

Summary
Programmed death‐1 (PD‐1) plays an important role in protecting against inflammation and myocyte damage in T‐cell‐mediated myocarditis. To understand whether fibrinogen‐like protein‐2 (FGL2) can affect the role of the PD‐1/PD‐L1 pathway in experimental autoimmune myocarditis (EAM), we investigated cardiac function in EAM rats over‐expressing FGL2. Over‐expression of FGL2 significantly decreased PD‐1 and deteriorated cardiac function in rats with autoimmune myocarditis. Histopathology revealed increased inflammatory cell infiltrate in EAM‐FGL2 rats compared with the control groups (EAM, EAM‐GFP and NC). Notably, transcription factor forkhead box P3 (Foxp3) and retinoic acid‐related orphan receptor γt (ROR γt) protein and mRNA levels were statistically (P < 0·05) increased in EAM rats. We also found that interferon‐γ, interleukin‐6, interleukin‐17 and brain natriuretic peptide levels were profoundly increased in serum of FGL2 over‐expressing EAM rats. Hence, FGL2 plays an important role in the pathogenesis of autoimmune myocarditis that also involves the PD‐1/PD‐L1 pathway. Our findings may provide novel therapeutic targets for the treatment of immune‐induced heart injury.
Keywords: cardiac function, experimental autoimmune myocarditis, fibrinogen‐like protein‐2, inflammatory cytokines, programmed death‐1
Abbreviations
- BNP
brain natriuretic peptide
- EAM
experimental autoimmune myocarditis
- FGL2
fibrinogen‐like protein‐2
- Foxp3
transcription factor forkhead box P3
- IFN‐γ
interferon‐γ
- IL‐6
interleukin‐6
- PD‐1
programmed death‐1
- RORγt
retinoic acid‐related orphan receptor γt
- Th17
T helper type 17
- Treg
regulatory T
- VMC
viral myocarditis
Introduction
Viral myocarditis (VMC) is an important cause of dilated cardiomyopathy that leads to significant mortality in young people.1, 2 Studies have shown that T helper type 17 (Th17)/ regulatory T (Treg) cell imbalance plays an important role in the pathogenesis of the immune‐cell‐mediated myocardial injury observed in VMC.3 Targeting immune cells to prevent and treat myocardial injury in VMC and dilated cardiomyopathy is an active area of research.
Programmed death factor 1 (PD‐1) is a member of the co‐stimulatory molecule CD28, cytotoxic T‐lymphocyte‐associated antigen 4 superfamily. The PD‐1 signalling pathway plays an extensive and complex role in immune regulation.4 PD‐1 and its ligand PD‐L1, can negatively regulate the balance between T‐cell activation and immune tolerance. This pathway has been an important therapeutic target in various cancers and immune‐cell‐mediated diseases. Loss of the PD‐1 gene can cause lethal dilated cardiomyopathy in mice due to the formation of autoantibodies against cardiac troponin I.5, 6 In CD4+ T‐cell‐dependent experimental autoimmune myocarditis (EAM) rat model, PD‐1 knockout aggravated the severity of myocarditis.7 Therefore, PD‐1 plays an important role in T‐cell‐mediated EAM.
Fibrinogen‐like protein 2 (FGL2), a member of the fibrinogen family, is expressed either as a pro‐coagulative membrane protein or as an immunosuppressive secreted protein.8 FGL2 over‐expression resulted in deterioration of heart function in rats with acute EAM, increased the inflammatory cell infiltrations, and increased the level of brain natriuretic peptide (BNP), tumour necrosis factor‐α, interleukin‐6 (IL‐6) and IL‐179 Increased expression of PD‐1/PD‐L1 in ischaemia re‐perfused and cryoinjured hearts was associated with a marked increase in IL‐17.10 In our current study we explored the involvement of the PD‐1/PD‐L1 pathway in immunotolerance as a potential mechanism for the pathogenesis of EAM mediated by FGL2.
