killer cell immunoglobulin‐like receptors and CD57 compared with the CD56bright CD16−/dim NK cell subset, which in turn exhibit better ability to proliferate and secrete IFN‐γ and TNF‐α in response to pro‐inflammatory cytokine stimulation.
Several lines of evidence have demonstrated the numerical, phenotypic and functional alteration of NK cells during human sepsis. Most of the studies have focused on the initial phase of sepsis by examining NK cells in the peripheral blood of patients at the time of or shortly after the diagnosis of sepsis or admission to the intensive care unit. In several studies, blood NK cells in patients with sepsis increased in numbers, and exhibited an activated phenotype with higher levels of TLR‐2/4/9 and CD69 expression as well as greater plasma concentrations of granzyme A and B, IFN‐γ and IL‐12p40, compared with healthy volunteers.107, 108, 109 The increased number of activated NK cells in the blood was consistent with observations in septic mice. The mechanism underlying this phenomenon is not completely clear, but it could possibly be caused by increased NK cell haematopoiesis, enhanced recruitment from secondary lymphoid tissue and self‐proliferation in response to microbial and cytokine stimulation.
As noted, NK cells might represent a dual‐edged sword, either by rapidly eliminating infections resulting in resolved inflammation, or by propagating excessive systemic inflammation. The multi‐faceted effects of NK cells have led to contradictory outcomes in patients with sepsis, correlating with either improved or worsened prognosis. Hence, it remains unclear what effect circulating NK cells may impose on the pathogenesis of acute sepsis in patients. The correlation results are incomplete in studies that only examined NK cell numbers but not effector functions. In addition, the conflicting conclusion drawn from each study might depend on the size and heterogeneity of included patients with sepsis, the nature of the infecting microorganisms, sites of infections, and the magnitude and complexity of pro‐inflammatory and anti‐inflammatory responses.
Conversely, a significant number of studies reported a decrease in NK cell number and IFN‐γ production, in combination with dysfunctions of other immune cells during early sepsis, indicating the possible development of immunosuppression in the host.107, 110 Boomer et al. reported decreased numbers of NK cells in the blood of patients within 24 hr after the onset of sepsis.111 The finding was consistent with previous observations by Holub et al.,112 who reported sustained decreases of NK cell numbers in patients with sepsis for the first 14 days after hospitalization, with a more significant drop during Gram‐negative compared with Gram‐positive sepsis. The mechanisms underlying the drop in NK cell numbers are not entirely understood, but increased apoptotic death might contribute to the phenomenon. Animal studies from our group have documented a rapid migration of NK cells to the site of infection within 4–6 hr. Hence, the migration of NK cells from peripheral blood to sites of infection may represent another mechanism of NK cell decline in peripheral blood during infection.
Comprehensive phenotypic and functional observations of all immune cells from multiple studies on patients with sepsis have shown consistent loss and dysfunction of dendritic cells, neutrophils, monocytes, myeloid‐derived suppressor cells, CD4+ T cells, CD8+ T cells, regulatory T cells and B cells during sepsis.32, 113 Leucocytes displayed increased apoptotic death, reduced cell proliferation, contracted antigen presentation, augmented inhibitory signalling, and persistent dysfunctions during the course of sepsis. NK cells have exhibited a similar pattern of loss and dysfunction. Hence, NK cell defects and dysfunctions may be a feature of global immunosuppression during the late phase of sepsis. As NK cells are the main cytotoxic and IFN‐γ‐producing cell type, persistent NK cell deficiency and dysfunction could cause impaired host defence against the primary pathogen and render patients more susceptible to secondary pathogen infections or latent viral reactivation, leading to worsened outcomes. NK cells are central effector cells controlling cytomegalovirus (CMV) infection. In a study by Chiche et al. in critically ill patients with seropositive CMV infection, impaired IFN‐γ secretion by NK cells was responsible for reactivation of CMV, which is a hallmark of sepsis‐induced immunosuppression.107 The reduction in the number of NK cells was not only observed in adult sepsis, but also in paediatric sepsis, during which CD56dim CD16+ cytotoxic NK cells significantly decreased in numbers during the acute phase.114, 115 Interestingly, NK cells may not always exhibit a consistent activation profile, as one study from Souza‐Fonseca‐Guimaraes et al.72 showed that both CD56bright and CD56dim NK cells increased TLR‐2/4/9 and CD69 expression but decreased IFN‐γ secretion in response to ex vivo stimulation with TLR‐4/9 agonists or whole bacteria in synergy with IL‐15 and IL‐18.
