

Summary
The use of whole blood gene expression to derive diagnostic biomarkers capable of distinguishing between phenotypically similar diseases holds great promise but remains a challenge. Differential gene expression analysis is used to identify the key genes that undergo changes in expression relative to healthy individuals, as well as to patients with other diseases. These key genes can act as diagnostic, prognostic and predictive markers of disease. Gene expression ‘signatures’ in the blood hold the potential to be used for the diagnosis of infectious diseases, where current diagnostics are unreliable, ineffective or of limited potential. For diagnostic tests based on RNA signatures to be useful clinically, the first step is to identify the minimum set of gene transcripts that accurately identify the disease in question. The second requirement is rapid and cost‐effective detection of the gene expression levels. Signatures have been described for a number of infectious diseases, but ‘clinic‐ready’ technologies for RNA detection from clinical samples are limited, though existing methods such as RT‐PCR are likely to be superseded by a number of emerging technologies, which may form the basis of the translation of gene expression signatures into routine diagnostic tests for a range of disease states.
Keywords: bacterial, bioinformatics, infection, transcriptomics, viral
Introduction
The global analysis of the genome, the epigenome, the transcriptome, the proteome and the metabolome, in the context of various diseases, has led to the improvement of our understanding of disease pathology and has already started reforming disease diagnostics, with the identification of a number of disease‐specific ‘‐omic’ signatures. Analysing data in a high‐throughput quantitative manner has highlighted the way in which the host responds to a number of pathogens. In this review, we will focus on gene expression profiling and biomarker signatures, specifically in the context of infectious diseases, which remain among the leading causes of mortality and disability worldwide. Globally, approximately 15 million of 57 million (over 25%) annual deaths are estimated to be related directly to infectious diseases.1 Newly emerging and re‐emerging pathogens constitute an urgent and ongoing threat to public health throughout the world, while large‐scale, unnecessary use of antibiotics driven by fear of missing severe bacterial infection contributes to the growing problem of antimicrobial resistance. The main focus of this review is diagnostic whole blood gene expression signatures, and their translation into future bedside point‐of‐care (POC) diagnostic tests.
Gene expression is the link between the genotype and the phenotype of an organism, using the information stored in the DNA to produce functional products through transcription (functional RNA species) and translation (proteins).2 Even though information stored as DNA is the same across the cells of an organism, the pattern of genes that are expressed, their level of expression and their isoforms differ between cells according to conditions, so defining the physiological state of each cell. Only a fraction of the approximately 30 000 genes encoded in the human genome are expressed in a given cell at a given time, defining the cell's state. A variety of mechanisms are employed to define which genes are transcribed into RNA and which messenger RNAs (mRNAs) are translated into proteins. The expression levels of a specific gene can be quantified by detecting the presence and measuring the abundance of the final product (either the protein or functional RNA species) or its precursor (typically mRNA). Measuring the amount of mRNA can act as a ‘proxy’ for the overall cellular activity at the molecular level, enabling the elucidation of a patient's response to external stimuli, such as infection, and furthering our understanding of the molecular regulatory mechanisms that underlie disease.
Measuring gene expression
There are various methods for cellular RNA quantification, each associated with a range of advantages and disadvantages. Depending on the nature of the experiment and the actual scientific questions posed, one method may be preferable over the other. Reverse transcription‐quantitative polymerase chain reaction (RT‐qPCR) is the method of preference when a relatively small previously identified set of genes are to be studied. It is still considered the ‘gold standard’ for RNA quantification, making it preferable for validation studies. On the other hand, microarrays and RNA‐sequencing (RNA‐seq) have made whole transcriptome analysis possible, with arrays examining only the transcripts that correspond to probes that are printed a priori on a chip. The opportunities for novel transcript discovery and splicing isoform detection, along with its wider dynamic range, are the main biological reasons why RNA‐seq is superseding microarrays, despite its requirements for ample storage space, high‐level data management, as well as powerful computational infrastructure.3 Comparative analyses between RNA‐seq and microarray techniques have demonstrated that although a larger proportion of genes identified as differentially expressed by RNA‐seq can be subsequently validated by RT‐qPCR, the two methods complement each other in transcriptome profiling.4, 5 Soon after next‐generation sequencing (NGS) was established, the third‐generation sequencing methods emerged (also known as single molecule sequencing methods and next‐next‐generation sequencing). These methods are revolutionizing the life science field in an unprecedented way by reducing the sequencing error rate, the time to results from days to hours, and the overall cost per run.6, 7 As far as the field of transcriptomics is concerned, the impact is anticipated to be particularly high because third‐generation sequencing allows for direct sequencing of RNA molecules while negating the need for cDNA synthesis and amplification steps. Even though sequencing methods confer a natural advantage, offering a wealth of information and the possibility for deeper exploration, gene expression microarrays have been addressing the identification of differentially expressed genes successfully for many years. They have enabled the elucidation of patients’ response to pathogens, so furthering our understanding of the molecular regulatory mechanisms that underlie infection and disease (Fig. 1).
Figure 1.