Material and methods
Reagents
Lentivirus vector and GFP plasmids were prepared by the Jikai Gene Chemical Technology Corporation (Shanghai, China). Paracoccidioidomycosis and complete Freund's adjuvant were purchased from Sigma Corporation (St. Louis, MO, USA). Applied antibodies were purchased from Abcam Trading Co. Ltd. (Cambridge, UK). The ELISA kit was obtained from Senxiong Technology Corporation (Shanghai, China).
Animals
Thirty‐two SPF Lewis rats (6 weeks old) were purchased from Weitonglihua Experimental Animal Technology Corporation (quality conformance number: SCXK 2012‐0001, Beijing, China). Padding and feed were provided by laboratory animal centre in Nanchang University.
Establishment of EAM rat models
The study protocol was approved by the Institutional Review Board of the First Affiliated Hospital of Nanchang University. All participants signed the informed consent before study participation. Generation of EAM rat models was described previously.9, 11 Briefly, purified porcine myosin was dissolved in 0·15 m PBS at a concentration of 2 g/l. The solution was mixed with complete Freund's adjuvant in 1 : 1 volume and emulsified completely under sterile conditions. Paracoccidioidomycosis (2·0 mg/ml) was mixed and emulsified with complete Freund's adjuvant in the same volume, and subcutaneously injected at multiple points – bilateral groin, footpad and oxter of 24 rats at days 1 and 8 (200 μl/rat). Then, the rats were randomly divided into four groups with eight rats in each group: EAM rats that received lentivirus carrying FGL2 (EAM‐FGL2 group); EAM rats that received lentivirus carrying GFP (EAM‐GFP group); EAM rats that received PBS buffer (EAM group); rats that were not injected to induce EAM but received PBS buffer (NC group). Lentivirus at a concentration of 5 × 107 Transducing Units(TU)/ml and PBS buffer were injected into the tail vein in each group of rats (1 ml).
Echocardiogram
At day 40 post inoculation, rats were anaesthetized with 7% chloral hydrate. Diasonograph (Philips, Amsterdam, the Netherlands) was used to detect the ultrasonic cardiogram of each rat with a transducer frequency of 37 MHz. Detection indices included heart rate, left ventricular end‐diastolic dimension, left ventricular end‐systolic diameter, left ventricular fractional shortening and left ventricular ejection fraction. Each index was examined three times to obtain an average.
Histopathology
Rats were killed by cervical dislocation, and hearts were extracted and fixed with 10% formalin. Each fixed cardiac tissue was embedded, sectioned and stained with haematoxylin & eosin. Inflammation of myocardial tissues was evaluated as previously described12. Five high‐power fields were randomly chosen in each slice. According to the percentage of infiltration of inflammatory cells and myocardial necrosis area compared with the total area, a scoring system was calculated: 0 for no lesion, 1 point for < 25%, 2 points for 25–50%, 3 points for 50–75% and 4 points for > 75%.
ELISA
Rat blood was centrifuged at 1000 g for 15 min after 20 min standing. The serum was separated and examined using an ELISA kit to detect the BNP, IL‐6, IL‐17, and interferon‐γ (IFN‐γ) according to the specification of the ELISA kit.
RT‐PCR
Total RNA was extracted from myocardial tissue with TRIzol according to the manufacturer's instructions. Samples were subjected to RT‐PCR with a First Strand cDNA Synthesis Kit. The primers were synthesized by Invitrogen Biotechnology Co., Ltd (Carlsbad, CA, USA), and their base sequence was displayed below.
Western blotting
Total protein was extracted from myocardial tissue, and loaded onto SDS–PAGE at a dose of 40 μg. Electrophoretic deposition was performed under constant voltage of 80 V in spacer gel and 120 V in separating gel. After electrophoresis, proteins were transferred onto PVDF membranes. The membranes were blocked in 5% skimmed milk, and incubated with appropriate antibody in 4° overnight. Then the proteins were detected by enhanced chemiluminescence colouration. GAPDH served as internal control.
Statistical analysis
Data were presented as mean ± SD, and analysed with spss 19.0 software (SPSS, Inc., Chicago, IL, USA). Comparison between various groups was analysed by one‐way analysis of variance, while intragroup comparison was performed by Student's t‐test. Values of P < 0·05 represented statistical difference.