Taken together, it is not completely clear what alternations and adaptations human NK cells develop in response to sepsis. To our current knowledge, NK cells behave differently during sepsis according to the type and severity of underlying infections. In a study by Gogos et al.,108 NK cell number significantly increased during sepsis caused by community‐acquired pneumonia in which Gram‐positive Streptococcus pneumoniae was the main causative pathogen. Alternatively, increased apoptotic death of NK cells was observed in patients with sepsis with ventilator‐associated pneumonia and hospital‐acquired pneumonia, which were mainly caused by Gram‐negative Pseudomonas aeruginosa. Another route for addressing this question is to evaluate the susceptibility of patients with congenital NK cell deficiency to sepsis in the clinical setting. Congenital NK cell deficiency is associated with recurrent and disseminated viral infections and various types of tumour virus‐induced malignancies.116 Studies on the incidence of bacterial sepsis in patients with inborn NK cell defects are limited. Recently, a large cohort study of patients with common variable immunodeficiency launched by Ebbo et al.117 showed that a higher incidence of sepsis/bacteraemia was found in patients with severe NK cell lymphopenia, indicating an important role of NK cells in controlling bacterial infections at the early phase. However, this cohort study could not exclude the potential deleterious effects of NK cells, if present, in mediating inflammatory pathology during acute sepsis.
Limitations of studying NK cells in animals and humans
A wide array of genetic animal models result in NK cell deficiency, including genetic disruption of IL‐15, IL‐2, IL‐15Rα, IL‐2Rβ or γc. However, all of those models result in modifications to multiple leucocyte populations. There are no genetic models in which NK cells are selectively depleted. Instead, depletion of NK cells in vivo has so far relied on treatment with NK cell‐depleting monoclonal antibodies such as anti‐asialoGM1, anti‐NK1.1 or anti‐NKp46. However, there are some drawbacks regarding their specificity, efficiency and suitability across all mouse strains. A recent study by Nishikado et al. reported that anti‐asialoGM1 antibody not only depletes NK cells, but also exhibits a lethal off‐target effect on basophils.118 In another study, tissue‐resident NK cells that mediate ischaemic kidney injury fail to be depleted by anti‐asialoGM1 antibody. Like NK cells, NKT cells also express NK1.1 antigen on the surface and can be depleted simultaneously by treatment with anti‐NK1.1 antibody.93 Furthermore, several commonly used laboratory mouse strains such as BALB/c, CBA, C3H and A mice do not express the NK1.1 antigen, in which NK cell depletion depends on the use of anti‐NKp46 antibody.
Using NKp46 promoter, Walzer and colleagues generated transgenic mice expressing the diphtheria toxin (DT) receptor in NK cells. Injection with DT resulted in specific and complete ablation of NK cells in mice.119 Most recently, a novel Ncr1‐Cre mouse model that expresses Cre recombinase under the control of the Ncr1 (p46) promoter in NK cells provided a platform from which to study NK cell biology.120 Eckelhart et al. crossed mice floxed with signal transducer and activator of transcription 5 (Stat5) to Ncr1‐Cre mice to generate a new Stat5flop/flop Ncr1‐iCreTg mouse that was largely devoid of NK cells in peripheral lymphoid organs and only contains NK cell precursor cells in the bone marrow. It would be of great interest to induce sepsis in this transgenic mouse model to demonstrate the role of NK cells during sepsis. However, NKp46‐DTR mice and Stat5 Ncr1‐Cre mice have not been readily available for study.
Although assessment of human NK cells advances the potential application of NK cell therapy for the treatment of sepsis, none of the current studies provide mechanistic insights into NK cell function during sepsis. Moreover, the examination of blood NK cells from patients with sepsis only provides us with limited understanding of the phenotypic and functional alternations of NK cells from all tissues and compartments. In addition, available data from patients with sepsis is largely inadequate due to small sample size, and focused measurement of parameters at the onset or early phase of sepsis. Data interpretation is also limited as some studies failed to include evaluation of NK cell functions and included patients with high heterogeneity in terms of types of underlying infections, site of infections and severity of clinical progression. Given these limitations, the role of NK cells in the pathogenesis of sepsis is still an ongoing enigma. This challenge could be addressed with future studies with larger sample size, well‐controlled patient heterogeneity, increased end points and extended study period.