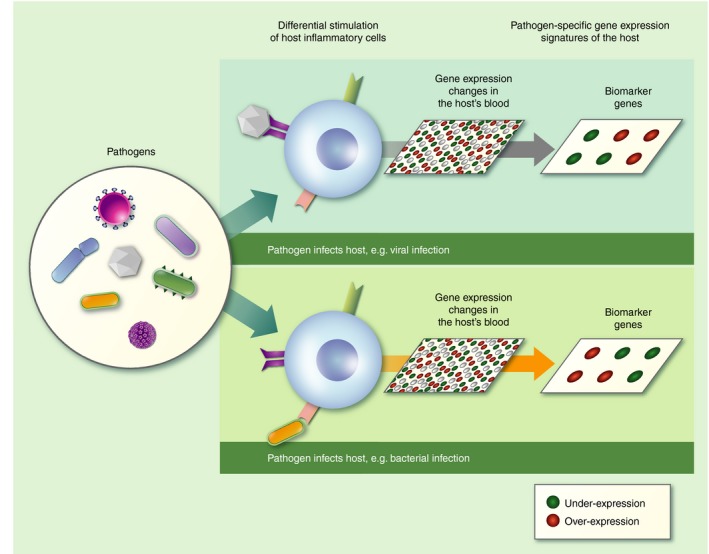
Different pathogens elicit different host transcriptomic responses that can be measured from whole blood.
Sample composition
The aforementioned methods for measuring gene expression allow the identification of differentially expressed genes between cells, tissues, disease states and treatments. They are invaluable tools for shedding light on biological processes and identifying the key molecules that allow discrimination between different disease states.8 Various models are employed to study the response to infection, spanning from cell‐line in vitro experiments and animal models to studies of multi‐level human responses.9 In vitro host‐response studies can monitor cells after the exposure to the pathogen to unravel cell‐specific mechanisms underlying the host response to the pathogen. Cells of preference can either be critical components of the immune system (e.g. natural killer cells, T cells) or pathogen‐specific target cell types and cell lines. Even though in vitro approaches may not be able to fully describe the transcriptional response of the host, they offer a controlled environment and allow for examining changes in expression over time.10, 11, 12, 13 Other studies focus on analysing the transcriptome of human tissue samples from the site of the primary pathogen infection,14 with the capacity to preserve the histological context of the affected tissue.15
These approaches can be of high value when the interest is unravelling the biology or the identification of disease stages. However, to identify biomarkers that can be of clinical significance, it is important to determine patterns of gene expression in easily accessible bodily fluids, such as nasopharyngeal secretions and peripheral whole blood from human patient samples. As distinct patterns of gene expression have been associated with different infectious diseases and disease stages, these patterns allow distinction between patients affected by a disease from healthy controls or between patients with different diseases.16
Blood is not only an accessible tissue, permitting investigation of candidate biomarkers, but blood cells interact with many other tissues of the body, playing a key role in transportation of oxygen, nutrients and waste, as well as in immunity, inflammation, signalling and defence. Molecular profiling of circulating blood cells reflects physiological and pathological events occurring in various different tissues of the body. Hence, whole blood gene expression profiling is not only a means of exploring multiple physiological processes, but also a means of identification of gene expression patterns that offers a broad picture of the organism's health state and overall immunity. Peripheral blood cells share > 80% of the transcriptome with brain, colon, heart, kidney, liver, lung, prostate, spleen and stomach.17 Hence, it has been feasible to derive distinct host response signatures for a variety of diseases from transcriptional profiling of peripheral blood.
Populations and patient characterization
The most pivotal components of human biomarker studies are the experimental design and the clinical recruitment. A widely practiced approach to ensure robustness and reproducibility is to identify biomarkers in a ‘discovery’ patient group, and reproduce findings in an independent ‘validation’ group. Rigorous patient phenotyping in both the discovery and validation groups, using carefully curated data based on the best available tests, is required because a small proportion of false assignment, particularly in the discovery group, can skew correct biomarker identification.18, 19 Although strict inclusion criteria can increase the likelihood that signatures are trained on accurately phenotyped patients, this can be difficult to achieve, particularly where the need for novel biomarkers is made pressing by the lack of an available perfect ‘gold‐standard’ test. This is particularly true for tuberculosis, which is associated with non‐specific symptoms and diagnostic tests that yield positive results that do not necessarily rule out latent tuberculosis infection.20 Recruitment for these studies should be conducted among the most relevant populations, to ensure that the discovery cohort is representative of the population at which the biomarker tests are aimed. In cases where a gene expression signature should be applicable regardless of background endemic infections or HIV infection, both rural and urban populations should be recruited, as well as HIV‐infected and ‐uninfected participants.21, 22 These should then be stratified between groups in addition to age, gender and other factors that may influence clinical presentation, diagnostic workflows and the sample's cellular composition, which should be considered in the analytical pipeline.