Results
FGL2 causes a deterioration of cardiac function in autoimmune myocarditis rats
At day 40 post inoculation, the cardiac function from each group of rats was examined by ultrasonic cardiogram. In EAM groups, left ventricular end‐diastolic dimension and left ventricular end‐systolic diameter were significantly increased, but left ventricular ejection fraction, left ventricular fractional shortening and heart rate were dramatically decreased, compared with the NC group (P < 0·05). Notably, left ventricular end‐diastolic dimension and left ventricular end‐systolic diameter in the EAM‐FGL2 group were dramatically decreased compared with those in both EAM and EAM‐GFP groups, suggesting that FGL2 could lead to deterioration of cardiac function in autoimmune myocarditis in rats (Table 1 and Fig. 1).
Table 1.
Result of rat colour Doppler ultrasound examination
| NC | EAM | EAM‐GFP | EAM‐FGL2 | |
|---|---|---|---|---|
| LVEDd (cm) | 0·490 ± 0·062 | 0·589 ± 0·050* | 0·603 ± 0·038* , *** | 0·647 ± 0·059* , *** |
| LVEDs (cm) | 0·274 ± 0·031 | 0·377 ± 0·028* | 0·395 ± 0·036* , *** | 0·459 ± 0·056* , ** |
| LVEF (%) | 80·71 ± 3·545 | 71·66 ± 3·228* | 69·53 ± 5·807* , *** | 62·24 ± 3·648* , ** |
| LVFS (%) | 43·87 ± 3·772 | 35·99 ± 2·623* | 34·52 ± 4·309* , *** | 29·30 ± 2·257* , ** |
| HR (bpm) | 342 ± 26·786 | 294 ± 18·375* | 300 ± 14·990* , *** | 255 ± 19·506* , ** |
Abbreviations: HR, heart rate; LVEDd, left ventricular end‐diastolic distance; LVEDs, left ventricular end‐systolic diameter; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening.
*Indicates P < 0·05 while comparing with NC group.
**P < 0·05 while comparing with EAM group.
***P > 0·05 while comparing with EAM group.
Figure 1.
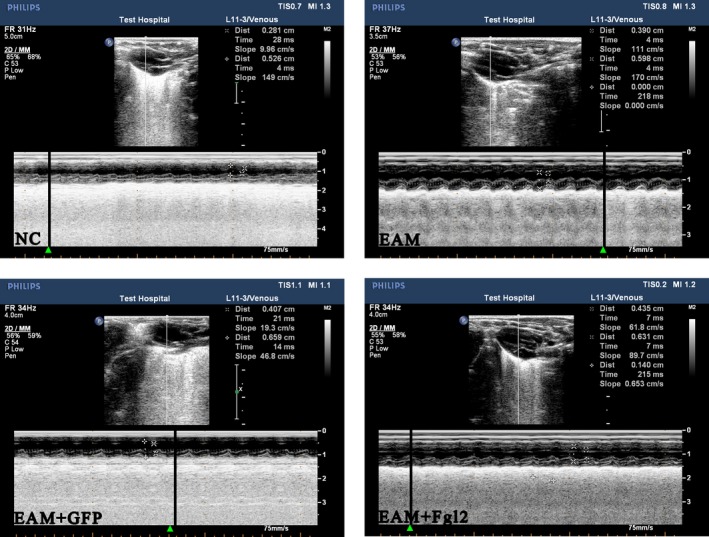
Rat cardiac ultrasound graph. Ultrasonic cardiogram detection has been performed to examine the cardiac function of each rat. Comparing with the control, NC, group, left ventricular end‐diastolic distance (LVEDd) and left ventricular end‐systolic diameter (LVEDs) of rats in experimental groups increased while left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS) with statistical difference. The left ventricular end‐diastolic distance (LVEDd) and left ventricular end‐systolic diameter (LVEDs) of rats in experimental groups were significantly increased compared to those in the normal control group (NC group). However, the left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS) in the EAM groups were decreased statistically (P < 0·05). There was no significant difference between EAM‐GFP group and EAM group (P > 0·05). Notably, LVEDs and LVEF in the EAM‐FGL2 groups were significantly changed compared to these indicators of EAM group, suggesting overexpression of FGL2 further aggravates cardiac function deterioration. [Colour figure can be viewed at wileyonlinelibrary.com]
FGL2 increases inflammatory cell infiltration in autoimmune myocarditis rats
Effective visualization of myocardial inflammatory cell infiltrates revealed an increase in inflammatory cell infiltrates in EAM‐FGL2 compared with EAM and EAM‐GFP (P < 0·05). No inflammation or fibrosis was detected in the NC group at day 40 after inoculation (Fig. 2).