NK cells and the future treatment of sepsis
As discussed above, there are multiple lines of evidence showing multifaceted roles of NK cells in both murine and human sepsis. Results from previous studies have expanded our knowledge regarding how NK cells mediate phenotypic and functional adaptations to septic insults and how they in turn affect the pathophysiology of sepsis. Nevertheless, available data so far were limited in a thorough quantitative and qualitative evaluation of NK cells and only included assessment of patients at the early phase of sepsis. Additionally, NK cells are heterogeneous in functions including cytotoxic, tolerance and regulatory subsets and are widely distributed in diverse tissues. Most of the published studies failed to evaluate the specific contributions of different NK cell subsets to the progression of sepsis. Therefore, a dynamic high‐throughput monitoring of NK cell‐specific molecular signatures relating to sepsis activity and development represents the first step of fully establishing a complete immunoprofiling of NK cells over the course of sepsis and illuminating the correlation of NK cell alterations with the final prognosis of sepsis.
During sepsis, dynamic changes in microbial, cellular and biomedical components constantly influence the functional characteristics of NK cells over the course. It appears that Gram‐negative sepsis is more likely to cause apoptotic death of NK cells compared with sepsis with Gram‐positive pathogens. In addition, a dysregulated pro‐inflammatory cytokine milieu at the acute phase may orchestrate uncontrolled activation of NK cells, facilitating the severity of sepsis. First, it is necessary to use similar high‐throughput approaches to identify clinical, transcriptional profiling of patients with sepsis as well as molecular correlates of disease progression. Next, all the information that is collected can guide the stratification of the patients and further assess the distinct signatures in response to NK cell‐based treatment of different patient subgroups.
Natural killer cells appear to be critical to eliminate pathogens during the early phase of sepsis and prevent patients from developing secondary infections or viral reactivation during the immunosuppressive phase. In this context, NK cell deficiency and dysfunction during sepsis could be rescued by adoptive transfer of NK cells, blockade of inhibitory receptors or treatment with NK cell activating cytokines such IL‐12, IL‐15 or IL‐18. However, during acute sepsis or septic shock, strategies aiming to neutralize specific inflammatory mediators produced by NK cells might be important to quench the systemic inflammatory responses. As the timing and duration of NK cell‐based therapy highly depends on the activity of sepsis and NK cells, the global immunomonitoring and immunoscoring of sepsis progression and patient response to NK cell‐based therapy might lead to a better understanding of the nature of sepsis and facilitate the optimization of clinical protocols of these immunotherapeutic agents.
Disclosure
The authors do not have any competing interests to disclose.
Acknowledgements
This work is supported by National Institutes of Health Grant R01 GM66885. JKB is supported by National Institutes of Health Grant R01 GM121711. NKP is supported by American Heart Association Postdoctoral Grant 16POST29920007.
Contributor Information
Yin Guo, Email: yin.guo1126@gmail.com.
Edward R. Sherwood, Email: edward.r.sherwood@vanderbilt.edu.
References
- 1. Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P et al Assessment of global incidence and mortality of hospital‐treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 2016; 193:259–72. [DOI] [PubMed] [Google Scholar]
- 2. Kahn JM, Le T, Angus DC, Cox CE, Hough CL, White DB et al The epidemiology of chronic critical illness in the United States*. Crit Care Med 2015; 43:282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA 2011; 306:2614–5. [DOI] [PubMed] [Google Scholar]
- 4. Bohannon J, Guo Y, Sherwood ER. The role of natural killer cells in the pathogenesis of sepsis: the ongoing enigma. Crit Care 2012; 16:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet 2005; 365:63–78. [DOI] [PubMed] [Google Scholar]
- 6. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M et al The third international consensus definitions for sepsis and septic shock (sepsis‐3). JAMA 2016; 315:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glaros T, Larsen M, Li L. Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front Biosci 2009; 14:3988–93. [DOI] [PubMed] [Google Scholar]