Bioinformatics analysis and signature identification
Gene expression microarrays and, more recently, RNA‐seq enable transcriptional profiling of large cohort sizes in a high‐throughput manner, providing highly dimensional data sets that require sophisticated bioinformatics analysis to process and understand. Biomarker signatures identified in the last few years have mostly been derived from microarray data; however, RNA‐seq is emerging as a technique that is rapidly replacing microarrays. It permits hypothesis‐free experimental design, detection of novel transcripts and alternative splicing, gene fusion and allele‐specific expression, while also allowing for simultaneous sequencing of pools of transcripts that may come from different organisms and coexist in the same environment, termed meta‐transcriptomics.23, 24 This is pivotal in studying infection because it allows unravelling of the dynamic interplay between the host and the interacting organisms by measuring their altered gene expression patterns simultaneously. 25
Four main steps constitute the analytical pipelines of gene expression biomarker studies, with many variations according to the experimental needs: (i) data quality control and pre‐processing, (ii) gene biomarker selection, (iii) prediction model implementation and (iv) performance evaluation. Although more complicated post‐experimental data analysis is needed for the RNA‐seq data,26 once the expression data are pre‐processed, machine‐learning methodology leverages the analytical biomarker identification pipeline and prediction assessment workflow downstream. Due to the large number of candidate biomarkers measured and the duplicated information for classification they may provide, the choice of feature (biomarker) selection algorithm is crucial. There is a plethora of feature selection and classification methods that are employed in combination by the computational community to address the problems that gene expression analysis poses.27 In some cases, feature selection can be embedded in the learning algorithms, for example in penalized regression models (elastic net, LASSO) and decision trees.
The goal remains the identification of the smallest possible set of non‐redundant genes with the best possible predictive performance that can be maximally reproducible. Therefore, different functions and metrics are used to evaluate the performance of biomarkers regarding their ability to discriminate between patient groups. Receiver operating characteristic curves are a fundamental tool for evaluating the signatures’ diagnostic performance, whereas positive and negative predictive values incorporate the prevalence of the disease in difference settings in the evaluation.28, 29, 30, 31 Importantly, as signature derivation can be influenced by population selection, sample handling, quantification approaches and analytical tools, external validation in different populations, using alternative quantification methods, is instrumental.
Translation of gene expression signatures into diagnostic, prognostic and theranostic tools
Gene expression signatures derived from whole blood have been reported for several diseases including bacterial and viral infections32, 33, 34 as well as pathogen‐specific diseases including malaria,35, 36 typhoid fever,37, 38 dengue virus infection,23 HIV infection,34 human respiratory syncytial virus infection,24 tularaemia39 and tuberculosis.21, 22, 40 Recently, new parsimonious approaches32 as well as multi‐cohort meta‐analyses41 have identified gene expression signatures comprising minimal numbers of genes, paving the way for easier translation into cost‐effective clinical diagnostic tests.41
It is envisaged that the gene expression signatures described above can be used to diagnose diseases, track disease progression and monitor treatment efficacy.40 Gene expression signatures could enhance earlier disease diagnosis, before the onset of disease symptoms. Earlier diagnosis of infectious diseases improves treatment outcomes and may prevent onwards transmission. Translating gene expression signatures into diagnostic, prognostic or theranostic (i.e. therapy guiding) biomarkers requires clinically useable technology for reliable and reproducible measurement of gene expression. In oncology, gene expression signatures are already being used to distinguish between tumour types, identify the stage of cancer, and predict the efficacy of administering certain therapeutics. For example, the MammaPrint Assay analyses the expression of a set of 70 genes, and can indicate the probability of breast cancer recurring in a given patient.42 This assay involves microarray analysis of tumour sample cDNA. However, this is a relatively costly assay that requires sophisticated laboratory infrastructure and highly trained personnel.
For gene expression signatures to be translated into diagnostic tools used in routine clinical practice, a reduction in cost and processing requirements is needed. One of the most likely ways of achieving this will involve a move from microarray‐based assays towards using RT‐qPCR and related technologies, combined with analysis software that will be able to give a simple readout indicating the disease probability score, based on the concentration of each of the transcripts that comprise a disease signature. Although measuring gene expression using RT‐qPCR on a routine basis would be costly and require highly skilled laboratory technicians, it may be appropriate for use in resource‐rich laboratory settings, provided it is of significant clinical value. Just as gene expression signatures are typically discovered using quantification relative to either a reference gene or other genes, relative quantification of the transcripts will be more feasible to implement than absolute quantification. In addition, the added information provided by absolute quantification will be of little clinical use. However, quantification relative to an appropriate reference gene will be important to normalize expression values between individuals and RNA extraction processes (Fig. 1).
A range of devices that allow for automation of PCR and RT‐PCR have been developed in recent years. Although most of these are designed for the detection of pathogen genomic DNA and RNA, they could potentially be developed for use in gene expression analysis, provided they allow for sensitive, quantitative detection of mRNA. An example of an automated PCR machine is Cepheid's Gene Xpert® system, which allows for the sensitive detection of pathogen genomic DNA, and now RNA.43 The advantages of this system include sample processing within each cartridge, fully automated PCR and a ‘plug and play’ system, where different samples and pathogen species can be examined simultaneously. Similarly, Roche's cobas® Liat RT‐PCR System has been used for the detection of viral RNA at the POC.44 This system involves bench‐top multiplex RT‐PCR analysis of, for example, nasopharyngeal swabs for respiratory pathogens. However, like Roche's cobas® Liat RT‐PCR System, this is not quantitative. FilmArray®, developed by bioMérieux (Marcy l'Etoile, France), is a multiplex PCR system that integrates sample extraction and preparation, amplification and detection. With a turnaround time of approximately 1 hr, and minor manual requirements, this represents a promising platform to which gene expression signatures could be applied. The automated sampling process involves extraction and purification of nucleic acids from unprocessed PAXgene Blood RNA Tube samples, which is followed by nested multiplex PCR. The reported sensitivity is 85–100%, depending on the nucleic acid target. However, this technology is currently used to detect pathogen DNA and has not been used for RT‐PCR, nor is it quantitative. bioMérieux has also developed two technologies that provide quantitative detection of RNA. Argene® performs RT‐PCR to detect viral RNA, and sample extraction can be automated. However, the time to result is 3·5 hr, and so this would not be applicable to POC settings. Although it can perform RT, it has not been used for the detection of mRNA, which may be less abundant than viral RNA. Although promising, the sensitivity and quantification capabilities of these technologies are currently unproven for clinical diagnostic testing using gene expression signatures.