Figure 2.
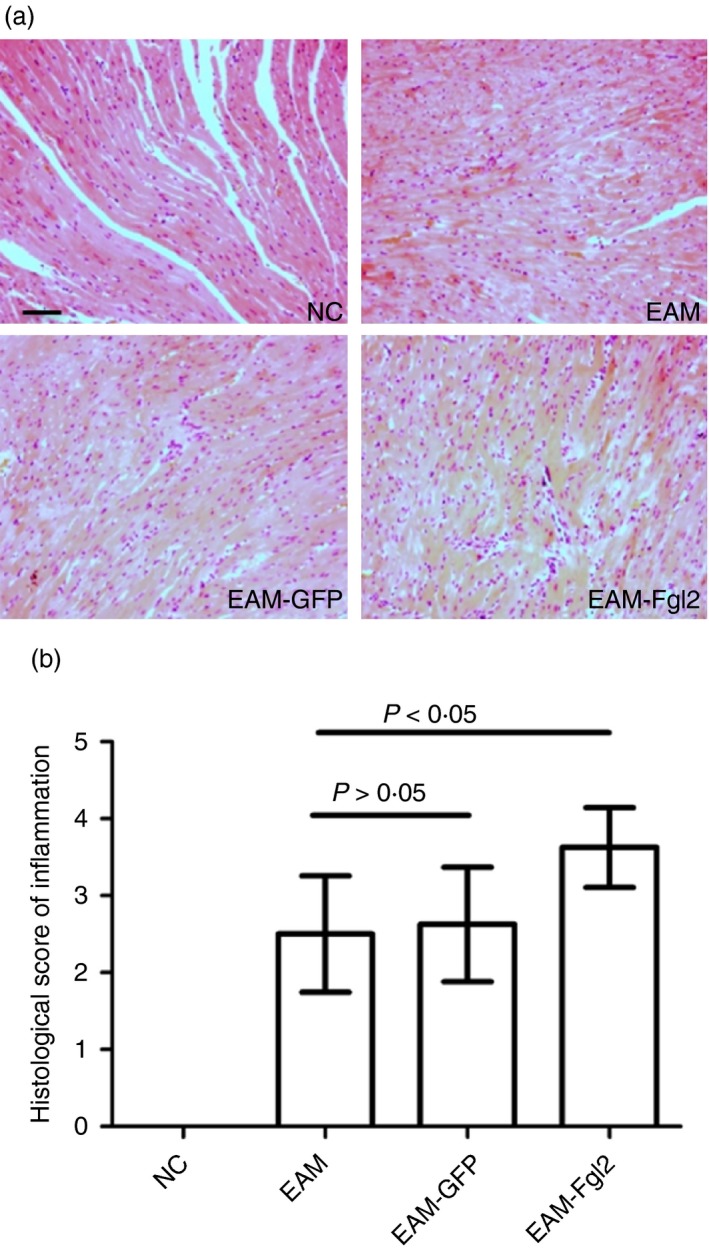
Histopathological observation (haematoxylin & eosin staining, × 200) in myocardial tissue in rats (a). Different extents of inflammatory cell infiltration and myocardial cellular degeneration were observed in myocardial tissue of EAM group, EAM‐FGL2 group and NC group (b) (P < 0·05). Scale bar 50 μm. [Colour figure can be viewed at wileyonlinelibrary.com]
FGL2 promotes the levels of BNP, IL‐6, IL‐17 and IFN‐γ in sera of autoimmune myocarditis rats
Levels of BNP in the EAM, EAM‐GFP and EAM‐FGL2 groups were much higher than in the NC group. BNP levels in the EAM‐FGL2 group were increased compared with those in the EAM and EAM‐GFP groups, which was consistent with the echo observations (P < 0·05). Furthermore, the levels of IL‐6, IL‐17 and IFN‐γ in the EAM‐FGL2 groups were also remarkably increased compared with those in the EAM and EAM‐GFP groups, suggesting that FGL2 causes the cardiac function to deteriorate by increasing the levels of inflammatory cytokines in EAM rats (P < 0·05) (Table 2).