- 8. Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348:138–50. [DOI] [PubMed] [Google Scholar]
- 9. Adib‐Conquy M, Cavaillon JM. Compensatory anti‐inflammatory response syndrome. Thromb Haemost 2009; 101:36–47. [PubMed] [Google Scholar]
- 10. Glauser MP. The inflammatory cytokines. New developments in the pathophysiology and treatment of septic shock. Drugs 1996; 52(Suppl 2):9–17. [DOI] [PubMed] [Google Scholar]
- 11. Okusawa S, Gelfand JA, Ikejima T, Connolly RJ, Dinarello CA. Interleukin 1 induces a shock‐like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclooxygenase inhibition. J Clin Invest 1988; 81:1162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Varma TK, Lin CY, Toliver‐Kinsky TE, Sherwood ER. Endotoxin‐induced γ interferon production: contributing cell types and key regulatory factors. Clin Diagn Lab Immunol 2002; 9:530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Romero CR, Herzig DS, Etogo A, Nunez J, Mahmoudizad R, Fang G et al The role of interferon‐γ in the pathogenesis of acute intra‐abdominal sepsis. J Leukoc Biol 2010; 88:725–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B et al Simultaneous targeting of IL‐1 and IL‐18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med 2014; 189:282–91. [DOI] [PubMed] [Google Scholar]
- 15. Sherwood ER, Toliver‐Kinsky T. Mechanisms of the inflammatory response. Best Pract Res Clin Anaesthesiol 2004; 18:385–405. [DOI] [PubMed] [Google Scholar]
- 16. De Backer D, Orbegozo Cortes D, Donadello K, Vincent JL. Pathophysiology of microcirculatory dysfunction and the pathogenesis of septic shock. Virulence 2014; 5:73–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Backer D, Creteur J, Preiser JC, Dubois MJ, Vincent JL. Microvascular blood flow is altered in patients with sepsis. Am J Respir Crit Care Med 2002; 166:98–104. [DOI] [PubMed] [Google Scholar]
- 18. Osterud B, Bjorklid E. The tissue factor pathway in disseminated intravascular coagulation. Semin Thromb Hemost 2001; 27:605–17. [DOI] [PubMed] [Google Scholar]
- 19. Ishihara H, Matsui A, Muraoka M, Tanabe T, Tsubo T, Matsuki A. Detection of capillary protein leakage by indocyanine green and glucose dilutions in septic patients. Crit Care Med 2000; 28:620–6. [DOI] [PubMed] [Google Scholar]
- 20. Levi M, ten Cate H, van der Poll T, van Deventer SJ. Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA 1993; 270:975–9. [PubMed] [Google Scholar]
- 21. Zeerleder S, Hack CE, Wuillemin WA. Disseminated intravascular coagulation in sepsis. Chest 2005; 128:2864–75. [DOI] [PubMed] [Google Scholar]
- 22. Bone RC, Sprung CL, Sibbald WJ. Definitions for sepsis and organ failure. Crit Care Med 1992; 20:724–6. [DOI] [PubMed] [Google Scholar]
- 23. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 2003; 101:3765–77. [DOI] [PubMed] [Google Scholar]
- 24. Schouten M, Wiersinga WJ, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol 2008; 83:536–45. [DOI] [PubMed] [Google Scholar]
- 25. Krishnagopalan S, Kumar A, Parrillo JE, Kumar A. Myocardial dysfunction in the patient with sepsis. Curr Opin Crit Care 2002; 8:376–88. [DOI] [PubMed] [Google Scholar]
- 26. Rudiger A, Singer M. Mechanisms of sepsis‐induced cardiac dysfunction. Crit Care Med 2007; 35:1599–608. [DOI] [PubMed] [Google Scholar]
- 27. Niederman MS, Fein AM. Sepsis syndrome, the adult respiratory distress syndrome, and nosocomial pneumonia. A common clinical sequence. Clin Chest Med 1990; 11:633–56. [PubMed] [Google Scholar]
- 28. Nesseler N, Launey Y, Aninat C, Morel F, Malledant Y, Seguin P. Clinical review: The liver in sepsis. Crit Care 2012; 16:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bagshaw SM, George C, Bellomo R, Committee ADM. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care 2008; 12:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Du B, Chen D, Liu D. [Prediction of prognosis of patients with multiple organ dysfunction syndrome by sepsis‐related organ failure assessment]. Zhonghua Yi Xue Za Zhi 2001; 81:78–81. [PubMed] [Google Scholar]
- 31. Delano MJ, Ward PA. Sepsis‐induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest 2016; 126:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013; 13:260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hotchkiss RS, Moldawer LL. Parallels between cancer and infectious disease. N Engl J Med 2014; 371:380–3. [DOI] [PubMed] [Google Scholar]
- 34. Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P et al Monocyte deactivation in septic patients: restoration by IFN‐γ treatment. Nat Med 1997; 3:678–81. [DOI] [PubMed] [Google Scholar]