Although the cost and time required for NGS has fallen significantly in recent years, few NGS technologies have been applied for clinical diagnostics, and even fewer for gene expression analysis. However, in contrast to previous technologies, NGS offers flexible open platforms for simultaneous detection of nucleic acids from both the host and the pathogen, which can facilitate even more accurate disease diagnosis. A key recent advance in NGS is the development of Oxford Nanopore's MinION sequencer, which allows for portable and affordable sequencing (Oxford Science Park, Oxford, UK). The MinION has been used for the epidemiological surveillance of the Ebola virus 45 and more recently the Zika virus in field settings.46 As results are semi‐quantitative, and Oxford Nanopore have now announced that it is possible to sequence RNA using the MinION, this technology holds great potential as a new contender in gene expression analysis in close to POC settings.
Isothermal amplification
Although RT‐qPCR is the reference standard method for gene expression analysis, its use at the POC is limited by the requirement for a thermocycler and complex data analysis. In contrast, isothermal amplification techniques, which are typically conducted at one specified temperature, do not require thermocycling and so have fewer equipment requirements. Isothermal amplification nucleic acid detection strategies are attractive for molecular POC diagnostics, and can be achieved in a number of ways, each exploiting the presence of target RNA or DNA to trigger certain processes, such as:
Primer extension, followed by a mechanism that frees the template (target), allowing it to be recycled and used again for primer extension (e.g. helicase displacement assay, strand displacement assay)
Toehold‐mediated strand displacement or hybridization to a hairpin loop or two DNA probes, that leads to a change in secondary structure (such as the formation of a G‐quadruplex, which has a peroxidase‐like catalytic function)
Hybridization of two DNA probes that can then be ligated (e.g. ligase chain reaction)
The aggregation of DNA probes, providing a ratiometric increase in signal (e.g. branched DNA assays)
Some isothermal amplification strategies, such as loop‐mediated isothermal amplification, rolling circle amplification, strand displacement amplification and helicase dependent amplification, require a DNA template to proceed. Detection of mRNA using these strategies would therefore require an initial reverse transcription step before the amplification reaction. Others, such as nucleic acid sequence‐based amplification and exponential amplification reaction are able to use RNA sequences as the template for amplification and therefore negate the requirement of the RT step. The Alere™ i‐technology uses a type of isothermal amplification called nicking enzyme amplification reaction, which uses RNA as a direct target and has a turnaround time of < 15 min.47 Isothermal amplification strategies have numerous advantages compared with PCR, but it should be noted that lack of specificity is a key issue in the design of such assays and should be a major consideration in their use for gene expression analysis.
Emerging technologies
Many emerging technologies could enable sensitive, yet also quantitative, detection of gene expression signatures for diagnostic purposes. Many emerging techniques and materials have been employed for the detection of nucleic acids, such as electrochemical detection,48, 49 microfluidic‐based lab‐on‐a‐chip devices,50, 51, 52 nanomaterials53, 54 and the recently introduced Oxford Nanopore MinION sequencing platform complemented by real‐time bioinformatics analytical tools.55, 56, 57 In addition, molecular based methods are rapidly emerging. For example, the re‐purposing of CRISPR‐based systems for highly specific and sensitive detection of RNA and DNA was recently demonstrated. The ‘SHERLOCK’ (Specific High‐Sensitivity Enzymatic Reporter unLOCKing) platform successfully detected low quantities of pathogen and human RNA/DNA, with single base mismatch specificity. It is not yet clear whether this system has the quantitative accuracy required to compare relative expression levels of multiple genes, which would be needed for the interrogation of host RNA signatures.58
Nanomaterials are a large and growing class of materials that are now being used to make extraordinarily sensitive diagnostic tests. There is a multitude of nanomaterials that can be used for the purposes of RNA sensing, including gold nanoparticles (AuNPs),59 silica nanoparticles,60 magnetic nanoparticles61 and electrochemically active nanomaterials such as zinc oxide.62 Of these, AuNPs are the best characterized and understood. The ease with which AuNPs can be functionalized with antibodies, and their strong red colour, has made them an important component of immunochromatography assays, also known as lateral flow tests. These are generally low‐cost, paper‐based devices that satisfy many of the ASSURED criteria for POC diagnostic tests.63, 64 However, although these types of test are appropriate for immunoassays, their application for nucleic acid detection is more challenging. Although isothermal amplification assays can be conducted on paper,65 these tests are generally for nucleic acid detection alone, whereas quantitative analysis of nucleic acid concentration has yet to be achieved.