Table 2.
Serum brain natriuretic protein (BNP), interleukin‐6 (IL‐6), IL‐17 and interferon‐γ (IFN‐γ) level of rats in each group
| Groups | BNP (μg/l) | IFN‐γ (ng/l) | IL‐6 (ng/l) | IL‐17 (pg/ml) |
|---|---|---|---|---|
| NC group | 14·73 ± 1·29 | 17·52 ± 2·61 | 12·53 ± 1·47 | 9·13 ± 0·93 |
| EAM‐GFP group | 25·73 ± 3·71* , *** | 38·81 ± 5·94* , *** | 17·11 ± 1·96* , *** | 23·82 ± 2·52* , *** |
| EAM group | 23·91 ± 3·92* | 40·23 ± 5·23* | 19·12 ± 2·18* | 21·19 ± 1·78* |
| EAM‐FGL2 group | 32·28 ± 3·34* , ** | 52·41 ± 7·26* , ** | 25·39 ± 2·94* , ** | 34·21 ± 4·1n2* , ** |
*P < 0·05 while comparing with NC group.
**P < 0·05 while comparing with EAM group.
***P > 0·05 while comparing with EAM group.
FGL2 down‐regulates PD‐1, but up‐regulates RORγt and Foxp3 mRNA and protein levels in autoimmune myocarditis rats
The mRNA levels of FGL2, transcription factor forkhead box P3 (Foxp3) and retinoic acid‐related orphan receptor γt (RORγt) in EAM, EAM‐GFP and EAM‐FGL2 groups were much higher than those in the NC group. The mRNA levels of FGL2, Foxp3 and RORγt in EAM‐FGL2 were the highest among all groups (P < 0·05). However, mRNA levels of PD‐1 in the EAM, EAM‐GFP and EAM‐FGL2 groups were reduced compared with that in NC group. Furthermore, mRNA levels of PD‐1 in the EAM‐FGL2 group were significantly decreased compared with those in the EAM and EAM‐GFP groups (P < 0·05) (Fig. 3).
Figure 3.
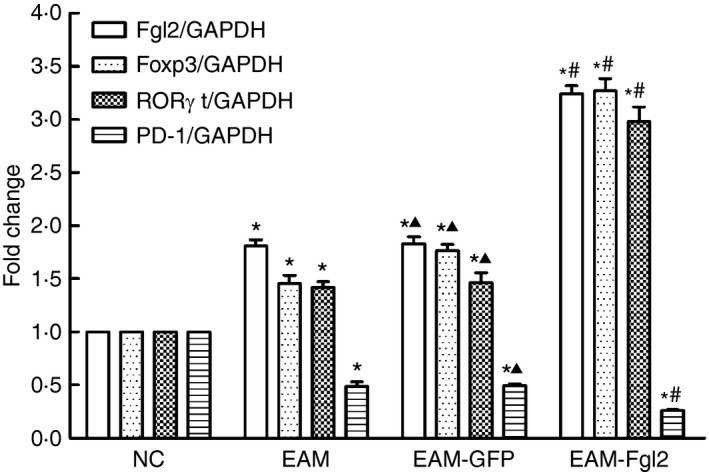
Data of fluorescent quantitative PCR. The increased mRNA expression of fibrinogen‐like protein‐2 (FGL2), transcription factor forkhead box P3 (Foxp3) and retinoic acid‐related orphan receptor γt (ROR γt). In the EAM group was significantly higher than those in the NC group (P < 0·05). In contrast, there was little PD‐1 mRNA expression observed in the NC group, and that in the EAM and EAM‐GFP groups was even less than in the NC group (P < 0·05). PD‐1 expression in the EAM‐FGL2 group was found to be a minimum in all four groups with a statistical significance of P < 0·05. *P < 0·05 while comparing with NC group; #P < 0·05 while comparing with EAM group; P > 0·05 while comparing with EAM group.