- 35. Pastille E, Didovic S, Brauckmann D, Rani M, Agrawal H, Schade FU et al Modulation of dendritic cell differentiation in the bone marrow mediates sustained immunosuppression after polymicrobial sepsis. J Immunol 2011; 186:977–86. [DOI] [PubMed] [Google Scholar]
- 36. Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest 1991; 88:1747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH et al Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 2011; 306:2594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Venet F, Chung CS, Kherouf H, Geeraert A, Malcus C, Poitevin F et al Increased circulating regulatory T cells (CD4+ CD25+ CD127−)) contribute to lymphocyte anergy in septic shock patients. Intensive Care Med 2009; 35:678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakusic A, Gajic O. Chronic critical illness: unintended consequence of intensive care medicine. Lancet Respir Med 2016; 4:531–2. [DOI] [PubMed] [Google Scholar]
- 40. Marchioni A, Fantini R, Antenora F, Clini E, Fabbri L. Chronic critical illness: the price of survival. Eur J Clin Invest 2015; 45:1341–9. [DOI] [PubMed] [Google Scholar]
- 41. Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J et al Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Intensive Care Med 2004; 30:536–55. [DOI] [PubMed] [Google Scholar]
- 42. Lei MG, Gao JJ, Morrison DC, Qureshi N. Pathogenesis of sepsis: current concepts and emerging therapies. Mo Med 2003; 100:524–9. [PubMed] [Google Scholar]
- 43. Patel GP, Balk RA. Systemic steroids in severe sepsis and septic shock. Am J Respir Crit Care Med 2012; 185:133–9. [DOI] [PubMed] [Google Scholar]
- 44. Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez‐Rodriguez A et al Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 2001; 344:699–709. [DOI] [PubMed] [Google Scholar]
- 45. Marti‐Carvajal AJ, Sola I, Lathyris D, Cardona AF. Human recombinant activated protein C for severe sepsis. Cochrane Database Syst Rev 2012; 3:CD004388. [DOI] [PubMed] [Google Scholar]
- 46. Patil NK, Bohannon JK, Sherwood ER. Immunotherapy: a promising approach to reverse sepsis‐induced immunosuppression. Pharmacol Res 2016; 111:688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK et al Role of G‐CSF in monophosphoryl lipid A‐mediated augmentation of neutrophil functions after burn injury. J Leukoc Biol 2016; 99:629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hernandez A, Bohannon JK, Luan L, Fensterheim BA, Guo Y, Patil NK et al The role of MyD88‐ and TRIF‐dependent signaling in monophosphoryl lipid A‐induced expansion and recruitment of innate immunocytes. J Leukoc Biol 2016; 100:1311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Luan L, Patil NK, Guo Y, Hernandez A, Bohannon JK, Fensterheim BA et al Comparative transcriptome profiles of human blood in response to the toll‐like receptor 4 ligands lipopolysaccharide and monophosphoryl lipid A. Sci Rep 2017; 7:40050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patil NK, Bohannon JK, Luan L, Fensterheim B, Hernandez A, Wang J et al Flt3 ligand treatment attenuates T cell dysfunction and improves survival in a murine model of burn wound sepsis. Shock 2017; 47:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK. The cytokine response to lipopolysaccharide does not predict the host response to infection. J Immunol 2017; 198:3264–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marshall JC. Sepsis: rethinking the approach to clinical research. J Leukoc Biol 2008; 83:471–82. [DOI] [PubMed] [Google Scholar]
- 53. Marshall JC. The staging of sepsis: understanding heterogeneity in treatment efficacy. Crit Care 2005; 9:626–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural‐killer‐cell surveillance and therapy of cancer. Nat Rev Cancer 2002; 2:850–61. [DOI] [PubMed] [Google Scholar]
- 55. Rabacal W, Pabbisetty SK, Hoek KL, Cendron D, Guo Y, Maseda D et al Transcription factor KLF2 regulates homeostatic NK cell proliferation and survival. Proc Natl Acad Sci USA 2016; 113:5370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Padro Dietz C, Luong A. Innate lymphoid cells: the innate counterpart to T helper cells. Adv Otorhinolaryngol 2016; 79:58–68. [DOI] [PubMed] [Google Scholar]
- 57. Montaldo E, Vacca P, Vitale C, Moretta F, Locatelli F, Mingari MC et al Human innate lymphoid cells. Immunol Lett 2016; 179:2–8. [DOI] [PubMed] [Google Scholar]
- 58. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M et al RAG‐2‐deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992; 68:855–67. [DOI] [PubMed] [Google Scholar]