Fluorescence‐based homogeneous assays that require no washing steps represent a promising alternative for quantitative detection of nucleic acids at the POC. Fluorescent nanoparticles, such as quantum dots (QDs), have been used for these purposes due to their bright fluorescence and amenability to multiplexing. A homogeneous, QD‐based RNA sensing assay was recently reported, which gave a fluorescent readout in proportion to the concentration of target RNA present.53 Traditionally, these signals would need to be recorded using costly fluorescence spectroscopy methods, which is not suitable for use in POC settings. However, recent advances in digital camera technology, as well as the increased availability of blue LEDs and lasers,66 have resulted in dramatically reduced costs for fluorescence imaging systems. In 2012, Xie et al.67 showed that it was possible to detect the Mycobacterium tuberculosis‐specific enzyme BlaC in sputum using the camera of a cell phone, using a simple LED as a light source. BlaC‐specific fluorogenic substrates were designed, which showed a 100‐fold increase in fluorescence intensity in the presence of BlaC. More recently, QD fluorescence was measured using an LED to excite the QDs, capturing emission with a smartphone camera and analysing fluorescence intensity with a custom‐made smartphone application.68 The powerful cameras of current mobile phone technology mean that the results of fluorescence‐based tests can be analysed by mobile phones and translated into user‐friendly, portable and low‐cost devices.69
A novel advance in gene expression analysis was performed by Geiss et al.,54 who reported in 2008 the development of the NanoString nCounter system to detect multiple mRNA transcripts. Colour‐coded probe pairs were able to hybridize with complementary mRNA, resulting in tripartite structures made up of the target mRNA, a specific reporter probe and a capture probe. Affinity purification results in capture on a surface. Following immobilization, fluorescence imaging was able to reveal the order of fluorescent segments in the colour‐coded reporter probes, allowing the calculation of the concentration of a particular mRNA transcript. Detection limits of below 0·5 fM were reported, and the linear dynamic range was over 500‐fold. NanoString technology has been used successfully in the diagnosis of patients with raised interferon response genes (Aicardi–Goutières syndrome),70 and for the profiling of children with septic shock.71 However, this method relies on sophisticated equipment, which would be inappropriate for use at or near to POC settings. It may, however, be useful for the detection of extremely low‐level transcripts where the technology is readily available, and for diagnosis of conditions without alternative accurate tests, where rapid turn‐around times are not required.
Various challenges remain for the nanodiagnostics field, particularly with regards to optimizing the surface chemistry of nanomaterials in order to achieve greater predictability of their behaviour and avoid undesirable interactions with other biomolecules that may be present in complex matrices. A key challenge for the QD field is the toxicity of the chemicals that make up QD cores, which are typically heavy metals, such as cadmium selenide, that consequently must be handled and disposed of with care. Risks are mitigated by the minute amounts required for diagnostic assays and by using newly developed QDs that do not contain heavy metals. These QDs are composed of small organic molecules and polymers (p‐dots) and so show lower toxicity than their heavy metal counterparts, yet retain their extraordinary optical properties.72 Given these advances, it is evident that combining the advantageous optical properties of nanomaterials with programmable molecular biology approaches such as isothermal amplification could enable the design of a new class of sensitive, robust and versatile diagnostic tests.
Sample to answer: a promising reality or a long way off?
As the majority of gene expression analysis studies are based on RNA isolated from whole blood, this is the most likely analytical sample that could be used for detecting gene expression signatures. Importantly, genomic DNA, which shares sequence homology with the transcripts comprising gene expression signatures, is removed, as well as many molecules that could interfere with mRNA detection assays. In addition, most mRNA is intracellular, and so a cell lysis step would be necessary for their detection. RNA is less stable than DNA and can be degraded by endogenous RNases, which are present on the skin as well as in blood and tissue. Careful handling of samples is therefore essential to conserve RNA integrity. RNA is usually purified from whole blood by first using a detergent to lyse cells and proteinase K to degrade proteins. Following the addition of ethanol or isopropanol, which precipitates nucleic acids, this lysed sample is typically applied to a column, which specifically binds to nucleic acids. After a washing step, a DNase is applied to the column, which hydrolyses the genomic DNA that has bound to it. The RNA can then be eluted by dissolving it in an appropriate solvent. RNase inhibitors are often present throughout this process to prevent RNA degradation.73
Current techniques for RNA purification are labour‐intensive and ill‐suited to POC or resource‐limited settings. However, available technologies that detect transcript abundance are amenable to use in diagnostic laboratories, and existing technologies including qPCR and NanoString are being adopted for diagnostic testing of infectious and non‐infectious conditions where the accurate rather than rapid diagnostic testing is needed. Recent studies have shown that microfluidic devices can be used to perform a one‐step RNA purification and RT‐PCR assay, with minimal input from laboratory personnel.52 In future, these sample preparation processes will require less time and handling and may prove to be sufficiently robust to be used routinely.