Consistent with the mRNA levels, the protein levels of FGL2, Foxp3 and RORγt could be barely detected in the myocardial tissue of the NC group, whereas the expression of these three proteins increased in the other three groups. Among them, protein expression in the EAM‐FGL2 group was the highest (P < 0·05), whereas EAM and EAM‐GFP groups showed no difference. PD‐1 expression in the three experimental groups was higher than in the NC group (P < 0·05). Likewise, PD‐1 expression in the EAM group and EAM‐GFP group displayed no difference, whereas it was lowest with a statistical significance (P < 0·05) in the EAM‐FGL2 group (Fig. 4).
Figure 4.
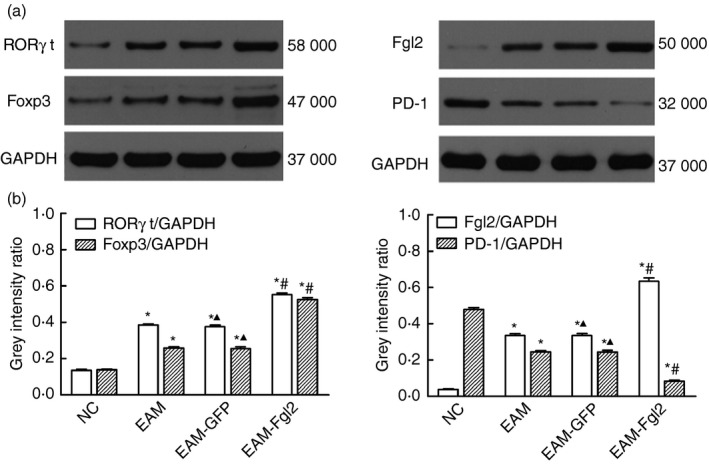
Protein expression assessed by Western blotting. (a) The expression of fibrinogen‐like protein‐2 (FGL2), transcription factor forkhead box P3 (Foxp3) and retinoic acid‐related orphan receptor γt (ROR γt) could be barely examined in the myocardial tissue of the NC group, whereas the expression of these three proteins increased in the other three groups. (b) Quantification of (a). *P < 0·05 while comparing with NC group, # P < 0·05 comparing with EAM group, ▲ P > 0·05 comparing with EAM group.
Discussion
The PD‐1/PD‐L1 pathway is known to regulate the delicate balance between immune tolerance and immune activation. Previous studies have shown that EAM rats in which PD‐1 was knocked down developed auto‐antibodies against much severe myocarditis. In the present study, we first created the EAM rat model by subcutaneous injection with porcine cardiac myosin. To over‐express FGL2 in these EAM rats, lentivirus‐FGL2 was administrated through the tail veins of EAM rats. We found that PD‐1 was decreased in the EAM group, and over‐expression of FGL2 further reduced PD‐1 levels in the EAM‐FGL2 group.
Iwai et al.13 reported an increase in proliferation and infiltration activity of effector T cells that eliminated the virus in adenovirus‐infected PD‐1−/− mouse compared with wild‐type mouse, demonstrating that blockade of the PD‐1 signal pathway enhanced antiviral immunity. However, Seko et al.14 found that PD‐1 inhibited T‐cell activation and thereby reduced myocardial inflammation in the CVB3‐induced VMC model. In the present study, FGL2 over‐expression down‐regulated PD‐1, aggravated myocardial inflammation and reduced cardiac function, which is consistent with the report of Seko et al.14 Given that we observed this phenomenon in the chronic stage of autoimmune myocarditis and given the known T‐cell activatory effects of PD‐1 in response to adenovirus, we hypothesized that PD‐1 plays distinct roles during various stages of VMC. Specifically, we hypothesized that during the acute stage of viral infection, PD‐1 suppresses effector T‐cell proliferation and infiltration thereby delaying the elimination of viruses, resulting in the development of autoimmune myocarditis. During the autoimmune myocarditis stage, we hypothesized that PD‐1 inhibits the development of myocarditis through its immunosuppressive activity.