- 59. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol 2016; 17:758–64. [DOI] [PubMed] [Google Scholar]
- 60. Lanier LL. NK cell receptors. Annu Rev Immunol 1998; 16:359–93. [DOI] [PubMed] [Google Scholar]
- 61. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 2011; 89:216–24. [DOI] [PubMed] [Google Scholar]
- 62. Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 1990; 11:237–44. [DOI] [PubMed] [Google Scholar]
- 63. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL et al Activation of NK cells and T cells by NKG2D, a receptor for stress‐inducible MICA. Science 1999; 285:727–9. [DOI] [PubMed] [Google Scholar]
- 64. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar‐Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol 1999; 17:189–220. [DOI] [PubMed] [Google Scholar]
- 65. Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I et al IL‐21 in synergy with IL‐15 or IL‐18 enhances IFN‐γ production in human NK and T cells. J Immunol 2003; 170:5464–9. [DOI] [PubMed] [Google Scholar]
- 66. Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M et al CD56bright natural killer cells are present in human lymph nodes and are activated by T cell‐derived IL‐2: a potential new link between adaptive and innate immunity. Blood 2003; 101:3052–7. [DOI] [PubMed] [Google Scholar]
- 67. Patil NK, Luan L, Bohannon JK, Guo Y, Hernandez A, Fensterheim B et al IL‐15 superagonist expands mCD8+ T, NK and NKT cells after burn injury but fails to improve outcome during burn wound infection. PLoS ONE 2016; 11:e0148452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H et al Activation of NK cell cytotoxicity. Mol Immunol 2005; 42:501–10. [DOI] [PubMed] [Google Scholar]
- 69. Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR et al Acquisition of murine NK cell cytotoxicity requires the translation of a pre‐existing pool of granzyme B and perforin mRNAs. Immunity 2007; 26:798–811. [DOI] [PubMed] [Google Scholar]
- 70. Farag SS, VanDeusen JB, Fehniger TA, Caligiuri MA. Biology and clinical impact of human natural killer cells. Int J Hematol 2003; 78:7–17. [DOI] [PubMed] [Google Scholar]
- 71. Takeda K, Hayakawa Y, Atsuta M, Hong S, Van Kaer L, Kobayashi K et al Relative contribution of NK and NKT cells to the anti‐metastatic activities of IL‐12. Int Immunol 2000; 12:909–14. [DOI] [PubMed] [Google Scholar]
- 72. Souza‐Fonseca‐Guimaraes F, Parlato M, Philippart F, Misset B, Cavaillon JM, Adib‐Conquy M. Toll‐like receptors expression and interferon‐γ production by NK cells in human sepsis. Crit Care 2012; 16:R206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sherwood ER, Lin CY, Tao W, Hartmann CA, Dujon JE, French AJ et al Beta 2 microglobulin knockout mice are resistant to lethal intraabdominal sepsis. Am J Respir Crit Care Med 2003; 167:1641–9. [DOI] [PubMed] [Google Scholar]
- 74. Tao W, Sherwood ER. Beta2‐microglobulin knockout mice treated with anti‐asialoGM1 exhibit improved hemodynamics and cardiac contractile function during acute intra‐abdominal sepsis. Am J Physiol Regul Integr Comp Physiol 2004; 286:R569–75. [DOI] [PubMed] [Google Scholar]
- 75. Sherwood ER, Enoh VT, Murphey ED, Lin CY. Mice depleted of CD8+ T and NK cells are resistant to injury caused by cecal ligation and puncture. Lab Invest 2004; 84:1655–65. [DOI] [PubMed] [Google Scholar]
- 76. Enoh VT, Fairchild CD, Lin CY, Varma TK, Sherwood ER. Differential effect of imipenem treatment on wild‐type and NK cell‐deficient CD8 knockout mice during acute intra‐abdominal injury. Am J Physiol Regul Integr Comp Physiol 2006; 290:R685–93. [DOI] [PubMed] [Google Scholar]
- 77. Heremans H, Dillen C, van Damme J, Billiau A. Essential role for natural killer cells in the lethal lipopolysaccharide‐induced Shwartzman‐like reaction in mice. Eur J Immunol 1994; 24:1155–60. [DOI] [PubMed] [Google Scholar]
- 78. Christaki E, Diza E, Giamarellos‐Bourboulis EJ, Papadopoulou N, Pistiki A, Droggiti DI et al NK and NKT cell depletion alters the outcome of experimental pneumococcal pneumonia: relationship with regulation of interferon‐γ production. J Immunol Res 2015; 2015:532717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Badgwell B, Parihar R, Magro C, Dierksheide J, Russo T, Carson WE 3rd. Natural killer cells contribute to the lethality of a murine model of Escherichia coli infection. Surgery 2002; 132:205–12. [DOI] [PubMed] [Google Scholar]
- 80. Goldmann O, Chhatwal GS, Medina E. Contribution of natural killer cells to the pathogenesis of septic shock induced by Streptococcus pyogenes in mice. J Infect Dis 2005; 191:1280–6. [DOI] [PubMed] [Google Scholar]