Conclusion
As we continue to explore the most reliable biomarkers and their translation into routine diagnostic tests, there is growing insight to consider from the initial study design to the level of molecular detection. In recent years, numerous methodologies have emerged for measuring gene expression biomarkers that make their translation into practice more feasible. Although there are still many hurdles to overcome in the introduction of gene expression biomarkers into mainstream clinical decision‐making, the next decade will probably see the advent of gene expression signatures as diagnostic biomarkers in clinical practice for a range of diseases.
Disclosures
No competing interests.
References
- 1. Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re‐emerging infectious diseases. Nature 2004; 430:242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. In: Science G, ed. Molecular Biology of the Cell. New York, NY: Garland Science, 2002: http://www.garlandscience.com/product/isbn/0815341059. [Google Scholar]
- 3. Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA‐seq. Nat Methods 2011; 8:469–77. [DOI] [PubMed] [Google Scholar]
- 4. Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA‐seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 2008; 18:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kogenaru S, Qing Y, Guo Y, Wang N. RNA‐seq and microarray complement each other in transcriptome profiling. BMC Genom 2012; 13:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lipson D, Raz T, Kieu A, Jones DR, Giladi E, Thayer E et al Quantification of the yeast transcriptome by single‐molecule sequencing. Nat Biotechnol 2009; 27:652–8. [DOI] [PubMed] [Google Scholar]
- 7. Schadt EE, Turner S, Kasarskis A. A window into third‐generation sequencing. Hum Mol Genet 2010; 19(R2):R227–40. [DOI] [PubMed] [Google Scholar]
- 8. Murray D, Doran P, MacMathuna P, Moss AC. In silico gene expression analysis – an overview. Mol Cancer, 2007; 6:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waddell SJ, Butcher PD, Stoker NG. RNA profiling in host–pathogen interactions. Curr Opin Microbiol 2007; 10:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, Belcher CE et al Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci USA 2002; 99:972–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran P, Ahmad R, Xu JW, Ahmad A, Menezes J. Host's innate immune response to fungal and bacterial agents in vitro: up‐regulation of interleukin‐15 gene expression resulting in enhanced natural killer cell activity. Immunology 2003; 109:263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pathan N, Hemingway CA, Alizadeh AA, Stephens AC, Boldrick JC, Oragui EE et al Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 2004; 363:203–9. [DOI] [PubMed] [Google Scholar]
- 13. Kim HS, Choi EH, Khan J, Roilides E, Francesconi A, Kasai M, Sein T et al Expression of genes encoding innate host defense molecules in normal human monocytes in response to Candida albicans . Infect Immun 2005; 73:3714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Q, Smith AJ, Schacker TW, Carlis JV, Duan L, Reilly CS, Haase AT. Microarray analysis of lymphatic tissue reveals stage‐specific, gene expression signatures in HIV‐1 infection. J Immunol 2009; 183:1975–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crosetto N, Bienko M, van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet 2015; 16:57–66. [DOI] [PubMed] [Google Scholar]
- 16. Liu M, Popper SJ, Rubins KH, Relman DA. Early days: genomics and human responses to infection. Curr Opin Microbiol 2006; 9:312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med 2006; 147:126–32. [DOI] [PubMed] [Google Scholar]
- 18. Cox JA, Lukande RL, Lucas S, Nelson AM, Van Marck E, Colebunders R. Autopsy causes of death in HIV‐positive individuals in sub‐Saharan Africa and correlation with clinical diagnoses. AIDS Rev 2010; 12:183–94. [PubMed] [Google Scholar]
- 19. Ansari NA, Kombe AH, Kenyon TA, Hone NM, Tappero JW, Nyirenda ST, Binkin NJ et al Pathology and causes of death in a group of 128 predominantly HIV‐positive patients in Botswana, 1997–1998. Int J Tuberc Lung Dis 2002; 6:55–63. [PubMed] [Google Scholar]
- 20. Deffur A, Wilkinson RJ, Coussens AK. Tricks to translating TB transcriptomics. Ann Transl Med 2015; 3(Suppl 1):S43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N et al Detection of tuberculosis in HIV‐infected and ‐uninfected African adults using whole blood RNA expression signatures: a case–control study. PLoS Med 2013; 10:e1001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anderson ST, Kaforou M, Brent AJ, Wright VJ, Banwell CM, Chagaluka G et al Diagnosis of childhood tuberculosis and host RNA expression in Africa. N Engl J Med 2014; 370:1712–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Westermann AJ, Gorski SA, Vogel J. Dual RNA‐seq of pathogen and host. Nat Rev Microbiol 2012; 10:618–30. [DOI] [PubMed] [Google Scholar]
- 24. Conesa A, Madrigal P, Tarazona S, Gomez‐Cabrero D, Cervera A, McPherson A et al A survey of best practices for RNA‐seq data analysis. Genome Biol 2016; 17:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li T, Zhang C, Ogihara M. A comparative study of feature selection and multiclass classification methods for tissue classification based on gene expression. Bioinformatics 2004; 20:2429–37. [DOI] [PubMed] [Google Scholar]