In the current study, we found that Foxp3 protein and mRNA expression in the experimental groups was higher than in the NC group with the maximum Foxp3 expression observed in the EAM‐FGL2 group. Previous studies have demonstrated that immunosuppressive Treg cells could improve cardiac remodelling.15, 16, 17, 18 This contrasts with our observation that high Foxp3 expression resulted in aggravated myocardial damage. Zhong et al.19 found that PD‐1 reduced the first signal required for T‐cell activation by binding with its ligands B7‐1 or B7‐2, thereby, negatively regulating the T‐cell receptor signalling pathway. Research has found that PD‐1 expression increased in Treg cells.20 In addition, another report found that oestrogen promoted the PD‐1 expression in Treg cells and increased the immunosuppressive action of Treg cells. In PD‐1 knockout mice, Treg cells expressed Foxp3 protein lacking its repressive activity,21 suggesting that PD‐1 played a regulatory role in immunosuppressive activity.
Expression of PD‐1 decreased while Foxp3 expression increased among the three EAM groups. We therefore speculated that reduced PD‐1 negatively regulated the immunosuppressive activity of Treg cells even when Foxp3 was highly expressed, and that FGL2 over‐expression aggravated myocardial damage in EAM rats through inhibition of PD‐1 and immunoreceptor tyrosine‐based inhibitory motif domain expression in Treg cells, which resulted in deteriorated cardiac function. We found that Foxp3 expression level increased with increasing severity of autoimmune myocarditis whereas Treg cell counts were known to be decreased in other autoimmune diseases. This indicated that the biological behaviour of Treg cells in VMC was much more complicated.
ELISA have been performed to determine the IL‐6, IL‐17 and IFN‐γ expression. All of these cytokines were highly expressed in the EAM‐FGL2 group (P < 0·05). Moreover, Western blotting and quantitative PCR assays revealed that RORγt protein and mRNA expression was highest in the EAM‐FGL2 group. These observations suggested that FGL2 over‐expression regulated Th17 cell differentiation at transcriptional level and promoted secretion of IL‐17. The IL‐17 secretion, inhibition of Treg cell immunosuppression, and down‐regulation of PD‐1, sequentially tilted the immune balance towards immunological damage.
To summarize, PD‐1 expression decreased and Foxp3 expression increased in EAM. FGL‐2 over‐expression further increased the expression of Foxp3 and IL‐17 and decreased the expression of PD‐1 in the hearts of these EAM rats. These expression patterns are known to increase the Treg and Th17 cell populations while reducing the T‐cell immunosuppressive activity of Treg cells. We believe that this tilted the immune balance towards immunological damage, mediated in part by the increased number of Th17 cells, whereas Treg cells are functionally impaired in suppressing these T cells thereby failing to resolve the inflammatory process. Therefore, we speculate that FGL2 gene silencing using a lentivirus can be used for the prevention and treatment of autoimmune myocarditis by blocking its chronic persistent period, and eventually preventing progression to dilated cardiomyopathy.
In conclusion, FGL2 plays a role in the progression of autoimmune myocarditis in rats potentially by accelerating Th17 cell proliferation and differentiation, However, the relationship between FGL2, PD‐1 and Th17/Treg cells, and their effects on immunological response during the pathogenesis of autoimmune myocarditis remains unclear, and needs to be further investigated.
Disclosure
The authors have no financial or commercial conflicts of interest.