- 81. Heinzel FP, Rerko RM, Ling P, Hakimi J, Schoenhaut DS. Interleukin 12 is produced in vivo during endotoxemia and stimulates synthesis of γ interferon. Infect Immun 1994; 62:4244–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jansen PM, van der Pouw Kraan TC, de Jong IW, van Mierlo G, Wijdenes J, Chang AA et al Release of interleukin‐12 in experimental Escherichia coli septic shock in baboons: relation to plasma levels of interleukin‐10 and interferon‐γ . Blood 1996; 87:5144–51. [PubMed] [Google Scholar]
- 83. Fehniger TA, Yu H, Cooper MA, Suzuki K, Shah MH, Caligiuri MA. Cutting edge: IL‐15 costimulates the generalized Shwartzman reaction and innate immune IFN‐γ production in vivo . J Immunol 2000; 164:1643–7. [DOI] [PubMed] [Google Scholar]
- 84. Ito H, Koide N, Hassan F, Islam S, Tumurkhuu G, Mori I et al Lethal endotoxic shock using α‐galactosylceramide sensitization as a new experimental model of septic shock. Lab Invest 2006; 86:254–61. [DOI] [PubMed] [Google Scholar]
- 85. Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A et al Essential role of IRF‐3 in lipopolysaccharide‐induced interferon‐β gene expression and endotoxin shock. Biochem Biophys Res Commun 2003; 306:860–6. [DOI] [PubMed] [Google Scholar]
- 86. Guo Y, Luan L, Patil NK, Wang J, Bohannon JK, Rabacal W et al IL‐15 enables septic shock by maintaining NK cell integrity and function. J Immunol 2017; 198:1320–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Guo YLL, Luan L, Patil NK, Sherwood ER. Immunobiology of the IL‐15/IL‐15Rα complex as an antitumor and antiviral agent. Cytokine Growth Factor Rev 2017; https://doi.org/10.1016/j.cytogfr.2017.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Guo Y, Luan L, Rabacal W, Bohannon JK, Fensterheim BA, Hernandez A et al IL‐15 superagonist‐mediated immunotoxicity: role of NK cells and IFN‐γ . J Immunol 2015; 195:2353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Inoue S, Unsinger J, Davis CG, Muenzer JT, Ferguson TA, Chang K et al IL‐15 prevents apoptosis, reverses innate and adaptive immune dysfunction, and improves survival in sepsis. J Immunol 2010; 184:1401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Herzig DS, Driver BR, Fang G, Toliver‐Kinsky TE, Shute EN, Sherwood ER. Regulation of lymphocyte trafficking by CXC chemokine receptor 3 during septic shock. Am J Respir Crit Care Med 2012; 185:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Herzig DS, Luan L, Bohannon JK, Toliver‐Kinsky TE, Guo Y, Sherwood ER. The role of CXCL10 in the pathogenesis of experimental septic shock. Crit Care 2014; 18:R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Marquardt N, Wilk E, Pokoyski C, Schmidt RE, Jacobs R. Murine CXCR3+ CD27bright NK cells resemble the human CD56bright NK‐cell population. Eur J Immunol 2010; 40:1428–39. [DOI] [PubMed] [Google Scholar]
- 93. Etogo AO, Nunez J, Lin CY, Toliver‐Kinsky TE, Sherwood ER. NK but not CD1‐restricted NKT cells facilitate systemic inflammation during polymicrobial intra‐abdominal sepsis. J Immunol 2008; 180:6334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Herzig DS, Guo Y, Fang G, Toliver‐Kinsky TE, Sherwood ER. Therapeutic efficacy of CXCR3 blockade in an experimental model of severe sepsis. Crit Care 2012; 16:R168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Herzig D, Fang G, Toliver‐Kinsky TE, Guo Y, Bohannon J, Sherwood ER. STAT1‐deficient mice are resistant to cecal ligation and puncture‐induced septic shock. Shock 2012; 38:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Anthony DA, Andrews DM, Chow M, Watt SV, House C, Akira S et al A role for granzyme M in TLR4‐driven inflammation and endotoxicosis. J Immunol 2010; 185:1794–803. [DOI] [PubMed] [Google Scholar]
- 97. Lim AI, Verrier T, Vosshenrich CA, Di Santo JP. Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol 2017; 44:61–8. [DOI] [PubMed] [Google Scholar]
- 98. Rasid O, Ciulean IS, Fitting C, Doyen N, Cavaillon JM. Local microenvironment controls the compartmentalization of NK cell responses during systemic inflammation in mice. J Immunol 2016; 197:2444–54. [DOI] [PubMed] [Google Scholar]