- 26. Altman DG, Bland JM. Diagnostic tests. 1: sensitivity and specificity. BMJ 1994; 308:1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Altman DG, Bland JM. Diagnostic tests 2: predictive values. BMJ 1994; 309:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altman DG, Bland JM. Diagnostic tests 3: receiver operating characteristic plots. BMJ 1994; 309:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deeks JJ, Altman DG. Diagnostic tests 4: likelihood ratios. BMJ 2004; 329:168–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herberg JA, Kaforou M, Wright VJ, Shailes H, Eleftherohorinou H, Hoggart CJ et al Diagnostic test accuracy of a 2‐transcript host RNA signature for discriminating bacterial vs viral infection in febrile children. JAMA 2016; 316:835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zaas AK, Chen M, Varkey J, Veldman T, Hero AO 3rd, Lucas J et al Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 2009; 6:207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahajan P, Kuppermann N, Mejias A, Suarez N, Chaussabel D, Casper TC et al Association of RNA biosignatures with bacterial infections in febrile infants aged 60 days or younger. JAMA 2016; 316:846–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffiths MJ, Shafi MJ, Popper SJ, Hemingway CA, Kortok MM, Wathen A et al Genomewide analysis of the host response to malaria in Kenyan children. J Infect Dis 2005; 191:1599–611. [DOI] [PubMed] [Google Scholar]
- 34. Colborn JM, Ylöstalo JH, Koita OA, Cissé OH, Krogstad DJ. Human gene expression in uncomplicated Plasmodium falciparum malaria. J Immunol Res 2015; 2015:162639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khoo SK, Petillo D, Parida M, Tan AC, Resau JH, Obaro SK. Host response transcriptional profiling reveals extracellular components and ABC (ATP‐binding cassette) transporters gene enrichment in typhoid fever‐infected Nigerian children. BMC Infect Dis 2011; 11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson LJ, Dunstan SJ, Dolecek C, Perkins T, House D, Dougan G et al Transcriptional response in the peripheral blood of patients infected with Salmonella enterica serovar Typhi. Proc Natl Acad Sci U S A 2009; 106:22433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andersson H, Hartmanová B, Bäck E, Eliasson H, Landfors M, Näslund L et al Transcriptional profiling of the peripheral blood response during tularemia. Genes Immun 2006; 7:503–13. [DOI] [PubMed] [Google Scholar]
- 38. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T et al An interferon‐inducible neutrophil‐driven blood transcriptional signature in human tuberculosis. Nature 2010; 466:973–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loke P, Hammond SN, Leung JM, Kim CC, Batra S, Rocha C et al Gene expression patterns of dengue virus‐infected children from Nicaragua reveal a distinct signature of increased metabolism. PLoS Negl Trop Dis 2010; 4:e710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mejias A, Dimo B, Suarez NM, Garcia C, Suarez‐Arrabal MC, Jartti T et al Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med 2013; 10:e1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sweeney TE, Braviak L, Tato CM, Khatri P. Genome‐wide expression for diagnosis of pulmonary tuberculosis: a multicohort analysis. Lancet Respiratory Medicine 2016; 4:213–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buyse M, Loi S, van’t Veer L , Viale G, Delorenzi M, Glas AM et al Validation and clinical utility of a 70‐gene prognostic signature for women with node‐negative breast cancer. J Natl Cancer Inst 2006; 98:1183–92. [DOI] [PubMed] [Google Scholar]
- 43. Dize L, West S, Williams JA, Van Der Pol B, Quinn TC, Gaydos CA. Comparison of the Abbott m2000 realtime CT assay and the cepheid GeneXpert CT/NG assay to the Roche Amplicor CT assay for detection of Chlamydia trachomatis in ocular samples from Tanzania. J Clin Microbiol 2013; 51:1611–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nolte FS, Gauld L, Barrett SB. Direct comparison of alere i and cobas® liat influenza A and B tests for rapid detection of influenza virus infection. J Clin Microbiol, 2016; 54:2763–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L et al Real‐time, portable genome sequencing for Ebola surveillance. Nature 2016; 530:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faria NR, Sabino EC, Nunes MR, Alcantara LC, Loman NJ, Pybus OG. Mobile real‐time surveillance of Zika virus in Brazil. Genome Med 2016; 8:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nie S, Roth RB, Stiles J, Mikhlina A, Lu X, Tang YW, Babady NE. Evaluation of Alere i Influenza A&B for rapid detection of influenza viruses A and B. J Clin Microbiol 2014; 52:3339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin M, Song P, Zhou G, Zuo X, Aldalbahi A, Lou X et al Electrochemical detection of nucleic acids, proteins, small molecules and cells using a DNA‐nanostructure‐based universal biosensing platform. Nat Protoc 2016; 11:1244–63. [DOI] [PubMed] [Google Scholar]