Acknowledgements
This work was supported by a grant from the National Nature Science Foundation of China (No. 81660067, 81260044) and the Technology Support project of Jiangxi province (No. 2010BSA12000) to Z. Zheng; AHA SDG grant 17SDG33410868 to H. Wu. ZZ and HW conceived and coordinated this study. Yu performed experiments, and ZZ, YY and HW analysed the data. ZZ, RP and HW wrote the paper.
Contributor Information
Zhenzhong Zheng, Email: greateful@163.com.
Hao Wu, Email: hao.wu3@childrens.harvard.edu.
References
- 1. Rose NR. Viral myocarditis. Curr Opin Rheumatol 2016; 28:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T et al Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol 2001; 2:739–45. [DOI] [PubMed] [Google Scholar]
- 3. Li Y, Shi Y, Liao Y, Yan L, Zhang Q, Wang L. Differential regulation of Tregs and Th17/Th1 cells by a sirolimus‐based regimen might be dependent on STAT‐signaling in renal transplant recipients. Int Immunopharmacol 2015; 28:435–43. [DOI] [PubMed] [Google Scholar]
- 4. Park HJ, Park JS, Jeong YH, Son J, Ban YH, Lee BH et al PD‐1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD‐L1 expressed on CD8+ T cells. J Immunol 2015; 194:5801–11. [DOI] [PubMed] [Google Scholar]
- 5. Tarrio ML, Grabie N, Bu DX, Sharpe AH. Lichtman AH.PD‐1 protects against inflammation and myocyte damage in T cell‐mediated myocarditis. J Immunol 2012; 188:4876–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J et al Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD‐1‐deficient mice. Nat Med 2003; 9:1477–83. [DOI] [PubMed] [Google Scholar]
- 7. Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol 2008; 181:2513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu J, Yan J, Rao G, Latha K, Overwijk WW, Heimberger AB et al The duality of FGL2 – secreted immune checkpoint regulator versus membrane‐associated procoagulant: therapeutic potential and implications. Int Rev Immunol 2016; 35:325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zheng Z, Huang H, Liang J. Overexpression of FGL2 deteriorates the heart function of experimental autoimmune myocarditis rats. Eur J Inflamm 2015; 13:66–71. [Google Scholar]
- 10. Baban B, Liu JY, Qin X, Weintraub NL, Mozaffari MS. Upregulation of programmed death‐1 and its ligand in cardiac injury models: interaction with GADD153. PLoS ONE 2015; 10:e0124059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hanawa H, Inomata T, Sekikawa H, Abo T, Kodama M, Izumi T et al Analysis of heart‐infiltrating T‐cell clonotypes in experimental autoimmune myocarditis in rats. Circ Res 1996; 78:118–25. [DOI] [PubMed] [Google Scholar]
- 12. Rezkalla S, Kloner RA, Khatib G, Khatib R. Beneficial effects of captopril in acute coxsackievirus B3 murine myocarditis. Circ 1990; 81:1039–46. [DOI] [PubMed] [Google Scholar]
- 13. Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD‐1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med 2003; 198:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seko Y, Yagita H, Okumura K, Azuma M, Nagai R. Roles of programmed death‐1 (PD‐1)/PD‐1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res 2007; 75:158–67. [DOI] [PubMed] [Google Scholar]
- 15. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L et al The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27:20–1. [DOI] [PubMed] [Google Scholar]
- 16. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003; 4:337–42. [DOI] [PubMed] [Google Scholar]
- 17. Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 2009; 119:2904–12. [DOI] [PubMed] [Google Scholar]
- 18. Dobaczewski M, Xia Y, Bujak M, Gonzalez‐Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol 2010; 176:2177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL. PD‐L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for VH11/VH12 and phosphatidylcholine binding. Eur J Immunol 2007; 37:2405–10. [DOI] [PubMed] [Google Scholar]
- 20. Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. Regulated compartmentalization of programmed cell death‐1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J Immunol 2006; 176:2808–16. [DOI] [PubMed] [Google Scholar]
- 21. Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Treg suppressive activity involves estrogen‐dependent expression of programmed death‐1 (PD‐1). Int Immunol, 2007; 19:337–43. [DOI] [PubMed] [Google Scholar]