- 99. Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D et al Tissue‐resident NK cells mediate ischemic kidney injury and are not depleted by anti‐Asialo‐GM1 antibody. J Immunol 2015; 195:4973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nilsson N, Bremell T, Tarkowski A, Carlsten H. Protective role of NK1.1+ cells in experimental Staphylococcus aureus arthritis. Clin Exp Immunol 1999; 117:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hirsh M, Carmel J, Kaplan V, Livne E, Krausz MM. Activity of lung neutrophils and matrix metalloproteinases in cyclophosphamide‐treated mice with experimental sepsis. Int J Exp Pathol 2004; 85:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pastille E, Pohlmann S, Wirsdorfer F, Reib A, Flohe SB. A disturbed interaction with accessory cells upon opportunistic infection with Pseudomonas aeruginosa contributes to an impaired IFN‐γ production of NK cells in the lung during sepsis‐induced immunosuppression. Innate Immun 2015; 21:115–26. [DOI] [PubMed] [Google Scholar]
- 103. Hiraki S, Ono S, Kinoshita M, Tsujimoto H, Takahata R, Miyazaki H et al Neutralization of IL‐10 restores the downregulation of IL‐18 receptor on natural killer cells and interferon‐γ production in septic mice, thus leading to an improved survival. Shock 2012; 37:177–82. [DOI] [PubMed] [Google Scholar]
- 104. Hall LJ, Murphy CT, Hurley G, Quinlan A, Shanahan F, Nally K et al Natural killer cells protect against mucosal and systemic infection with the enteric pathogen Citrobacter rodentium . Infect Immun 2013; 81:460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer‐cell subsets. Trends Immunol 2001; 22:633–40. [DOI] [PubMed] [Google Scholar]
- 106. Marquez ME, Millet C, Stekman H, Conesa A, Deglesne PA, Toro F et al CD16 cross‐linking induces increased expression of CD56 and production of IL‐12 in peripheral NK cells. Cell Immunol 2010; 264:86–92. [DOI] [PubMed] [Google Scholar]
- 107. Chiche L, Forel JM, Thomas G, Farnarier C, Vely F, Bl´ery M et al The role of natural killer cells in sepsis. J Biomed Biotechnol 2011; 2011:986491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Gogos C, Kotsaki A, Pelekanou A, Giannikopoulos G, Vaki I, Maravitsa P et al Early alterations of the innate and adaptive immune statuses in sepsis according to the type of underlying infection. Crit Care 2010; 14:R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Giannikopoulos G, Antonopoulou A, Kalpakou G, Makaritsis K, Panou C, Papadomichelakis E et al The functional role of natural killer cells early in clinical sepsis. Apmis 2013; 121:329–36. [DOI] [PubMed] [Google Scholar]
- 110. Forel JM, Chiche L, Thomas G, Mancini J, Farnarier C, Cognet C et al Phenotype and functions of natural killer cells in critically‐ill septic patients. PLoS ONE 2012; 7:e50446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Boomer JS, Shuherk‐Shaffer J, Hotchkiss RS, Green JM. A prospective analysis of lymphocyte phenotype and function over the course of acute sepsis. Crit Care 2012; 16:R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Holub M, Kluckova Z, Beneda B, Hobstová J, Huzicka I, Prazák J et al Changes in lymphocyte subpopulations and CD3+/DR+ expression in sepsis. Clin Microbiol Infect 2000; 6:657–60. [DOI] [PubMed] [Google Scholar]
- 113. Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM et al Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun 2010; 78:1582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Georgeson GD, Szony BJ, Streitman K, Kovacs A, Kovacs L, Laszlo A. Natural killer cell cytotoxicity is deficient in newborns with sepsis and recurrent infections. Eur J Pediatr 2001; 160:478–82. [DOI] [PubMed] [Google Scholar]
- 115. Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly‐Scumpia K, Barker T et al Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood 2008; 112:1750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Rai N, Thakur N. Congenital CMV with LAD type 1 and NK cell deficiency. J Pediatr Hematol Oncol 2013; 35:468–9. [DOI] [PubMed] [Google Scholar]
- 117. Ebbo M, Gerard L, Carpentier S, Vély F, Cypowyj S, Farnarier C et al Low circulating natural killer cell counts are associated with severe disease in patients with common variable immunodeficiency. EBioMedicine 2016; 6:222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Nishikado H, Mukai K, Kawano Y, Minegishi Y, Karasuyama H. NK cell‐depleting anti‐asialo GM1 antibody exhibits a lethal off‐target effect on basophils in vivo . J Immunol 2011; 186:5766–71. [DOI] [PubMed] [Google Scholar]
- 119. Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH et al Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci USA 2007; 104:3384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T et al A novel Ncr1‐Cre mouse reveals the essential role of STAT5 for NK‐cell survival and development. Blood 2011; 117:1565–73. [DOI] [PubMed] [Google Scholar]