- 49. Xiong E, Zhang X, Liu Y, Zhou J, Yu P, Li X, Chen J. Ultrasensitive electrochemical detection of nucleic acids based on the dual‐signaling electrochemical ratiometric method and exonuclease III‐assisted target recycling amplification strategy. Anal Chem 2015; 87:7291–6. [DOI] [PubMed] [Google Scholar]
- 50. Easley CJ, Karlinsey JM, Bienvenue JM, Legendre LA, Roper MG, Feldman SH et al A fully integrated microfluidic genetic analysis system with sample‐in‐answer‐out capability. Proc Natl Acad Sci U S A 2006; 103:19272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spizz G, Young L, Yasmin R, Chen Z, Lee T, Mahoney D et al Rheonix CARD® technology: an innovative and fully automated molecular diagnostic device. Point Care 2012; 11:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shaw KJ, Hughes EM, Dyer CE, Greenman J, Haswell SJ. Integrated RNA extraction and RT‐PCR for semi‐quantitative gene expression studies on a microfluidic device. Lab Invest 2013; 93:961–6. [DOI] [PubMed] [Google Scholar]
- 53. Gliddon HD, Howes PD, Kaforou M, Levin M, Stevens MM. A nucleic acid strand displacement system for the multiplexed detection of tuberculosis‐specific mRNA using quantum dots. Nanoscale 2016; 8:10087–95. [DOI] [PubMed] [Google Scholar]
- 54. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL et al Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol 2008; 26:317–25. [DOI] [PubMed] [Google Scholar]
- 55. Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T et al The potential and challenges of nanopore sequencing. Nat Biotechnol 2008; 26:1146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cao MD, Nguyen SH, Ganesamoorthy D, Elliott AG, Cooper MA, Coin LJ. Scaffolding and completing genome assemblies in real‐time with nanopore sequencing. Nat Commun 2017; 8:14515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC et al Decoding long nanopore sequencing reads of natural DNA. Nat Biotechnol 2014; 32:829–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re‐emerging infectious diseases. Nature 2004; 430:242–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Veigas B, Machado D, Perdigão J, Portugal I, Couto I, Viveiros M, Baptista PV. Au‐nanoprobes for detection of SNPs associated with antibiotic resistance in Mycobacterium tuberculosis . Nanotechnology 2010; 21:415101. [DOI] [PubMed] [Google Scholar]
- 60. Qin DL, He XX, Wang KM, Tan WH. Using fluorescent nanoparticles and SYBR Green I based two‐color flow cytometry to determine Mycobacterium tuberculosis avoiding false positives. Biosens Bioelectron 2008; 24:626–31. [DOI] [PubMed] [Google Scholar]
- 61. Lee WG, Kim YG, Chung BG, Demirci U, Khademhosseini A. Nano/Microfluidics for diagnosis of infectious diseases in developing countries. Adv Drug Deliv Rev 2010; 62:449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Das M, Sumana G, Nagarajan R, Malhotra BD. Application of nanostructured ZnO films for electrochemical DNA biosensor. Thin Solid Films 2010; 519:1196–201. [Google Scholar]
- 63. Mabey D, Peeling RW, Ustianowski A, Perkins MD. Diagnostics for the developing world. Nat Rev Microbiol 2004; 2:231–40. [DOI] [PubMed] [Google Scholar]
- 64. Kettler H, White K, Hawkes S. Mapping the landscape of diagnostics for sexually transmitted infections. Geneva: World Health Organization, 2004. [Google Scholar]
- 65. Liu M, Hui CY, Zhang Q, Gu J, Kannan B, Jahanshahi‐Anbuhi S et al Target‐induced and equipment‐free DNA amplification with a simple paper device. Angew Chem Int Ed Engl 2016; 55:2709–13. [DOI] [PubMed] [Google Scholar]
- 66. Yager P, Domingo GJ, Gerdes J. Point‐of‐care diagnostics for global health. Annu Rev Biomed Eng 2008; 10:107–44. [DOI] [PubMed] [Google Scholar]
- 67. Xie H, Mire J, Kong Y, Chang M, Hassounah HA, Thornton CN et al Rapid point‐of‐care detection of the tuberculosis pathogen using a BlaC‐specific fluorogenic probe. Nat Chem 2012; 4:802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Petryayeva E, Algar WR. Multiplexed homogeneous assays of proteolytic activity using a smartphone and quantum dots. Anal Chem 2014; 86:3195–202. [DOI] [PubMed] [Google Scholar]
- 69. Zhu H, Isikman SO, Mudanyali O, Greenbaum A, Ozca A. Optical imaging techniques for point‐of‐care diagnostics. Lab Chip 2013; 13:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Armangue T, Orsini JJ, Takanohashi A, Gavazzi F, Conant A, Ulrick N et al Neonatal detection of Aicardi–Goutières syndrome by increased C26:0 lysophosphatidylcholine and interferon signature on newborn screening blood spots. Mol Genet Metab, 2017; pii: S1096‐7192(17)30369‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wong HR, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, Bigham MT et al Developing a clinically feasible personalized medicine approach to pediatric septic shock. Am J Respir Crit Care Med 2015; 191:309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Massey M, Wu M, Conroy EM, Algar WR. Mind your P's and Q's: the coming of age of semiconducting polymer dots and semiconductor quantum dots in biological applications. Curr Opin Biotechnol 2015; 34:30–40. [DOI] [PubMed] [Google Scholar]
- 73. Feezor RJ, Baker HV, Mindrinos M, Hayden D, Tannahill CL, Brownstein BH et al Whole blood and leukocyte RNA isolation for gene expression analyses. Physiol Genomics 2004; 19:247–54. [DOI] [PubMed] [Google Scholar]